

Abstract
Three-dimensional (3D) tissue models have significantly improved our understanding of structure/function relationships and promise to lead to new advances in regenerative medicine. However, despite the expanding diversity of 3D tissue fabrication methods, approaches for functional assessment have been relatively limited. Here, we describe the fabrication of microtissue (μ-tissue) suspensions and their quantitative evaluation with techniques capable of analyzing large sample numbers and performing multiplexed parallel analysis. We applied this platform to 3D μ-tissues representing multiple stages of liver development and disease including: embryonic stem cells, bipotential hepatic progenitors, mature hepatocytes, and hepatoma cells photoencapsulated in polyethylene glycol hydrogels. Multiparametric μ-tissue cytometry enabled quantitation of fluorescent reporter expression within populations of intact μ-tissues (n≥102-103) and sorting-based enrichment of subsets for subsequent studies. Further, 3D μ-tissues could be implanted in vivo, respond to systemic stimuli, retrieved and quantitatively assessed. In order to facilitate multiplexed ‘pooled’ experimentation, fluorescent labeling strategies were developed and utilized to investigate the impact of μ-tissue composition and exposure to soluble factors. In particular, examination of drug/gene interactions on collections of 3D hepatoma μ-tissues indicated synergistic influence of doxorubicin and knockdown of the anti-apoptotic gene BCL-XL. Collectively, these studies highlight the broad utility of μ-tissue suspensions as an enabling approach for high n, populational analysis of 3D tissue biology in vitro and in vivo.
Introduction
Cellular functions in vivo are coordinately influenced by an interactive 3D framework of microscale signals, defined by interactions with nearby cells and the mechanical and chemical properties of the surrounding extracellular matrix and local soluble environment. Improved control over the 3D cellular microenvironment has been instrumental to the understanding of fundamental cellular processes and progress towards functional tissue models for drug development and regenerative therapies 1, 2. Studies utilizing natural or synthetic materials have begun to elucidate the role of tissue structure and 3D organization of niche cues on differentiation 3-9, proliferation and migration 10, matrix deposition 11, 12, development 13, 14 and pathogenesis 15, 16. New fabrication technologies 17, 18 and combinatorial material approaches 9, 19 have emerged to aid in the rational design and rapid development of engineered tissues. Despite these advances in biomaterials, investigators have been forced to rely on low-throughput technologies to assess biological sequelae in engineered tissues (e.g. fluorescent imaging, histological sections, or RNA extraction). Thus, while promising 3D tissues models are rapidly emerging, their implementation is hindered by an effective bottleneck, whereby capabilities for the evaluation of large sample numbers have been limited.
One approach to expand the capacity of tissue response assessment (number of replicates, unique conditions) is the fabrication of tissue microarrays 20, 21. For example, robotic spotting has been used to create arrays of cells in 2D and 3D configurations interfaced with extracellular matrix or soluble enzyme combinations 22-24. In addition, microfabrication technology has been adopted to array cell aggregates by electric field manipulation 25, 26 and to elucidate how mechanical forces influence the contractility and phenotype of 3D cell-hydrogel structures 27. Despite the improved throughput, these in vitro systems require a surface that is spatially templated, which restricts handling options and limits translation to the in vivo setting. Elsewhere, soft-lithography and photolithography-based techniques have enabled the fabrication of sub-mm sized cellular hydrogel structures, which can be manipulated and positioned for assessment using microfluidics or surface tension-driven assembly 28-32. These developments offer the capability of studying collections of communicating cells as well as the potential to scale-up such cellular units for tissue engineering. However, functional assessment of these tissues has still been predicated upon serial imaging, which can be laborious, time-consuming and highly variable within sample populations.
In this report, we describe the development of a platform that facilitates the high n, quantitative, multiplex assessment of suspensions of miniaturized 3D encapsulated cellular constructs (μ-tissues). 3D μ-tissues are designed to represent small-scale units of multicellular tissue and are engineered by photoencapsulating 500-1000 cells in encoded polyethylene glycol biomaterial hydrogels. Suspensions of 3D μ-tissues are uniquely amenable to functional analyses via complementary high-throughput ‘readouts’ (e.g. flow analysis, flow sorting, optical multiplexing), and within multiple formats (e.g. multi-well, in vivo) (Fig. 1a). In a process analogous to flow cytometry for single cell biology, this platform enables populational analysis and sorting of multicellular engineered μ-tissues. Furthermore, the multiplexing strategies developed in this study permit pooled experimentation that lay the groundwork for large-scale screens of 3D phenotypes. In these studies, we illustrate the utility of this platform in a set of liver biology applications currently in need of expanded 3D analysis capabilities, including: cellular toxicity, stem cell differentiation and cancer responsiveness to gene/drug interactions.
Figure 1.
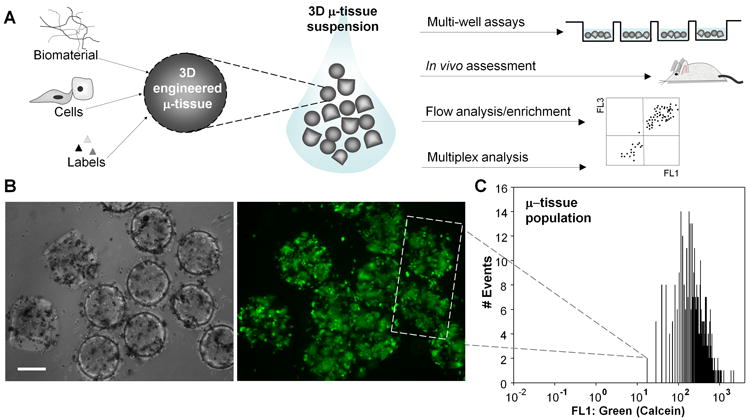
3D μ-tissue suspensions. A) Schematic depicting the manipulation of 3D μ-tissue suspensions incorporating modular components of the microenvironment (biomaterial, cellular, identifying labels). Examination of μ-tissue populations enables 3D μ-tissue assessment in multiple formats (multiwell, in vivo) and integration with flow cytometric and multiplex analysis. Cytometric measurement and manipulation capabilities for intact μ-tissues are further outlined in SI Fig. 1. B) Liver (primary hepatocyte/J2-3T3 fibroblast) μ-tissue stained with the cellular calcein AM viability dye and imaged by phase contrast (left) and epifluorescent (right) microscopy. C) Viability profile of a representative 3D liver μ-tissue population, based on quantitative flow analysis of n=338 calcein-labeled liver μ-tissues. (Scale bar, 200 μm.)
Results
Quantitative assessment of 3D μ-tissue suspensions
To fabricate populations of μ-tissues, we adapted a photopatterning procedure in which a solution of photocrosslinkable synthetic pre-polymer (polyethylene glycol-diacrylate, PEG-DA) containing cells is exposed to UV light through a mask with defined features, and μ-tissues are subsequently harvested into media. We chose a PEG-based system due to its biocompatibility, hydrophilicity and highly tunable chemical and mechanical properties 33. We and others have previously studied chemical and structural PEG-DA modifications in order to influence the viability and function of numerous encapsulated cell types for tissue engineering 7, 34-36. In order to evaluate the capabilities of the μ-tissue platform for 3D biology, we focused on the liver as a case study and explored the responses of μ-tissues representing multiple stages of hepatic development and disease. As a first demonstration, we fabricated μ-tissues comprised of primary rat hepatocytes co-cultured with mouse embryonic fibroblasts known to support hepatocyte function 7, and confirmed survival of encapsulated cells (Figure 1b). Each hepatocyte μ-tissue was loaded with ∼500 mixed cells, to represent individual functional units of liver. UV intensities up to 30 mW/cm2 and exposure times ranging from 30-70s were required to polymerize distinct structures, maintain cell viability, and preserve fidelity of the harvested μ-tissues. Microtissue size, which is determined by the photomask used for polymerization, ranged from 250-350 μm diameter. At the dimensions and cell densities used in this study, no central necrosis was observed suggesting that nutrient transport was not limiting.
The size and suspension properties of these μ-tissues allow for facile manipulation by pipette and flow (as shown in SI Movies 1-3) and integration with technologies designed for examining ‘large’ objects (i.e.>250 μm). For instance, a pneumatically-driven flow analyzer and sorter, capable of fluorescence detection from 500 μm diameter particles, was used to analyze viability for a population of intact calcein-labeled hepatocyte μ-tissues, n=338 (Fig 1c). In addition to the quantitative data acquired by this approach, the large sample size enables an enhancement in the statistical power of biological comparisons 37. For example, in order to detect a TC50 (toxic concentration of drug that reduces viability to 50%) for the population in Figure 1c, a minimum sample size of n=30 would be required for statistical significance (p<0.05). With the availability of n=338, power analysis of the population in Figure 1c reveals that mean differences in μ-tissue viability as low as 1.24-fold could be detected with high confidence (power=0.9). These results suggest that populational μ-tissue analysis could improve statistical confidence in the functional assessment of engineered tissue constructs, especially for ‘small’ but important effects.
Multiparametric flow analysis and enrichment of 3D embryonic stem and progenitor cell μ-tissues in vitro and in vivo
3D hydrogel scaffolds are promising platforms for stem cell culture in vitro, with the ability to influence proliferation and differentiation 3. However, these processes are regulated by an interplay of external signals 38, 39 and stochastic elements 40, 41; thus they are inherently ‘noisy’. Approaches for increasing the capacity of quantitative analysis would permit detailed measurements of heterogeneity within a large sample of stem cell-derived engineered tissues. Furthermore, enrichment capacities would allow for subsequent examination of distinct tissue sub-populations, including analysis of the multicellular components present within individual μ-tissues. Therefore, we explored the potential of the μ-tissue platform for the study, quantification and enrichment of stem cell differentiation within biomaterials. We examined the differentiation of mouse embryonic stem (ES) cells towards the endoderm lineage, an important step in the in vitro differentiation of hepatic cells from pluripotent precursors 42. 3D ES cell μ-tissues expressing a fluorescent reporter (dTomato) for expression of the endoderm marker sox1743 were fabricated and cultured in basal differentiation media (-LIF) for 6 days. Microtissue flow analysis demonstrated a small, but reproducible, percentage of tissues which exhibited increased red fluorescence indicative of reporter expression (Fig. 2a). Utilizing the sorting capabilities of this system, enrichment from n=4,076 3D ES-derived μ-tissues was used to isolate high-FL3:Red (dTomato)-expressing tissues (1.71% of total μ-tissues, equaling 69 in a representative experiment), as well as low-FL3:Red (dTomato)-expressing tissues (20.2% of total μ-tissues) (Fig. 2a). Concurrent measurement of red fluorescence “peak height” (maximum point fluorescence) aided in the identification and enrichment of samples with sub-tissue fluorescence, and correlated well with FL3 detection of total μ-tissue fluorescence. In order to confirm that fluorescence-based enrichment reflected reporter activity, we performed imaging and RT-PCR analysis and found an 86-fold enrichment of sox17 mRNA in sorted high-red samples (Fig. 2b, c). Additional inspection of the mRNA profile revealed elevated expression of other endoderm markers, foxa2 (18-fold) and gata4 (22-fold), but not of the neuroectoderm marker, pax6 (Fig. 2d), further corroborating the endoderm profile. These data illustrate the ability to analyze large sample sizes and perform enrichment, which promises to be useful for the analysis of low frequency events such as lineage-specific differentiation. Future experiments could perturb the microenvironment (biomaterial, soluble exposures, cellular composition and cellular interactions) to study differentiation in a 3D context.
Figure 2.
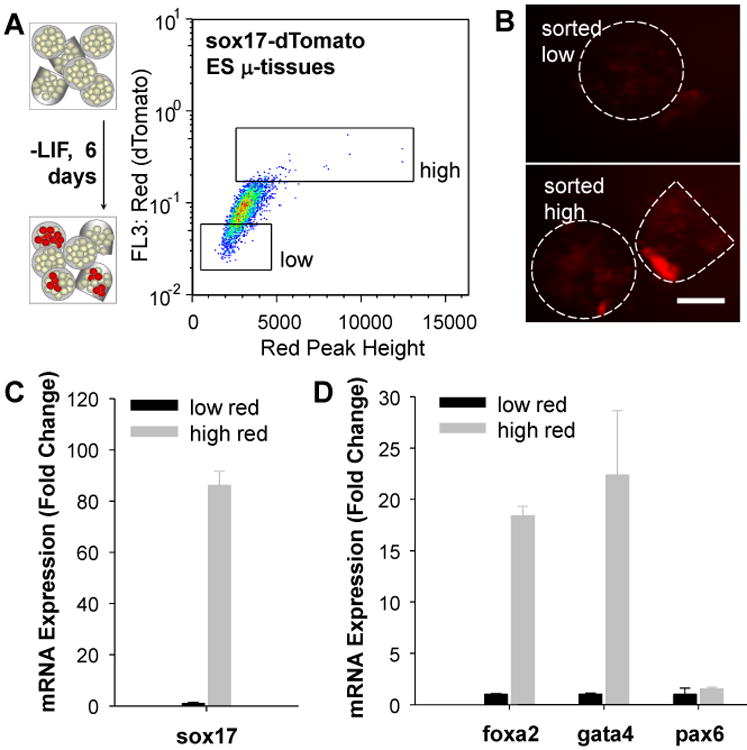
Flow analysis and enrichment of reporter 3D μ-tissues. A) 3D sox17-dTomato ES cell μ-tissues cultured in differentiation media (-LIF) for 6d and analyzed by flow cytometry based on red peak height vs. total red fluorescence. Fluorescence-activated sorting was used to enrich for high-red (dTomato)-expressing ES μ-tissues, compared to low-red controls. B,C) Gated high and low-red ES μ-tissues collected into a multiwell plate. Fluorescence imaging for dTomato intensity (B) and quantitative RT-PCR analysis of sox17 expression (C) confirmed reporter fidelity. D) Gene expression analysis of post-sort μ-tissues revealed additional endoderm marker (foxa2 and gata4) enrichment, without enrichment for the neuroectoderm marker, pax6. Error bars represent s.d. of the mean (n=3). (Scale bar, 200 μm.)
To evaluate potential multiparametric analysis and in vivo translation, we created μ-tissues containing bipotential mouse embryonic liver (BMEL) cells, which exhibit a liver progenitor phenotype further along hepatocyte specification than endoderm 44. These were manipulated by adenovirus to express fluorescent proteins and, like the ES μ-tissues, could be efficiently enriched by flow-activated sorting (SI Fig.2). Additionally, cytometric assessment of μ-tissues permitted multiparametric (absorbance & fluorescence) measurements in a process analogous to flow cytometry of individual cells. For instance, BMEL cell μ-tissues (n=451) constitutively expressing both GFP and RFP (Fig. 3a) were analyzed in a single run to assess four parameters simultaneously: time-of-flight (TOF) as a measure of axial length (i.e. size), extinction (EXT) or optical density which correlates with material properties and cellular density, and green (FL1) versus red (FL3) fluorescence to measure reporter expression (Fig. 3b). Gating based on TOF and EXT signals enabled exclusion of any multi-tissue aggregates and debris from the analysis.
Figure 3.
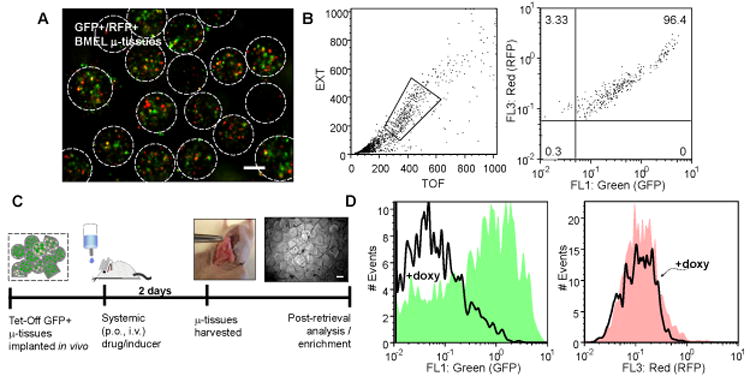
Responsiveness of dual reporter 3D μ-tissues to systemic stimuli in vivo. A,B) 3D GFP+/RFP+ BMEL μ-tissue population characterized by epifluorescence microscopy imaging (A), and quantitative flow analysis along multiple parameters, including time-of-flight (TOF), extinction (EXT), green and red fluorescence (B). C) Timeline of study probing 3D tet-inducible (TetOff-GFP+/RFP+) μ-tissue responsiveness in vivo. 3D μ-tissues are implanted via semi-permeable membrane into the peritoneal cavity of replicate mice, and mice are systemically administered doxycycline (doxy, 2 mg/mL) in 5% sucrose or sucrose only for 2d, prior to membrane harvest and μ-tissue retrieval. Phase micrograph depicts harvested μ-tissues in culture. D) Flow cytometry histograms for green and red fluorescence of n=2,242 3D TetOff-GFP+/RFP+ μ-tissues retrieved from duplicate doxy-treated or control mice. (Scale bars, 200 μm.)
We employed such a dual reporter system to test the utility of μ-tissue suspensions for in vivo studies. In particular, we sought to determine the accessibility of implanted μ-tissues to systemic stimuli such as orally administered drugs. Microtissues were loaded into a semi-permeable membrane, implanted in the peritoneal cavity of mice, exposed to systemic stimuli, retrieved and then analyzed. We delivered BMEL μ-tissues expressing TetOff-GFP and constitutive RFP peritoneally to replicate mice and systemically administered doxycycline (doxy, 2 mg/ml) in the drinking water for two days prior to μ-tissue retrieval (Fig. 3c). Quantitative analysis of n=2,242 μ-tissues retrieved from duplicate doxy-treated or untreated mice showed selective down-regulation of μ-tissue GFP expression by ∼6.6-fold in doxy-treated (solid line) versus control mice (filled green histogram) and RFP controls (Fig. 3d). Notably, while there is a distribution of GFP expression among individual μ-tissues, the large sample sizes for the μ-tissue populations yield a quantitative measurement that is accompanied by high power (0.94) and strong statistical significance (p<0.000000001), and are further capable of detecting mean shift differences as small as ∼1.5-fold for power=0.9 and p<0.01. Therefore, we have demonstrated a capacity to systemically administer factors and analyze μ-tissues following retrieval. Such a capacity could serve as the basis for a range of future investigations, including the study of tissue or tumor toxicity upon in vivo drug exposure or the response to induction of tetracycline-responsive elements (i.e. gene promoters, shRNA).
Microtissue encoding for parallel analysis and tracking in suspension-phase
In order to extend our system to assess 3D responses in parallel and within pooled μ-tissue samples, we developed a multiplexing encoding strategy. Specifically, incorporation of fluorescent nano- or microparticle labels into μ-tissues during their assembly (Fig. 4a) permitted optical identification of μ-tissue subsets within pooled populations. Microtissues could be identified based on both fluorescent particle identity (i.e. emission wavelength) and intensity of the labeled tissues (Fig. 4c) 45. Based on subsequent image analysis, encoded μ-tissues could be enriched to a purity of 98.0-98.4%, (Fig. 4d). These data indicate that μ-tissues could be tagged a priori during assembly to reflect biomaterial composition or cellular constituents, and utilized for real-time flow analysis and purification. In addition to encoding μ-tissue composition, we sought to develop a method to encode μ-tissue exposure (e.g. treatments with soluble factors). We therefore developed a method for post-hoc labeling of μ-tissue populations to enable the capacity to store information regarding post-assembly treatment conditions. For this approach, we focused on establishing an orthogonal labeling and identification scheme. 3D μ-tissues were embedded with biotinylated particles and exposed to solutions containing streptavidin conjugated near infrared (NIR) emitting molecules, which diffuse into the hydrogels and are retained through interaction with the biotin tethers (Fig. 4b). Labeling μ-tissues post-hoc with NIR fluorophores enabled resolution of identifiers within a detection window (∼700-900 nm) distinct from our fluorescent tags. Figure 4C illustrates 18 unique μ-tissue ‘codes’ (subset of 27 possible at the wavelengths selected here) generated by combinatorial labeling. The orthogonal detection of NIR-based codes was further demonstrated by the enumeration of sorted NIR-labeled μ-tissues, leveraging rapid high-resolution NIR plate-scanning techniques (SI Fig.3). In general, the quantification of labeled μ-tissues following flow-based enrichment could supplement cytometric fluorescent measures and greatly augment the number of codes and scope of multiplexing options accessible. Taken together, these results demonstrate the potential of encoding strategies (including a priori, and post-hoc orthogonal labels) as the foundation for parallel examination of 3D μ-tissues within pooled populations.
Figure 4.
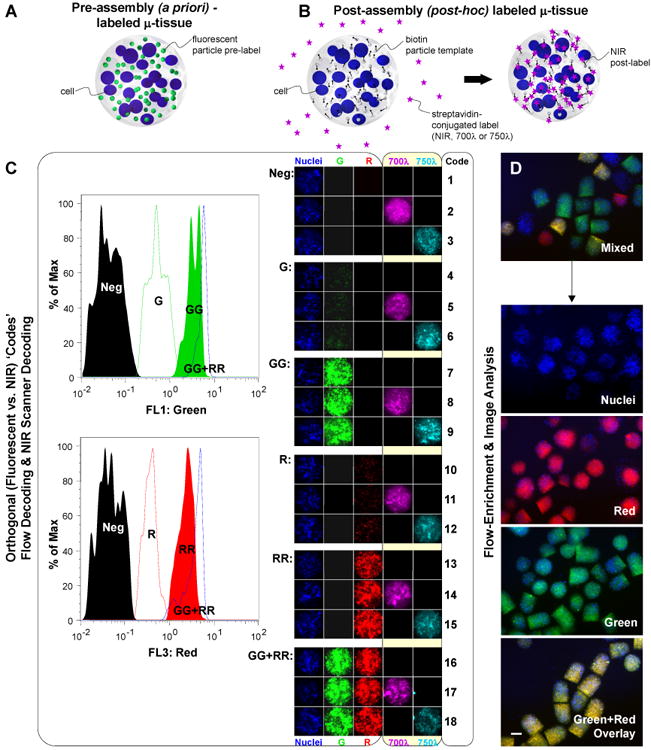
Encoding μ-tissues for parallel analysis and tracking. A) Schematic of a pre-assembly (a priori) labeled μ-tissue comprised of encapsulated cells and fluorescent particle tags. B) Schematic of a post-assembly (post-hoc) labeled μ-tissue comprised of encapsulated cells and biotin particle templates, and reacted in solution with diffusible streptavidin-conjugated NIR tags. C) Orthogonal detection of 18 pre-determined μ-tissue ‘code’ combinations, using flow analysis for μ-tissue fluorescence wavelength and intensity (left panel) and microscopy imaging for μ-tissue NIR emission (right panel). Flow cytometry histograms (left panel) demonstrate the decoding of labels varying in intensity along a single channel λ=510 (Neg, G, GG), λ=610 (Neg, R, RR), or combined channels (GGRR). Images of representative μ-tissue ‘codes’ (right panel) illustrate the label combinations possible when expanding fluorescent particle tags with additional NIR tags. D) 3D μ-tissues pre-assembled with Cell Tracker Blue-labeled BMEL progenitors and quantum dot codes QD525 (green), QD605 (red), QD525+605, or no QD, are fluorescence-activated sorted to enrich a mixed multiplexed μ-tissue sample for distinct decoded μ-tissue populations. Representative epifluorescence images of mixed and decoded/purified samples are shown. (Scale bar, 200 μm.)
Multiplex assessment of drug/gene interactions on pooled 3D carcinoma μ-tissues
We next explored whether multiplexed μ-tissues could be integrated with populational flow analysis to enable pooled assessment of 3D responses. Pooled assessment approaches such as bar-coded RNA interference (RNAi) and one-bead/one-compound libraries are gaining favor as tools that reduce analysis time, include internal controls to minimize error propagation, facilitate iterative enrichment, and translate to in vivo contexts 46-48. Accordingly, the study of the impact of genetic elements on 3D cellular responses to drugs 49 is one area poised to benefit from this pooled screening approach. Many cancers, including hepatocellular carcinomas, exhibit aberrant gene regulation which promotes survival and decreased sensitivity to chemotherapy 50-52; therefore, treatments utilizing RNAi to silence gene expression have been tested either alone or in combination with drugs 53. Large scale screens typically either test factors individually using various high-throughput technologies (e.g. robotic-aided multiwell platforms, microarrays) or use pooled libraries from which targets are identified following enrichment or selective pressure 54. Here, we tested the ability to encode 3D μ-tissues along multiple experimental axes (drug vs. gene), and examined the effects of chemotherapy and RNAi-mediated gene knockdown on 3D hepatoma μ-tissues, incorporating both real-time and enrichment-based analysis as a demonstration of scalable multiplexing strategies.
We assembled 3D hepatoma μ-tissues with a fluorescent-particle tag for identifying the genetic condition (BCL-XL siRNA or control vehicle only, LIPO), after pre-treating the human hepatoma cell line, HepG2, with siRNA targeting the anti-apoptotic gene BCL-XL or with lipofectamine alone. Concurrently, biotin particles were co-encapsulated to act as scaffolds for post-hoc labels identifying the ensuing drug treatment condition. Pooled, labeled μ-tissue populations (total n=738) were then exposed to solutions of low (20 nM) or high-dose (2 μM) doxorubicin (DOXO) containing NIR-emitting streptavidin-700λ or streptavidin-750λ molecules (Fig. 5a). The response of hepatoma μ-tissues along both treatment axes was assessed by combining orthogonal label identification techniques (flow analysis, NIR scanning) with viability assessment. Specifically, μ-tissues found below a red calcein viability threshold were first enriched by flow, and simultaneously decoded for siRNA treatment condition (Fig. 5b, SI Figs. 4, 5). NIR-imaging and evaluation of the relative contributions of distinct DOXO treatment conditions in the sorted populations then provided a measure of the chemotherapeutic effect of each drug/gene combination (SI Fig. 4, 5). We observed a ∼1.8-fold synergistic influence of 2 μM DOXO and BCL-XL siRNA on 3D hepatoma μ-tissues viability over DOXO or BCL-XL siRNA alone (Fig. 5c) which was statistically significant and was qualitatively corroborated by imaging (Fig. 5a). In addition to demonstrating the utility of multiplex assessment of 3D μ-tissues conditions, these results suggest that the BCL-XL anti-apoptotic protein protects hepatoma cancers from doxorubicin-induced apoptosis and may be a target for combination therapy (Fig. 5c). Furthermore, the combined strategy of pooling encoded μ-tissues gene conditions with encoded drug exposure will enable coincident assessment of multiple 3D conditions which could be integrated with small molecule screens.
Figure 5.
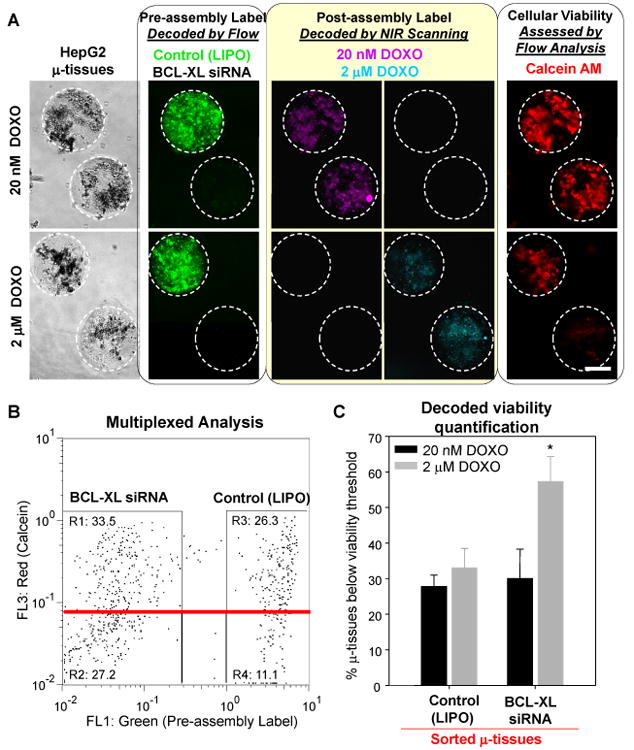
Multiplex assessment of drug/gene interactions on pooled 3D hepatoma μ-tissues. A) Representative phase and epifluorescence images of lipofectamine (LIPO)-treated or BCL-XL siRNA-treated hepatoma μ-tissues containing green fluorescent or unlabeled pre-assembly tags (left panel). Labeled μ-tissue mixtures were exposed to solutions of doxorubicin (DOXO) containing post-assembly NIR labels (middle panel) and stained with the red calcein AM live dye (right panel). B) Flow cytometry analysis of n=738 pooled hepatoma μ-tissues for green vs. red fluorescence shows real-time decoding of pre-assembly condition (BCL-XL siRNA vs. LIPO) and quantitative viability assessment based on calcein intensity. Enrichment for μ-tissue ‘responders’ was performed by collecting samples with viability below a pre-determined threshold (red line). C) NIR analysis of μ-tissue ‘responders’ enabled identification of post-assembly labels and quantification of the combined effect of BCL-XL siRNA and doxorobucin on hepatoma μ-tissues (see SI Figs. 4, 5 and Experimental). Error bars represent s.d of the mean (n=3). Statistical significance was determined using Student's paired t-test (p<0.05). (Scale bar, 200 μm.)
Discussion
We describe a platform based on 3D μ-tissue suspensions that can be fabricated in large ensembles, analyzed and sorted, cultured in vitro or implanted in vivo, and encoded in a multiplexed fashion for more rapid and thorough investigation of 3D cellular responses. In the development of this system, we utilized a range of liver cell types and experimental designs selected to demonstrate these unique capabilities (including 3D viability/toxicity measurements, 3D stem cell differentiation analysis and enrichment, and parallel elucidation of 3D drug/gene effects on cancer).
At the most fundamental level, quantitative analysis of large sample sizes of μ-tissues improves study power and helps to identify ‘noisy’ biological events, without relying on arrays, serial imaging or spatial addressing. Our initial sample size demonstration measured viability in hepatic μ-tissues in vitro and suggested that statistically significant viability differences as small as 1.24-fold could be detected with n∼102 suspended samples. Enrichment capabilities analogous to flow cytometry could also isolate the low ‘signal’ from the noise, as demonstrated by the 60-fold enrichment of differentiated endoderm marker expression from a large 3D ES μ-tissue population. Further, by leveraging the ability to transfer μ-tissue suspensions between environments (i.e. in vitro to in vivo), high n, quantitative analysis or enrichment of implanted 3D μ-tissues could be performed. The retrieval and quantitative assessment of n >103 living tissue units from an individual mouse, demonstrated here, represents a novel approach to testing 3D conditions in vivo with potentially broad utility in tissue engineering and toxicology. Furthermore, this approach could help control for animal variability and reduce overall animal requirements thereby improving cost, time and ethical constraints.
In addition to high n measurements and enrichment, analysis of pooled 3D μ-tissue samples enables parallel assessment of multiple 3D conditions, and could be ultimately combined with small molecule libraries in a manner analogous to one-bead/one-compound screens. Thus, we developed novel strategies for a priori and/or post-hoc μ-tissue encoding, which facilitated tracking and multiplexing of a pooled population of therapeutically-treated 3D hepatoma μ-tissues. Our findings demonstrating synergy between BCL-XL siRNA and doxorubicin treatments are consistent with other reports suggesting the involvement of BCL-XL in chemotoxin-induced apoptosis protection for various cancer cell types in monolayer culture 51, 55. Interestingly, although the synergy we observed in 3D μ-tissues at high-dose (2 μM) DOXO was of a similar magnitude to the synergistic effect in standard 2D culture (Fig. 5c, SI Fig. 6), the dose-dependent effect of doxorubicin exposure alone was attenuated in our 3D- compared to 2D- models. Enhanced drug resistance within 3D spheroids has been previously observed for other cancer cell types and treatment protocols 56, 57. Doxorubicin, utilized here, is known to inhibit cellular proliferation as well as trigger apoptosis 58, 59, and differences in proliferation potential in 2D versus 3D culture could contribute directly or indirectly (i.e. increased drug sensitivity) to the distinct dose profiles identified. As many cancer cell types have been shown to exhibit a range of unique phenotypes within 3D environments 13, 16, a platform enabling rapid and quantitative 3D assessment may help elucidate the mechanisms underlying cancer responses to therapies and provide insights into discrepancies observed between in vitro and in vivo studies.
Although these μ-tissue samples were exclusively PEG hydrogel-based, the fabrication and assessment of 3D μ-tissues is extensible to the inclusion of different biomaterials, cellular constituents, or encapsulated species. In particular, the extension to degradable or reversible materials could allow for single-cell recovery and analysis from enriched μ-tissues, thereby adding a new dimension of information to the small-scale tissue data shown here. Degradable materials may also facilitate in vivo studies which probe for more direct interactions with the host than achievable using implant membranes. Given our finding that small molecule transport and dynamic regulation of μ-tissues within membranes was possible, future in vivo studies will explore alternative methods to recover directly injected μ-tissues. Finally, in further versions of μ-tissue screens, incorporation of nucleic acid-based ‘barcoding’ technologies will assist in the expansion of multiplexing capabilities 60, 61.
Conclusion
As the field of regenerative medicine progresses and 3D tissue models gain wide adoption, methods to obtain statistical data will be critical to spur the field forward. We expect that the μ-tissue platform described here will be an effective new tool for probing the function of communities of cells in 3D environments with applications in stem cell biology, toxicology, and tissue engineering.
Experimental
Fabrication and Culture of μ-tissue Suspensions
Cellular μ-tissues were fabricated using a hydrogel polymerization apparatus previously described 7, 36 and harvested into solution for flow manipulation and analysis. Briefly, pre-polymer solution of cells, 10% w/v PEGDA (MW20 kDa; Laysan Bio, Inc.), 0.1% w/v Irgacure 2959 photoinitiator (Ciba) was loaded into a well with thickness defined by a 250 μm silicone spacer, and the solution exposed to UV light (320-390 nm, 30 mW/cm2, 30-70s; EXFO Lite) through an emulsion mask exhibiting a spotted pattern ranging from 250-350 μm in diameter, with 2.5 mm center-to-center spacing. Final μ-tissue encapsulated cell concentrations were: 60×106/mL BMEL aggregates, 100×106/mL ES aggregates, 20×106/mL HepG2 cells or 10×106/mL hepatocytes/mL pre-polymer solution. Hepatocyte-fibroblast μ-tissues included 15 μmol/mL acrylate-PEG-RGDS to promote hepatocyte survival and functions 7, 36.
Prior to μ-tissue encapsulation, all cells were cultured in a 5% CO2 humidified incubator at 37°C. HepG2 cells (ATCC) were cultured in HepG2 medium comprised of high glucose DMEM, 10% bovine serum, and 1% penicillin-streptomycin. Fresh hepatocytes from 2-3 month old adult female Lewis rats (Charles River Laboratories) were isolated and purified as previously described and cultured in hepatocyte medium comprised of high glucose DMEM (Invitrogen), 10% fetal bovine serum (FBS, Invitrogen), 0.5 U/mL insulin (Lilly), 7 ng/mL glucagons (Bedford Laboratories), 7.5 mg/mL hydrocortisone (Sigma), 10 U/mL penicillin (Invitrogen), and 10 mg/mL streptomycin (Invitrogen). 3T3-J2 fibroblasts provided by Dr. Howard Green (Harvard Medical School, Cambridge, MA) were cultured at less than 16 passages, in fibroblast medium comprised of high glucose DMEM, 10% bovine serum, and 1% penicillin-streptomycin. To create hepatocyte-fibroblast co-cultures, hepatocytes were seeded in hepatocyte medium at a density of 5×105 cells per well, in 34-mm tissue-culture wells adsorbed with 0.14 mg/mL Collagen-1 extracted from rat-tail tendons. Twenty-four hours later, fibroblasts were seeded at 5×105 cells per well in fibroblast medium. Medium was replaced daily with hepatocyte medium for 7-10 days prior to harvest by trypsinization and μ-tissue photo-encapsulation.
The BMEL cell line 9A1 was provided by Dr. Mary Weiss (Institut Pasteur) 44, 62 and cultured in BMEL medium comprised of RPMI 1640 with glutamax (Invitrogen), 30 ng/mL human IGF-II (Peprotech), 50 ng/mL human EGF (Peprotech), and 10 mg/mL recombinant human insulin (Invitrogen). BMEL cells were passaged every 2-4 days in flasks adsorbed with 0.5 mg/mL Collagen-1. GFP+/RFP+ BMEL cells were created by transducing cells with 15 MOI Ad5-CMV-RFP and 10 MOI Ad5-CMV-GFP adenoviral vectors (Gene Vector Core, U.Iowa), in the presence of 4 μg/mL polybrene (Chemicon). TetOff-GFP+/RFP+ BMEL cells were similarly created by transducing cells with 15 MOI Ad5-CMV-RFP, 5 MOI Ad5-TRE-GFP and 5 MOI Ad5-CMV-tTA adenoviral vectors, in the presence of 4 μg/mL polybrene. BMEL cell aggregates were formed by culturing 2×106 cells in 100 mm non-adhesive dishes for 2-4h prior to concentrating by mild centrifugation and μ-tissue photo-encapsulation.
The Sox17/dTomato reporter mouse embryonic stem (ES) cell line was provided by Dr. Douglas Melton's laboratory and cultured on mitomycin-C growth arrested mouse embryonic fibroblast (MEF) feeder layers in Knockout-DMEM (GIBCO) media supplemented with 15% ES-grade fetal bovine serum (Millipore), 2 mM L-glutamine (GIBCO), 1 mM nonessential amino acids (GIBCO), 1.1 mM β-mercaptoethanol (Sigma), 1 × penicillin/streptomycin (GIBCO), and 1000 units/mL LIF (ESGRO, Millipore) and passaged every 2-3 days. ES cells were depleted of MEFs by 30 min culture on gelatin-treated dishes and transferred to fresh gelatin-treated plates in full ES media plus LIF for 1 day. ES-Sox17 cell aggregates were then formed by culturing 8×106 cells in 100 mm non-adhesive dishes for 4-6h prior to concentrating by mild centrifugation and μ-tissue photo-encapsulation. Following encapsulation, ES μ-tissues were cultured under basal differentiation conditions, in ES media in the absence of LIF.
Animal Studies
RFP+/TetOff GFP+ BMEL μ-tissues were loaded at a concentration of ∼20,000/mL into cellulose ester dialysis tubing (MWCO 100 kDa, Spectrapor), the ends heat-sealed, and the implant equilibrated in medium for 18h prior to implantation into the peritoneum of mice. BALB/c nude/nude mice (Taconic) were anesthetized using 2.5% Isoflurane with 100% oxygen flow at 1.0 liter/min. Mice were injected intraperitoneally with analgesic buprenorphine (0.1 mg/kg), and skin was prepped using Betadine/isopropanol. A <1.25 cm incision was made in the skin, abdominal wall and peritoneum, and the implant comprised of ∼20,000 μ-tissues/mL in cellulose ester dialysis tubing (0.8 cm×2 cm, MWCO 100 kDa, Spectrapor), was placed in the peritoneum wrapped in intraperitoneal fat. The abdominal wall was closed with silk sutures and the skin closed with staples. Mice were monitored for return to normal activity then dosed with analgesic every 12-24h post-surgery. Mice were supplied with drinking water containing 2 mg/mL doxycycline in 5% sucrose or sucrose only until sacrificed by CO2 euthanization, at which time the implant was retrieved and μ-tissues harvested into solution for flow analysis. The Committee for Animal Care in the Department of Comparative Medicine at MIT approved all housing and surgical procedures.
Multiplex Codes
Pre-assembly labels added to pre-polymer solution were: red quantum dots (QDs) (Qdot 655 nm ITK amino PEG, 0.8 μM), green QDs (Qdot 605 nm ITK amino PEG, 1.2 μM), green (0.2 μm FluoSpheres 505/515, 0.02%) or red (1.0 μm FluoSpheres 580/605, 0.02%) fluorescent polystyrene microspheres. For post-assembly labels, biotinylated polystyrene particles (1.0 μm FluoSpheres-Biotin, 0.02%) were added to the pre-polymer solution and embedded within μ-tissues. Encoding was performed by incubating μ-tissue solutions with NIR dye-conjugated streptavidin molecules (streptavidin-Alexa Fluor -700 or -750, 10 μg/mL) at 37°C for 30 min, then washing μ-tissues at RT in 20 mL PBS. Microtissues were sorted into a clear-bottom, multiwell plate, and labeling verified using fluorescence microscopy. To quantify enriched NIR-labeled samples, the Odyssey IR imaging system (LI-COR) was used to scan multiwell plates, and μ-tissues were counted manually from replicate IR scans. Hoescht 33258 (2 μg/mL) was used to visualize cell nuclei within HepG2 μ-tissues. CellTracker Blue CMAC (5 μM) was used to visualize BMEL μ-tissues without QD codes, and was incubated with cells at 37°C for 45 min prior to encapsulation. Green or red calcein AM ‘live’ (5 μg/mL) and ethidium homodimer ‘dead’ (2.5 μg/mL) fluorescent staining was performed at 37°C for 30 min. All code reagents were from Invitrogen and were thoroughly washed and exchanged into distilled H2O with 100k Amicon Ultra filters (Millipore) to remove sodium azide prior to use.
Microtissue Characterization, Cytometry and Sorting
Fluorescent images were acquired using a Nikon Ellipse TE200 inverted fluorescence microscope and CoolSnap-HQ Digital CCD Camera. Reporter gene levels and viability were quantified using a complex object parametric analyzer for handling 500 μm objects (COPAS Select, Union Biometrica). Solutions of μ-tissues were analyzed for length (time of flight, TOF), optical density (extinction, EXT), and fluorescence (488/510±20-25 or 488/610±20-25 nm ex/em). Raw data were filtered for whole, dispersed μ-tissues using a TOF gate region to exclude populations of cell debris or μ-tissue aggregates. TOF/EXT-gated μ-tissues were analyzed for fluorescence and sorted at a rate of ∼20 μ-tissues per second into multiwell plates containing 100% FBS. Statistical analysis of co-culture μ-tissues was based on cytometry data using a two-sample t-test power analysis tool (NCSS Statistical Software).
Gene Expression Analysis of Enriched μ-tissue Populations
Total RNA was isolated from sorted Sox-17-ES μ-tissue populations using Trizol (Invitrogen) according to the manufacturer's instructions. Total RNA was incubated with RNase-free DNase (New England Biolabs) at 37°C for 40-60 min, and subsequently cleaned using RNeasy spin columns (Qiagen). cDNA from total RNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad), with reactions performed in the absence of reverse-transcriptase enzyme used as negative controls. RT-PCR was performed with cDNA templates using iQ SYBR green supermix and the the MyiQ real-time PCR detection system, set with cycling parameters: 95°C for 3 min, 40 cycles of 95°C for 10 s and 60°C for 45 s. Primers for sox17, foxa2, gata 4, pax6 and the housekeeping gene HPRT were used at 100 nM and the sequences are listed below (Integrated DNA Technologies). HPRT mRNA expression was used as a normalization control, and the mean mRNA expression of “sorted hi” μ-tissues is shown relative to “sorted low” μ-tissue expression for each gene of interest.
Mouse sox17
5′-AACCTCAGCATGTCACCTCATGGA-3′
5′-AGATGTCTGGAGGTGCTGCTCATT-3′
Mouse foxa2
5′-AAGTATGCTGGGAGCCGTGAAGAT-3′
5′-CGCGGACATGCTCATGTATGTGTT-3′
Mouse gata4
5′-AGGGTGAGCCTGTATGTAATGCCT-3′
5′-AGGACCTGCTGGCGTCTTAGATTT-3′
Mouse pax6
5′-GCCCTTCCATCTTTGCTTGGGAAA-3′
5′-TAGCCAGGTTGCGAAGAACTCTGT-3′
Mouse hprt
5′-GGAGTCCTGTTGATGTTGCCAGTA-3′
5′-GGGACGCAGCAACTGACATTTCTA-3′
Multiplex Assessment of Gene Knockdown and Drug Treatment
HepG2 cells were transfected with 100 nM BCL-XL ON-TARGET plus SMARTpool siRNA (NM_001191, Dharmacon) using Lipofectamine RNAi max (Invitrogen), 5h prior to harvest by trypsinization and μ-tissue photo-encapsulation with biotinylated particles. Mixtures of pre-assembly labeled BCL-XL siRNA and LIPO μ-tissue suspensions were supplemented with doxorubicin at 20nM or 2μM and cultured for 48h, at which time post-assembly streptavidin-NIR codes were added to each solution and incubated at 37°C for 60 min. Calcein AM staining and analysis of pooled μ-tissue solutions on the complex object analyzer and sorter were used to identify μ-tissues with viability below a pre-determined threshold, and to select for μ-tissues exhibiting the greatest relative response to DOXO±siRNA combination treatment. Quantification of viability in decoded samples was performed as described in SI Fig. 4.
Supplementary Material
Acknowledgments
This work was supported by funding from the NIH NIDDK, NIBIB, NSF CAREER, and David & Lucile Packard Foundation (1999-1453). A.A.C acknowledges support from the NDSEG and NSF fellowship programs. We are grateful to Julia Thompson (Union Biometrica) for helpful insights and assistance, Steven Katz for rat hepatocyte isolation, Dr. Rene Maehr and Dr. Douglas Melton (Harvard University) for providing the sox17 dTomato mES line, and Dr. Mary Weiss and Dr. Helene Strick-Marchand (Institut Pasteur) for providing the BMEL 9A1 line.
References
- 1.Schmeichel KL, Bissell MJ. J Cell Sci. 2003;116:2377–2388. doi: 10.1242/jcs.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kong HJ, Mooney DJ. Nat Rev Drug Discov. 2007;6:455–463. doi: 10.1038/nrd2309. [DOI] [PubMed] [Google Scholar]
- 3.Saha K, Pollock JF, Schaffer DV, Healy KE. Curr Opin Chem Biol. 2007;11:381–387. doi: 10.1016/j.cbpa.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burdick JA, Vunjak-Novakovic G. Tissue Eng Part A. 2009;15:205–219. doi: 10.1089/ten.tea.2008.0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lund AW, Yener B, Stegemann JP, Plopper GE. Tissue Engineering Part B-Reviews. 2009;15:371–380. doi: 10.1089/ten.teb.2009.0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang NS, Kim MS, Sampattavanich S, Baek JH, Zhang Z, Elisseeff J. Stem Cells. 2006;24:284–291. doi: 10.1634/stemcells.2005-0024. [DOI] [PubMed] [Google Scholar]
- 7.Underhill GH, Chen AA, Albrecht DR, Bhatia SN. Biomaterials. 2007;28:256–270. doi: 10.1016/j.biomaterials.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 8.Gerecht S, Burdick JA, Ferreira LS, Townsend SA, Langer R, Vunjak-Novakovic G. Proc Natl Acad Sci U S A. 2007;104:11298–11303. doi: 10.1073/pnas.0703723104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benoit DS, Schwartz MP, Durney AR, Anseth KS. Nat Mater. 2008;7:816–823. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gobin AS, West JL. Biotechnol Prog. 2003;19:1781–1785. doi: 10.1021/bp0341390. [DOI] [PubMed] [Google Scholar]
- 11.Mann BK, Schmedlen RH, West JL. Biomaterials. 2001;22:439–444. doi: 10.1016/s0142-9612(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 12.Shah DN, Recktenwall-Work SM, Anseth KS. Biomaterials. 2008;29:2060–2072. doi: 10.1016/j.biomaterials.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ. Proc Natl Acad Sci U S A. 1998;95:14821–14826. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosines E, Sampogna RV, Johkura K, Vaughn DA, Choi Y, Sakurai H, Shah MM, Nigam SK. Proc Natl Acad Sci U S A. 2007;104:20938–20943. doi: 10.1073/pnas.0710428105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, Richardson A, Weinberg RA. Proc Natl Acad Sci U S A. 2004;101:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischbach C, Chen R, Matsumoto T, Schmelzle T, Brugge JS, Polverini PJ, Mooney DJ. Nat Methods. 2007;4:855–860. doi: 10.1038/nmeth1085. [DOI] [PubMed] [Google Scholar]
- 17.Khetani SR, Bhatia SN. Curr Opin Biotechnol. 2006;17:524–531. doi: 10.1016/j.copbio.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Bryant SJ, Ratner BD. Annual Review of Biomedical Engineering. 2004;6:41–75. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- 19.Mei Y, Goldberg M, Anderson D. Curr Opin Chem Biol. 2007;11:388–393. doi: 10.1016/j.cbpa.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castel D, Pitaval A, Debily MA, Gidrol X. Drug Discovery Today. 2006;11:616–622. doi: 10.1016/j.drudis.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 21.Chen DS, Davis MM. Curr Opin Chem Biol. 2006;10:28–34. doi: 10.1016/j.cbpa.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Flaim CJ, Chien S, Bhatia SN. Nat Methods. 2005;2:119–125. doi: 10.1038/nmeth736. [DOI] [PubMed] [Google Scholar]
- 23.Lee MY, Kumar RA, Sukumaran SM, Hogg MG, Clark DS, Dordick JS. Proc Natl Acad Sci U S A. 2008;105:59–63. doi: 10.1073/pnas.0708756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jongpaiboonkit L, King WJ, Murphy WL. Tissue Eng Part A. 2009;15:343–353. doi: 10.1089/ten.tea.2008.0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albrecht DR, Underhill GH, Wassermann TB, Sah RL, Bhatia SN. Nat Methods. 2006;3:369–375. doi: 10.1038/nmeth873. [DOI] [PubMed] [Google Scholar]
- 26.Albrecht DR, Underhill GH, Mendelson A, Bhatia SN. Lab Chip. 2007;7:702–709. doi: 10.1039/b701306j. [DOI] [PubMed] [Google Scholar]
- 27.Legant WR, Pathak A, Yang MT, Deshpande VS, McMeeking RM, Chen CS. Proc Natl Acad Sci U S A. 2009;106:10097–10102. doi: 10.1073/pnas.0900174106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh WG, Itle LJ, Pishko MV. Anal Chem. 2003;75:5783–5789. doi: 10.1021/ac034773s. [DOI] [PubMed] [Google Scholar]
- 29.McGuigan AP, Sefton MV. Proc Natl Acad Sci U S A. 2006;103:11461–11466. doi: 10.1073/pnas.0602740103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong AP, Perez-Castillejos R, Christopher Love J, Whitesides GM. Biomaterials. 2008;29:1853–1861. doi: 10.1016/j.biomaterials.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du Y, Lo E, Ali S, Khademhosseini A. Proc Natl Acad Sci U S A. 2008;105:9522–9527. doi: 10.1073/pnas.0801866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam MT, Huang YC, Birla RK, Takayama S. Biomaterials. 2009;30:1150–1155. doi: 10.1016/j.biomaterials.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 33.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen KT, West JL. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 35.Liu VA, Bhatia SN. Biomedical Microdevices. 2002;4:257–266. [Google Scholar]
- 36.Liu Tsang V, Chen AA, Cho LM, Jadin KD, Sah RL, DeLong S, West JL, Bhatia SN. Faseb J. 2007;21:790–801. doi: 10.1096/fj.06-7117com. [DOI] [PubMed] [Google Scholar]
- 37.Machin D, Campbell MJ, Fayers PM, Pinol AY. Sample Size Tables for Clinical Studies. Blackwell Publishers; Malden, MA: 1997. [Google Scholar]
- 38.Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen CS. Cell Stem Cell. 2009;5:17–26. doi: 10.1016/j.stem.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Discher DE, Mooney DJ, Zandstra PW. Science. 2009;324:1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Nature. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann M, Chang HH, Huang S, Ingber DE, Loeffler M, Galle J. PLoS One. 2008;3:e2922. doi: 10.1371/journal.pone.0002922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kubo A, Shinozaki K, Shannon JM, Kouskoff V, Kennedy M, Woo S, Fehling HJ, Keller G. Development. 2004;131:1651–1662. doi: 10.1242/dev.01044. [DOI] [PubMed] [Google Scholar]
- 43.Borowiak M, Maehr R, Chen S, Chen AE, Tang W, Fox JL, Schreiber SL, Melton DA. Cell Stem Cell. 2009;4:348–358. doi: 10.1016/j.stem.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strick-Marchand H, Weiss MC. Hepatology. 2002;36:794–804. doi: 10.1053/jhep.2002.36123. [DOI] [PubMed] [Google Scholar]
- 45.Han M, Gao X, Su JZ, Nie S. Nat Biotechnol. 2001;19:631–635. doi: 10.1038/90228. [DOI] [PubMed] [Google Scholar]
- 46.Kainkaryam RM, Woolf PJ. Curr Opin Drug Discov Devel. 2009;12:339–350. [PMC free article] [PubMed] [Google Scholar]
- 47.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, Mermel C, Barbie DA, Awad T, Zhou X, Nguyen T, Piqani B, Li C, Golub TR, Meyerson M, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE. Proc Natl Acad Sci U S A. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meacham CE, Ho EE, Dubrovsky E, Gertler FB, Hemann MT. Nat Genet. 2009;41:1133–1137. doi: 10.1038/ng.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grossman I. Expert Opinion on Drug Metabolism & Toxicology. 2009;5:449–462. doi: 10.1517/17425250902902322. [DOI] [PubMed] [Google Scholar]
- 50.Folmer Y, Schneider M, Blum HE, Hafkemeyer P. Cancer Gene Ther. 2007;14:875–884. doi: 10.1038/sj.cgt.7701082. [DOI] [PubMed] [Google Scholar]
- 51.Takehara T, Liu X, Fujimoto J, Friedman SL, Takahashi H. Hepatology. 2001;34:55–61. doi: 10.1053/jhep.2001.25387. [DOI] [PubMed] [Google Scholar]
- 52.Brenes O, Arce F, Gatjens-Boniche O, Diaz C. Biomed Pharmacother. 2007;61:347–355. doi: 10.1016/j.biopha.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Pai SI, Lin YY, Macaes B, Meneshian A, Hung CF, Wu TC. Gene Ther. 2006;13:464–477. doi: 10.1038/sj.gt.3302694. [DOI] [PubMed] [Google Scholar]
- 54.Bernards R, Brummelkamp TR, Beijersbergen RL. Nat Methods. 2006;3:701–706. doi: 10.1038/nmeth921. [DOI] [PubMed] [Google Scholar]
- 55.Luo D, Cheng SCS, Xie H, Xie Y. Biochem Cell Biol. 2000:119–126. [PubMed] [Google Scholar]
- 56.Desoize B, Gimonet D, Jardiller JC. Anticancer Res. 1998;18:4147–4158. [PubMed] [Google Scholar]
- 57.Kobayashi H, Man S, Graham CH, Kapitain SJ, Teicher BA, Kerbel RS. Proc Natl Acad Sci U S A. 1993;90:3294–3298. doi: 10.1073/pnas.90.8.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arai M, Kondoh N, Imazeki N, Hada A, Hatsuse K, Matsubara O, Yamamoto M. Liver Int. 2009;29:55–62. doi: 10.1111/j.1478-3231.2008.01792.x. [DOI] [PubMed] [Google Scholar]
- 59.Saleh EM, El-Awady RA, Abdel Alim MA, Abdel Wahab AH. Cell Biochem Biophys. 2009 doi: 10.1007/s12013-009-9058-3. [DOI] [PubMed] [Google Scholar]
- 60.Pregibon DC, Toner M, Doyle PS. Science. 2007;315:1393–1396. doi: 10.1126/science.1134929. [DOI] [PubMed] [Google Scholar]
- 61.Xu Q, Schlabach MR, Hannon GJ, Elledge SJ. Proc Natl Acad Sci U S A. 2009;106:2289–2294. doi: 10.1073/pnas.0812506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strick-Marchand H, Morosan S, Charneau P, Kremsdorf D, Weiss MC. Proc Natl Acad Sci U S A. 2004;101:8360–8365. doi: 10.1073/pnas.0401092101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.