

Abstract
Membrane depolarization and ion fluxes are events that have been studied extensively in biological systems due to their ability to profoundly impact cellular functions, including energetics and signal transductions. While both fluorescent and electrophysiological methods, including electrode usage and patch-clamping, have been well developed for measuring these events in eukaryotic cells, methodology for measuring similar events in microorganisms have proven more challenging to develop given their small size in combination with the more complex outer surface of bacteria shielding the membrane. During our studies of death-initiation in Streptococcus pneumoniae (pneumococcus), we wanted to elucidate the role of membrane events, including changes in polarity, integrity, and intracellular ion concentrations. Searching the literature, we found that very few studies exist. Other investigators had monitored radioisotope uptake or equilibrium to measure ion fluxes and membrane potential and a limited number of studies, mostly in Gram-negative organisms, had seen some success using carbocyanine or oxonol fluorescent dyes to measure membrane potential, or loading bacteria with cell-permeant acetoxymethyl (AM) ester versions of ion-sensitive fluorescent indicator dyes. We therefore established and optimized protocols for measuring membrane potential, rupture, and ion-transport in the Gram-positive organism S. pneumoniae. We developed protocols using the bis-oxonol dye DiBAC4(3) and the cell-impermeant dye propidium iodide to measure membrane depolarization and rupture, respectively, as well as methods to optimally load the pneumococci with the AM esters of the ratiometric dyes Fura-2, PBFI, and BCECF to detect changes in intracellular concentrations of Ca2+, K+, and H+, respectively, using a fluorescence-detection plate reader. These protocols are the first of their kind for the pneumococcus and the majority of these dyes have not been used in any other bacterial species. Though our protocols have been optimized for S. pneumoniae, we believe these approaches should form an excellent starting-point for similar studies in other bacterial species.
Keywords: Immunology, Issue 84, Streptococcus pneumoniae, pneumococcus, potential-sensitive dyes, DiBAC, Propidium Iodide, acetoxymethyl (AM) ester, membrane rupture, Ion transport, bacterial ion concentrations, ion-sensitive fluorescence
Introduction
Our lab has identified a protein-lipid complex from human milk named HAMLET (for Human Alpha-lactalbumin Made LEthal to Tumor cells) that induces apoptosis in tumor cells, but is also able to kill a variety of bacterial species1,2. The species that were found to be particularly sensitive were those that target the respiratory tract, with Streptococcus pneumoniae (the pneumococcus) displaying the greatest sensitivity and an apoptosis-like phenotype of death2,3. Membrane depolarization and specific ion transport events are well-described and crucial events during apoptosis in eukaryotic cells, particular in the mitochondria, where radioactive TPP+ ions and fluorescent dyes including JC-1 and TMRE have been used to demonstrate depolarization of the mitochondrial membrane3-5. Thus, we sought to learn more about the effect of HAMLET on these membrane-related features in the pneumococcus as we focused our efforts to develop a better understanding of the mechanistic components of the apoptosis-like phenotype in bacteria, with great potential for identifying novel antibacterial therapies or vaccine candidates in the process.
In seeking to establish protocols for our mechanistic studies, we discovered that in contrast to the well-described methodology in eukaryotic systems, there are very few published studies detailing electrophysiology and ion transport mechanisms of the bacterial membrane6,7. This is primarily attributed to the smaller size of microorganisms and their surface architecture, particularly the presence of cell wall, that restricts accessibility of the membrane for use of conventional eukaryotic methods like patch clamping, although some studies using giant protoplasts have been performed with mixed success8,9. As work with these giant protoplasts is not an ideal or even practical method for most bacterial species since it requires manipulated bacteria in an unnatural and abiotic state, the limited studies of bacterial membrane polarity that have been performed have primarily employed flow cytometry and the use of cyanine and oxonol fluorescent dyes10-16.
Instead of flow cytometry, which gathers individual fluorescence readings from one bacterium at a single time point, we chose to use a fluorescence detection plate reader to detect fluorescence intensity of bacterial suspensions in a 96-well plate format over time. This enabled us to treat a population of bacteria at various time points with much greater simplicity and ease, and continuously monitor the fluorescence kinetics of the entire population for extended periods of time, which is difficult to achieve using flow cytometry. After testing a wide variety of the potential-sensitive fluorescent dyes (including those mentioned above for use with the mitochondria), we achieved the best technical and practical success using the bis-oxonol dye called DiBAC4(3) (bis-(1,3-dibutylbarbituric acid) trimethine oxonol) to monitor changes in polarity.
We also found it valuable to concurrently monitor disruptions in the integrity of the membrane using propidium iodide (PI). This dye fluoresces upon binding to nucleic acid, but is only able to do so when the integrity of the bacterial membrane is compromised, making it the popular component used to detect dead cells in live-dead staining assays. In addition to PI, SYTOX green and TO-PRO-1 are fluorescent dyes that are similar in action and have been previously used for bacteria in a few studies using flow cytometry detection methods17. We chose to use PI due to its excitation wavelength that allowed us to monitor its fluorescence concurrently with DiBAC in a given sample.
In our studies, we have observed that HAMLET, as well as another related protein-lipid complex with bactericidal activity known as ELOA, induced depolarization and rupture of the bacterial membrane as indicated by increases in the fluorescence of both dyes upon treatment of the pneumococci3,18,26. For both complexes, we observed that the fluorescence intensity of DiBAC4(3) increased prior to the increase in intensity of PI, indicating that depolarization occurred prior to rupture and is, therefore, a specific event induced by our bactericidal protein-lipid complexes of interest. This distinction is important to make, as rupture of the membrane can itself cause nonspecific depolarization. Measuring and analyzing both DiBAC4(3) and PI fluorescence kinetics concurrently allowed us to examine this relationship between the two membrane events, which is an additional advantage of using fluorometry instead of flow cytometry.
To monitor bacterial ion flux, there has been some previous success with using radioisotopes, including measuring uptake of 45Ca2+ in the pneumococcus19,20, which we have also used in our recent studies18,21. However, working with these radioactive ions has several drawbacks. They can be expensive, time-consuming, and messy, and can also expose the individual performing the experiment to harm, depending on the isotope of interest. Additionally, it is difficult to monitor rapid changes over time. Thus, we turned towards an alternative method of measurement that employs acetoxymethyl (AM) ester versions of ion-sensitive fluorescent indicator dyes. By itself, the indicator dye is charged and does not pass through the membrane easily, but with the addition of the lipophilic ester group, the now uncharged molecule can pass through the membrane of the bacterium. Upon entering the interior, the bacterial esterases cleave the ester group, leaving the dye free inside the cell and charged again, significantly slowing its ability to exit the cell and allowing the dye to accumulate inside over time. However, use of these ester dyes has only been described in a few bacterial species to detect changes in intracellular Ca2+ 22-24 and H+ 16, with varying methods of loading, detection, and success.
With a desire to monitor changes in intracellular Ca2+ and also K+ and H+ levels in S. pneumoniae upon treatment with HAMLET and other compounds, we successfully created protocols to efficiently load fluorescent indicator dyes into bacterial cells. Effective loading into bacteria required both probenecid that increases dye retention by blocking anion-transporters and PowerLoad, a proprietary compound from Life Technologies that increases loading efficiency. Fura-2/AM (detecting Ca2+), PBFI/AM (detecting K+), and BCECF/AM (detecting H+) were successfully loaded into both unencapsulated and encapsulated pneumococcal strains enabling measurement of the resulting fluorescence patterns after addition of ionophores, such as ionomycin (Ca2+ uncoupler), valinomycin (K+ uncoupler) and CCCP (H+ uncoupler) using a fluorescence detection plate reader18,21.
Protocol
1. Preparing Bacterial Cultures
- Growing culture for use in experiments
- In a 37 °C heating block, thaw a frozen stock-vial of S. pneumoniae and add its content to 9 ml of fresh, prewarmed Todd-Hewitt broth with 0.5% yeast extract (THY) for a total volume of 10 ml in a glass culture tube.
- Incubate statically at 37 °C until the culture reaches mid-log phase (Abs600nm ≈0.5-0.6).
2. Detecting Membrane Depolarization and Rupture
- Preparing reagents
- Prepare a 50 µM stock of DiBAC4(3) in 100% DMSO, and a 2 mg/ml stock of PI in ddH2O. The DiBAC4(3) stock is stable at -20 °C for at least 6 months. The PI stock is stable at 4 °C for at least 6 months.
- Wrap stocks in foil to protect from the light.
- Loading bacteria
- Pellet the bacterial cells from step 1.2.2 by centrifugation at 2,400 x g for 10 min at room temperature and wash twice by removing the supernatant, resuspending the pellet in 10 ml of 1x Phosphate-Buffered Saline (PBS), and centrifuge again at 2,400 x g for 10 min.
- Remove supernatant and resuspend pellet in PBS to the original volume. This will provide for approximately 108 colony-forming units of bacteria per ml. Remove 1 ml (enough for 5 samples) of the washed cells and place into a microcentrifuge tube.
- To these cells, add 25 µl of a 1 M filter-sterilized stock solution of glucose (prepared in ddH2O and filtered through a 0.45 µm filter) for a final concentration of 25 mM glucose, and incubate in a 37 °C heating block for fifteen min. NOTE: This provides energy for all ATP-dependent membrane channels and cellular components. The need for this step may vary, depending on the experiment.
- Add 5 µl of the 50 µM DiBAC4(3) stock and 10 µl of the 2 mg/ml PI stock. This provides for final concentrations of 250 nM and 20 µg/ml, respectively. Mix thoroughly by pipetting up and down several times. Add 200 µl of this cell suspension to each sample well of a clear 96-well plate.
- Detecting fluorescence
- Place plate in a prewarmed (37 °C) fluorescence detection plate reader.
- Ensure that appropriate filters are in place for DiBAC4(3) (490 nm excitation, 516 nm emission) and PI (535 nm excitation, 617 nm emission) detection.
- To allow for (and concurrently monitor) equilibration of DiBAC4(3) over the membrane, set the reader to take fluorescence measurements every min for about 30-40 min, or until reading stabilizes; this serves as a “pretreatment” reading.
- Eject plate and add the experimental agent of choice and immediately place the plate back in the reader to continue monitoring DiBAC4(3) and PI fluorescence for the desired length of time. If treating several samples at once, using a multichannel pipette can be helpful to add the agent simultaneously to all wells.
- Export the readings to a data analysis program such as Microsoft Excel. Plot fluorescence over time to evaluate membrane depolarization and rupture. Increasing fluorescence of DiBAC and PI indicates that depolarization and rupture are occurring.
3. Detecting Changes in Intracellular Ca2+ or K+ Concentrations
- Preparing reagents and loading buffer
- Prepare the following reagents: 5 mM stock of the ion-sensitive fluorescent dye (Fura-2/AM or PBFI/AM; add the appropriate amount of DMSO to a tube containing 50 µg of the dye), “100x” probenecid (add 1 ml of PBS to a 77 mg vial). The solubilized dyes can be frozen at -20 °C once and are stable in solution for 3 months, whereas the probenecid is stable in solution at -20 °C for 6 months.
- Prepare 1 ml of 2x Loading Buffer as follows: In the bottom of a 15 ml plastic conical tube, dispense 20 µl of 100x PowerLoad concentrate (stable at 2-8 °C for 6 months), followed by 2 µl of the 5 mM ion-sensitive dye directly into the PowerLoad. Vortex briefly to mix. Add 960 µl of PBS followed by 20 µl of 100x probenecid. Vortex the mixture one final time. Wrap in foil to protect from light.
- Loading the bacteria with dye
- Pellet and wash the mid-log phase bacterial culture as described in 2.2.1. Remove supernatant and resuspend pellet in 5 ml of PBS to make a “2x” cell suspension.
- Add 1 ml of the 2x cell suspension to the 2x Loading Buffer. This will provide for a final volume of 2 ml, and final concentrations of 1x PowerLoad, 5 µM fluorescent dye, and 1x probenecid. NOTE: This suspension provides enough volume for 10 samples since each sample well for measurement will contain 200 µl.
- Incubate in a 37 °C water bath for 75 min, protected from light. NOTE: The dye is loaded into the bacterial cells in the absence of any glycolytic substrates and in the presence of probenecid to minimize efflux of the dye.
- WASH 1: Pellet the cells by centrifugation at 2,400 x g for 10 min, remove supernatant, and resuspend in 2 ml PBS containing 1x probenecid. WASH 2, 3: Repeat step 3.2.4 2x. Incubate at 37 °C for another 30 min. This will allow time for bacterial esterases to hydrolyze the AM ester group away from the internalized fluorescent dyes.
- WASH 4: Pellet the cells by centrifugation at 2,400 x g for 10 min, remove supernatant, and resuspend in 2 ml of PBS containing 1x probenecid. NOTE: Depending on the experiment, glucose can be added back here to re-energize the bacteria.
- Detecting fluorescence
- Prewarm the fluorescence detection plate-reader plate reader to 37 °C.
- For each desired sample, add 200 µl of the loaded cells (from step 3.2.8) to the wells of a 96-well plate. (Perform the following steps to completion for one well/sample at a time).
- To establish a baseline reading for a well, place plate in the fluorescence detection plate-reader to measure the fluorescence (340 nm excitation / 510 nm emission for “Value 1” and 380 nm excitation / 510 nm emission for “Value 2”) at every sec for one min.
- Eject plate, add the treatment of choice to that well (in a small volume of approximately 5 µl), quickly pipette up and down a few times to mix, and immediately place back in the fluorescence detection plate-reader to read every sec for desired length of time. (If the injectors associated with the plate reader are available, these could also be used to seamlessly add the desired treatment, eliminating the need to stop the read and eject the plate).
- Export the readings to a data analysis program such as Microsoft Excel. Calculate the ratio of the fluorescence values for each time-point by dividing Value 1 by Value 2, and plot the ratio values on a graph.
4. Detecting Changes in Intracellular pH
- Preparing reagents and calibration buffers
- Prepare the following reagents: 5 mM stock of BCECF/AM (add the appropriate amount of DMSO to a tube containing 50 µg of the dye. This stock is stable at -20 °C for 3 months); 100x probenecid (add 1 ml of PBS to a 77 mg vial); 1 mM stock preparation of nigericin in ethanol (required to equilibrate the intracellular pH (pHi) of the cells to the pH of the surrounding buffer. This stock is stable at -20 °C for 6 months); 0.5 mM stock of Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) in DMSO (protonophore to uncouple the proton motive force. This stock is stable at -20 °C for 6 months)
- Prepare 10 ml of the following high potassium buffers: 135 mM KH2PO4 / 20 mM NaOH (monobasic) and 110 mM K2HPO4 / 20 mM NaOH (dibasic). Filter sterilize using a 0.45 µm filter. (These buffers can be stored at 4 °C until use).
- Mix the high potassium buffers at the necessary proportions to create high potassium buffers with at least 5-6 pH values ranging from 6.5 to 8.0.
- Prepare 1 ml of 2x Loading Buffer as follows: In the bottom of a 15 ml plastic conical tube, dispense 40 µl of 100x PowerLoad, followed by 20 µl of the 5 mM ion-sensitive dye directly into the PowerLoad. Vortex briefly to mix. Add 900 µl of PBS followed by 40 µl of 100x probenecid. Vortex the mixture one final time. Wrap in foil to protect from light.
- Loading the bacteria with dye
- Pellet and wash the 8 ml of the mid-log phase bacterial culture as described in 2.2.1. Remove supernatant, resuspend pellet in 2 ml of PBS to make a “4x” cell suspension.
- Add 1 ml of the 4x cell suspension to the 2x Loading Buffer. This will provide for a final volume of 2 ml, and final concentrations of 2x cells, 2x PowerLoad, 50 µM fluorescent dye, and 2x probenecid.
- Incubate in a 30 °C water bath for 40 min, protected from light. NOTE: The dye is loaded into the bacterial cells at the lower temperature of 30 °C and in the absence of any glycolytic substrates to minimize efflux of the dye.
- WASH 1: Pellet the cells by centrifugation at 2,400 x g for 10 min, remove supernatant to get rid of excess dye, and resuspend in 4 ml PBS containing 2x probenecid and 1 mM glucose (to reenergize the bacteria). WASH 2, 3: Repeat step 4.2.7 twice, with final resuspension in 4 ml PBS (for a final concentration of 1x cells) containing 2x probenecid and 10 mM glucose.
- Incubate at 37 °C for 5 min. This will allow the cells to fully energize and allow time for bacterial esterases to cleave the AM ester group away from the internalized BCECF.
- Establishing background fluorescence and in vivo calibration curve
- Prewarm the fluorescence detection plate-reader plate reader to 37 °C.
- To determine background fluorescence, filter-sterilize about 500 µl of the loaded and energized bacterial suspension (from step 4.2.7) to remove the bacteria, and add 200 µl of filtrate to the well of the 96-well plate.
- Place plate in the fluorescence detection plate-reader to measure the fluorescence (490 nm excitation / 530 nm emission for “Value 1” and 440 nm excitation / 530 nm emission for “Value 2”) for 1 min. This background fluorescence should be subtracted before calculation of the ratios for both the calibration curve and the experimental samples.
- To obtain an in vivo calibration curve, wash 500 µl aliquots of the loaded and energized cells from step 4.2.7 once and resuspend in high potassium buffers at different pH values (ranging from 6.5-8.0).
- Add 20 µM nigericin to the samples to equilibrate the intracellular pH of the cells to the pH of the surrounding buffer and incubate at 37 °C for 5 min. Add 200 µl of bacteria/pH buffer combinations to the wells of a 96-well plate.
- Place plate in the fluorescence detection plate-reader to measure the fluorescence for a few min to establish a steady reading for each well. Export the readings to a data analysis program such as Microsoft Excel.
- After subtracting the background fluorescence values, calculate the ratio of the fluorescence values by dividing Value 1 by Value 2. Plot the ratio values for each pH buffer to create the calibration curve. (A calibration curve should be generated for every new batch of loaded cells, as the amount of fluorescent dye that is loaded into the bacteria can vary each time).
- Measuring changes in intracellular pH
- For each desired sample, add 200 µl of the loaded and energized cells (from step 4.2.7) to the wells of a 96-well plate.
- Place plate in the fluorescence detection plate-reader and measure the fluorescence of the samples every 5 sec for 5 min.
- Eject the plate and add 10 µM CCCP to one well (serves as the positive control for decreasing intracellular pH) and the experimental agent(s) of interest to the other wells. To accurately compare their effects over time, add the CCCP and agents of choice at the same time using a multichannel pipette.
- Immediately return the plate to the plate-reader and continue measuring fluorescence every 5 sec for 10 min. As an additional control, eject the plate and add 20 µM Nigericin to all samples to equilibrate the pHi of the bacteria to the pH of the surrounding buffer, and read for 5 min.
- After subtracting the background fluorescence values, calculate the ratios of fluorescence for each sample. Interpolate the pHi for each reading from the calibration curve.
Representative Results
For all experiments, there is one sample and set of conditions present in each well. Thus, each tracing represents the fluorescence intensity of an entire population of bacteria over time. The results should be easily interpretable, with a clear distinction between the fluorescence of the treated samples and that of the untreated controls. The kinetics and degree of an observed change in fluorescence could provide information about the possible mechanism and extent of the event being monitored.
When exploring membrane polarity, the bacteria need to be incubated with DiBAC for about 40 min to allow for equilibration of the dye over the membrane, as indicated by the steady decrease and subsequent leveling of fluorescence in Figure 1A prior to treatment. As demonstrated in Figure 1B, PI does not require equilibration, as its fluorescent signal is steady throughout the first 40 min incubation, but its presence along with DiBAC is helpful to monitor membrane rupture concurrently with polarity. Depolarization and rupture of the bacteria is indicated by a rise in fluorescence intensity of both dyes. Other agents capable of depolarization and rupture, such as detergents (sodium deoxycholate18), uncouplers, or ionophores (CCCP), can be also be added to demonstrate these events using this methodology.
For Fura-2/AM, PBFI/AM, and BCECF/AM, the ratio of two fluorescent signals is calculated, and an increase or decrease in this ratio corresponds to the intracellular Ca2+ (Figure 2), K+ (Figure 3), and H+ (Figure 4) concentrations, respectively. Upon addition of the calcium ionophore ionomycin, Ca2+ flows into the cell causing an increase in the fluorescence ratio (Figure 2). Addition of valinomycin has the opposite effect, causing K+ to flow out of the cell and decrease the intracellular concentration, which is indicated by a decrease in the fluorescence ratio (Figure 3). BCECF allows for the measurement of intracellular pH by first creating a calibration curve (Figure 4A) in the presence of various buffers of known pH. A decrease in the fluorescence ratio of the dye corresponds to a decrease in pH, as seen following addition of the protonophores CCCP or nigericin, which collapses the proton gradient (Figure 4B).
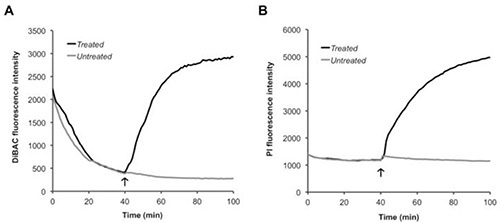 Figure 1. Monitoring membrane perturbations.S. pneumoniae was incubated with (A) DiBAC and (B) PI simultaneously for 40 min to allow for equilibration of DiBAC over the membrane. At the end of this incubation (arrow), PBS (untreated) or HAMLET (treated) was added and the samples were read for an additional hour. Click here to view larger image.
Figure 1. Monitoring membrane perturbations.S. pneumoniae was incubated with (A) DiBAC and (B) PI simultaneously for 40 min to allow for equilibration of DiBAC over the membrane. At the end of this incubation (arrow), PBS (untreated) or HAMLET (treated) was added and the samples were read for an additional hour. Click here to view larger image.
 Figure 2. Detecting changes in intracellular calcium levels. Pneumococci were loaded with Fura-2/AM and treated (arrow) with PBS (untreated) or the calcium ionophore ionomycin (positive control). The ratio of fluorescence values is presented. Click here to view larger image.
Figure 2. Detecting changes in intracellular calcium levels. Pneumococci were loaded with Fura-2/AM and treated (arrow) with PBS (untreated) or the calcium ionophore ionomycin (positive control). The ratio of fluorescence values is presented. Click here to view larger image.
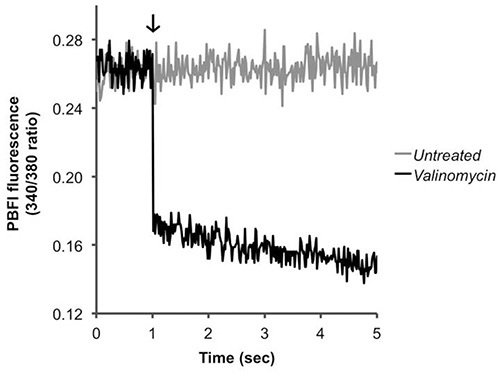 Figure 3. Detecting changes in intracellular potassium levels. Pneumococci were loaded with PBFI/AM and treated (arrow) with PBS (untreated) or the potassium ionophore valinomycin (positive control). The ratio of fluorescence values is presented. Click here to view larger image.
Figure 3. Detecting changes in intracellular potassium levels. Pneumococci were loaded with PBFI/AM and treated (arrow) with PBS (untreated) or the potassium ionophore valinomycin (positive control). The ratio of fluorescence values is presented. Click here to view larger image.
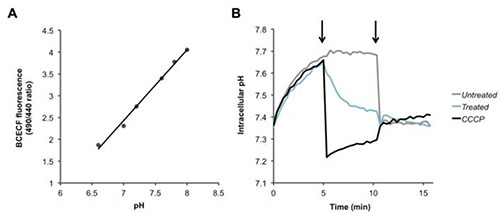 Figure 4. Detecting changes in intracellular proton levels. (A) Standard curve for intracellular pH calibration. (B) Pneumococci were loaded with the pH sensitive dye BCECF-AM, and were washed and resuspended in PBS + glucose. After recording baseline readings, at the first arrow, PBS (black), the protonophore CCCP (100 µM), or HAMLET (100 µg/ml or 6 µM), were added to the bacteria and fluorescence was measured over time. At the second arrow nigericin (20 µM) was added to completely dissipate the transmembrane proton gradient of all of the samples. Click here to view larger image.
Figure 4. Detecting changes in intracellular proton levels. (A) Standard curve for intracellular pH calibration. (B) Pneumococci were loaded with the pH sensitive dye BCECF-AM, and were washed and resuspended in PBS + glucose. After recording baseline readings, at the first arrow, PBS (black), the protonophore CCCP (100 µM), or HAMLET (100 µg/ml or 6 µM), were added to the bacteria and fluorescence was measured over time. At the second arrow nigericin (20 µM) was added to completely dissipate the transmembrane proton gradient of all of the samples. Click here to view larger image.
Discussion
Despite the limitation that size presents for using classic electrophysiology methods to detect changes in polarity and integrity of the membrane and changes in ion concentrations within bacteria, we have described a way to measure these events in S. pneumoniae using fluorescent dyes. Our protocols are the first of their kind described for the pneumococcus and one of the few described for bacterial species in general. By using a fluorescence detection plate reader, these events can be measured in small, 200 µl volume samples of bacteria, with fluorescence detected from the individual population of bacteria over time. The kinetics and changes in fluorescence intensity can be used to qualitatively determine what is happening to membrane polarity or integrity, or levels of Ca2+, K+, or H+ within the bacteria. Quantitatively speaking, the fluorescence intensity values can also be used to calculate the degree of depolarization, rupture, calcium uptake, potassium efflux, or pH change observed in one sample compared to another, providing critical information about a mechanism of interest. Additionally, our protocol should be useful as a starting point for loading other AM fluorescent dyes, allowing for the study of a variety of other cellular events in a variety of bacterial species. We have already been successful in applying some of these protocols in Staphylococcus aureus21.
When using the AM dyes, ample incubation time is important for dye loading and achieving the subsequent hydrolysis necessary for the dye to respond to the target ions within the bacteria and yield the corresponding fluorescence changes. The loading kinetics may vary between bacterial strains due to several factors including differences in bacterial surface architecture (capsule presence, membrane fluidity, presence of efflux pumps, etc.) and in the chemical structure of the dye. To achieve maximal loading for the bacterial strain and dye of interest, there are several parameters of our protocol that may require modulation including: incubation duration, incubation temperature, nutrient availability, PowerLoad concentration, and probenecid concentration. To obtain successful loading, fluorescence signal intensity is less important than showing that positive controls, such as addition of ionophores, provide a signal in the correct direction and/or showing linearity in the calibration curve. To optimize loading an increase in PowerLoad and Probenicide concentration followed by a lowering of the temperature to avoid extrusion of the dye is preferable to increasing dye concentration that may result in extracellular staining that will confound the signal. During the development of our protocols, we were able to successfully load our dyes of interest in both the wild type encapsulated pneumococcal strain D39 and the unencapsulated strain R36A25. However, we found that we could achieve optimal loading as indicated by fluorescent readings leading to increased ratios using R36A for Fura-2 and PBFI experiments using 37 °C incubation temperature, while a lower incubation temperature coupled with higher PowerLoad and probenecid concentrations worked better for loading of BCECF into D39. Although specific signal intensity may vary between experiments, the ratiometric nature of the measurements provided results with high reproducibility and little spread.
For fluorescence measurements of samples, use of the software accompanying most advanced plate readers allows for adjustments of a variety of detection parameters, including detection speed, detection sensitivity, and read duration, to name a few. This allows the researcher to find the optimal sensitivity for detection of the particular event that is being monitored. For optimal interpretation of results, it is critical that stable baselines of fluorescence and linear standard curves are established, as there can be inter-assay variations in the amount of dye that is present, equilibrated over the membrane, or loaded into the bacteria and hydrolyzed. Allowing time for dye equilibration over the bacterial membrane prior to treatment of any kind is particularly crucial when using DiBAC4(3) to allow for a stabilized fluorescence level prior to treatment. Additionally, including all of the proper controls in each individual analysis is essential for accurate data analysis, as the experimental additives that are introduced into the bacterial suspension to be tested may themselves autofluoresce at the particular wavelengths of detection or may unspecifically modify the fluorescence of the indicator dye through direct interactions.
We recognize a few potential areas of concern associated with the methods that we have described here. Unlike the carbocyanine dye DiOC2(3), DiBAC4(3) is a bisoxonol dye that is not ratiometric, so while the carbocyanine dye can account for changes in cell volume, DiBAC4(3) cannot. Thus, there may be changes in the level of fluorescence that correspond to changes in cell volume. We, however, did not notice this to be a significant problem, as the pneumococcus has a rigid cell wall that does not permit significant changes in volume without rupturing the bacteria. For examining bacterial ion fluxes, the sensitivity of the ion-sensitive dyes can vary depending on the ion of interest and the degree of change in its concentration. Additionally, the use of fluorescent dyes is experimentally not as direct a method as the use of radioisotopes. However, depending on the ion of interest, examining fluorescence is a much more practical option when considering both financial and safety-related limitations involved with the use of radioisotopes. Thus, the methodologies described in this manuscript provide new and consistent approaches to better study membrane polarity, integrity, and transport events and in bacterial systems.
Disclosures
The authors have no competing financial interests to declare.
Acknowledgments
This work was supported by the Bill and Melinda Gates Foundation (Grant 53085), the JR Oishei Foundation, and The American Lung Association (Grant RG-123721-N) to APH, and NIH (NIDCD) fellowship F31DC011218 to EAC.
References
- Hakansson A, Zhivotovsky B, Orrenius S, Sabharwal H, Svanborg C. Apoptosis induced by a human milk protein. Proc. Natl. Acad. Sci. U.S.A. 1995;92(17):8064–8068. doi: 10.1073/pnas.92.17.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson A, et al. A folding variant of alpha-lactalbumin with bactericidal activity against Streptococcus pneumoniae. Mol. Microbiol. 2000;35(3):589–600. doi: 10.1046/j.1365-2958.2000.01728.x. [DOI] [PubMed] [Google Scholar]
- Hakansson AP, Roche-Hakansson H, Mossberg AK, Svanborg C. Apoptosis-Like Death in Bacteria Induced by HAMLET, a Human Milk Lipid-Protein Complex. PLoS One. 2011;6(3) doi: 10.1371/journal.pone.0017717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C, Gogvadze V, Hakansson A, Svanborg C, Orrenius S, Zhivotovsky B. A folding variant of human alpha-lactalbumin induces mitochondrial permeability transition in isolated mitochondria. Eur. J. Biochem. 2001;268(1):186–191. doi: 10.1046/j.1432-1327.2001.01870.x. [DOI] [PubMed] [Google Scholar]
- Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (Δψ m) in apoptosis; an update. Apoptosis. 2003;8(2):115–128. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- Dominguez DC. Calcium signalling in bacteria. Mol. Microbiol. 2004;54(2):291–297. doi: 10.1111/j.1365-2958.2004.04276.x. [DOI] [PubMed] [Google Scholar]
- Corratge-Faillie C, Jabnoune M, Zimmermann S, Very AA, Fizames C, Sentenac H. Potassium and sodium transport in non-animal cells: the Trk/Ktr/HKT transporter family. Cell Mol. Life Sci. 2010;67(15):2511–2532. doi: 10.1007/s00018-010-0317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Petronilli V, Zoratti M. A patch-clamp study of Bacillus subtilis. Biochim. Biophys. Acta. 1992;1112(1):29–38. doi: 10.1016/0005-2736(92)90250-p. [DOI] [PubMed] [Google Scholar]
- Zoratti M, Petronilli V, Szabo I. Stretch-activated composite ion channels in Bacillus subtilis. Biochem. Biophys. Res. Commun. 1990;168(2):443–450. doi: 10.1016/0006-291x(90)92341-v. [DOI] [PubMed] [Google Scholar]
- Novo D, Perlmutter NG, Hunt RH, Shapiro HM. Accurate flow cytometric membrane potential measurement in bacteria using diethyloxacarbocyanine and a ratiometric technique. Cytometry. 1999;35(1):55–63. doi: 10.1002/(sici)1097-0320(19990101)35:1<55::aid-cyto8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Novo DJ, Perlmutter NG, Hunt RH, Shapiro HM. Multiparameter flow cytometric analysis of antibiotic effects on membrane potential, membrane permeability, and bacterial counts of Staphylococcus aureus and Micrococcus luteus. Antimicrob. Agents Chemother. 2000;44(4):827–834. doi: 10.1128/aac.44.4.827-834.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro HM. Membrane potential estimation by flow cytometry. Methods. 2000;21(3):271–279. doi: 10.1006/meth.2000.1007. [DOI] [PubMed] [Google Scholar]
- Shapiro HM. Microbial analysis at the single-cell level: tasks and techniques. J. Microbiol. Methods. 2000;42(1):3–16. doi: 10.1016/s0167-7012(00)00167-6. [DOI] [PubMed] [Google Scholar]
- Bashford CL, Chance B, Smith JC, Yoshida T. The behavior of oxonol dyes in phospholipid dispersions. Biophys. J. 1979;25(1):63–85. doi: 10.1016/S0006-3495(79)85278-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Wang Z-Y, Yamakoshi M, Kobayashi M, Nozawa T. Probing the transmembrane potential of bacterial cells by voltage-sensitive dyes. Anal. Sci. 2003;19(9):1239–1242. doi: 10.2116/analsci.19.1239. [DOI] [PubMed] [Google Scholar]
- Breeuwer P, Abee T. Assessment of the membrane potential, intracellular pH and respiration of bacteria employing fluorescence techniques. Mol. Microb. Ecol. Manual. 2004;8:1563–1580. [Google Scholar]
- Mortimer FC, Mason DJ, Gant VA. Flow cytometric monitoring of antibiotic-induced injury in Escherichia coli using cell-impermeant fluorescent probes. Antimicrob. Agents Chemother. 2000;44(3):676–681. doi: 10.1128/aac.44.3.676-681.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi EA, Marks LR, Duffey ME, Hakansson AP. A Novel Initiation Mechanism of Death in Streptococcus pneumoniae Induced by the Human Milk Protein-Lipid Complex HAMLET and Activated during Physiological Death. J. Biol. Chem. 2012;287(32):27168–27182. doi: 10.1074/jbc.M112.371070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombe MC, Laneelle G, Sicard AM. Characterization of a Streptococcus pneumoniae mutant with altered electric transmembrane potential. J. Bacteriol. 1984;158(3):1109–1114. doi: 10.1128/jb.158.3.1109-1114.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombe MC. Characterization of a calcium porter of Streptococcus pneumoniae involved in calcium regulation of growth and competence. J. Gen. Microbiol. 1993;139(3):433–439. doi: 10.1099/00221287-139-3-433. [DOI] [PubMed] [Google Scholar]
- Marks LR, Clementi EA, Hakansson AP. Sensitization of Staphylococcus aureus to Methicillin and Other Antibiotics In Vitro and In Vivo in the Presence of HAMLET. PLoS ONE. 2013;8(5) doi: 10.1371/journal.pone.0063158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futsaether CM, Johnsson A. Using fura-2 to measure intracellular free calcium in Propionibacterium acnes. Can. J. Microbiol. 1994;40(6):439–445. doi: 10.1139/m94-072. [DOI] [PubMed] [Google Scholar]
- Tisa LS, Adler J. Chemotactic properties of Escherichia coli mutants having abnormal Ca2+ content. J. Bacteriol. 1995;177(24):7112–7118. doi: 10.1128/jb.177.24.7112-7118.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werthen M, Lundgren T. Intracellular Ca2+ mobilization and kinase activity during acylated homoserine lactone-dependent quorum sensing in Serratia liquefaciens. J. Biol. Chem. 2001;276(9):6468–6472. doi: 10.1074/jbc.M009223200. [DOI] [PubMed] [Google Scholar]
- Avery OT, MacLeod CM, McCarty M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a dexoxyribonuceic acid fraction isolated from pneumococcus type III. J. Exp. Med. 1944;79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi EA, Wilhelm KR, Schleucher J, Morozova-Roche LA, Hakansson AP. A Complex of Equine Lysozyme and Oleic Acid with Bactericidal Activity against. PLoS One. 2013;8 doi: 10.1371/journal.pone.0080649. [DOI] [PMC free article] [PubMed] [Google Scholar]