

Abstract
Autism is a highly disabling neurodevelopmental disorder characterized by social deficits, language impairment, and repetitive behaviors. There are few effective biological treatments for this disorder, partly due to the lack of translational biomarkers. However, recent data suggest that autism has reliable electrophysiological endophenotypes, along with evidence that some deficits may be caused by NMDA receptor (NMDAR) dysfunction. Similarly, the NMDAR antagonist MK801 has been used in behavioral animal models of autism. Since MK801 has also been used as a model of schizophrenia, this paper examines the independent and overlapping ways in which MK801 recreates the electrophysiogical changes present in both diseases. Mouse EEG was recorded in response to auditory stimuli after either vehicle or MK801 and the dose-response relationship for each measure was determined. ERP component amplitude and latency analysis was performed along with time–frequency analysis of gamma frequency inter-trial coherence and evoked power. Evoked gamma power and ITC were decreased by MK801 at the highest dose. P1, N1 latency and gamma baseline power were increased in dose dependent fashion following MK801. There were no amplitude changes in P1 or N1. MK801 caused alterations in evoked gamma activity, gamma ITC, gamma baseline power, P1 and N1 latency similar to findings in autism. These data provide evidence indicating that NMDAR dysfunction may contribute to deficits specific to autism and some that overlap with other disorders such as schizophrenia. Such observations could be important for developing novel therapeutics, as electrophysiological endophenotypes associate with functional measures and may provide early biomarkers for efficacy in clinical trials.
Keywords: Autism, Electrophysiology, Endophenotype, Animal models, NMDA receptor antagonist
Autism spectrum disorders (ASD) are characterized by deficits in social, cognitive, and language function. Despite emerging evidence for genetic and molecular contributions to the disorder, behavioral measures remain the primary method of diagnosis. However, electrophysiological and neural biomarkers could potentially provide robust measures to quantify the degree of impairment and provide more focused guidance for treatment development [1]. These measures have advantages in their translatability between mice and humans. For instance, human auditory evoked responses have a P50 (P1) and N100 (N1) which display similar morphology and drug responses as the mouse P20 (P1) and N40 (N1) [2]. These measures can also be examined using magnetoencephalography (MEG) which has magnetic analogues including the M50 and M100 [3]. In addition, there is evidence that gamma inter-trial coherence and evoked power are related to similar cognitive and sensory processes across species [4].
Several recent findings provide guidance for pre-clinical model development for ASD. First, genetic and postmortem studies indicate that the pathophysiology of ASD may include a component of NMDA receptor (NMDAR) disruption [5]. Additional support for this idea has been provided by studies using MK801 to recreate autism-like behavioral deficits in mice [6,7]. Studies in children with ASD using MEG have elucidated novel neural biomarkers for the disorder which could be translated into animal EEG models [8]. Specifically, children with ASD show a delay in the M100 in superior temporal gyrus (STG), with this latency prolongation providing a high degree of accuracy for ASD classification. There is also evidence that neural synchrony is disturbed in ASD, particularly in the gamma-band [3]. Such oscillations are important for sensory integration and functional connectivity, which are disrupted in ASD [9]. There is also evidence that P1 and N1 latency are predictive of language impairment [10,11].
The current study examines a potential pre-clinical model for several types of electrophysiological biomarkers, P1 and N1 latency prolongation, reduced gamma inter-trial coherence and gamma evoked power, following exposure to a NMDAR antagonist, representative data shown in Fig. 1. We propose that quantifying the extent to which clinical ASD markers are recapitulated in this model will be a critical step toward evaluating its predictive validity for therapeutic development. This is important because NMDAR disruption models have also been closely linked to schizophrenia, another disabling but distinct disease. For example, gamma inter-trial coherence and evoked power disruptions have been shown in both diseases [3,4]. In contrast, there is evidence that P1 and N1 latency is increased in ASD, while most studies in schizophrenia have shown no change in either latency [12]. Alternatively, N1 amplitude is reduced in schizophrenia, but remains unaffected in ASD [8,13]. Therefore, parsing which aspects of the model map to ASD versus schizophrenia are important for determining the precise mechanisms of deficit, and corresponding therapeutic approaches in the different disorders.
Fig. 1.

Column A represents the raw EEG trace for four epochs. In column B, a representative EEG trace filtered between 30 and 80 Hz is presented, showing the gamma present in the raw signal. In column C, the ERP from the previous raw animal data is shown with the P1 and N1 components marked. In column D, a group inter-trial coherence plot is shown for each drug dose.
The MK801 dose-response relationship to schizophrenia- and autism-like symptoms may be related to the degree of glutamate signaling disruption in each disorder. We observed that low levels of MK801 caused EEG changes that are more autism-like with delays in P1 and N1 latency accompanied by reduced gamma coherence and evoked power. Alternatively, high dose MK801 caused reductions in N1 amplitude, gamma inter-trial coherence, and evoked power, similar to schizophrenia [14]. Such findings suggest a common glutamatergic circuit disruption in ASD and schizophrenia that varies in degree.
For our studies we used 15 male C57BL/6Hsd (B6) mice that were obtained from Jackson Laboratories at 7–8 weeks of age. All testing was conducted between 10 and 18 weeks of age. Mice were acclimated to the animal facility for at least 7 days before experimentation began and were housed four to five per cage until implantation of the recording electrode, after which they were single-housed for the remainder of the study. All subjects were maintained in a standard 12-h light/dark cycle with free access to food and water. Experiments were performed during the light phase between 9:00 a.m. and 4:00 p.m. All protocols were conducted in accordance with University Laboratory Animal Resources (ULAR) guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania. All efforts were undertaken to minimize the number of animals used in the experiment and their suffering.
The procedure for electrode implantation was as follows, at 12 weeks, animals underwent stereotaxic implantation of a stainless steel tripolar electrode assembly (Plastics1, Roanoke, Virginia) as published [15]. Animals were anesthetized with isoflurane, and a low-impedance (5k, 1000 Hz) macro-electrode was stereotaxically positioned between auditory cortex and auditory thalamus (1.8 mm posterior, 2.65 mm lateral, 2.75 mm deep relative to bregma) and referenced to frontal sinus. This configuration captures both early and late components of the auditory evoked potential (AEP), including the midlatency P20 (e.g., human P50/M50) and N40 (e.g., human N100/M100) [16]. The electrode pedestal was secured to the skull using ethyl cyanoacrylate (Loctite, Henkel, Germany) and dental cement (Ortho Jet, Lang Dental, Wheeling, Illinois). EEG was recorded 1 week later, as described below.
EEG was then recorded with Micro1401 hardware and Spike6 software (CED, Cambridge, UK) as published previously [17]. A total of 250 white noise clicks were presented with a 4 s interstimulus interval at 85 dB.
TheMK801 used was obtained from Sigma–Aldrich (St. Louis, MO). MK801 was used at doses of 0.1, 0.2, 0.3, and 0.4 mg/kg i.p. All drugs and saline controls were administered 5–10 min prior to initiation of recording. MK801 has a half-life of 2 h in rodents, ensuring consistent drug effect over a recording session.
ERP amplitudes and latencies were calculated for P1 (defined as the most positive deflection between 10 and 40 ms) and N1 (defined as the most negative deflection between 30 and 200 ms). These regions are important because the characteristic positive and negative deflections of the EEG recording occur at approximately 40% the latency but similar morphology to the equivalent human components [16,18]. Therefore, the P20 and N40 represent ERP deflections in mice analogous to the P50 and N100, respectively, in humans [2].
Inter-trial coherence (ITC), baseline (total power occurring −100 to −400 ms prestimulus), induced (non-phase locked power created post-stimulus with post-stimulus defined as 0–60 ms after each of the 200 stimuli) and evoked power (total power minus induced and baseline power), values were calculated with Morlet wavelet decomposition using EEGLab [19]. In particular, single-trial epochs −250 and 750 ms relative to the auditory stimulus were extracted from the continuous data. For each epoch (trial), the ITC, baseline power, and evoked power were calculated using 69 linearly-spaced frequencies from 12 to 80 Hz, with wavelet cycles increasing from 2 (at low frequencies) to 10 (at high frequencies). ITC is expressed as a unitless ratio between 0 and 1, where 1 represents complete phase synchrony at a given frequency and time across trials. Evoked and baseline were expressed in dB (10 log10). For each subject, the ITC and power measures were calculated as the average of a window around the peak gamma response, defined as the region from 0 to 60 ms poststimulus and from 30 to 80 Hz. In addition, P1 and N1 latency was calculated in milliseconds after stimulus.
Repeated measures ANOVAs were performed to investigate the effect of each pharmacologic agent on EEG measures. Significant effects were followed by Tukey’s post-tests where appropriate.
The following findings were observed, N1 latency changes had a significant positive association with dose of MK801 (Fig. 2 P < 0.0001, F(4,56) = 8.657). P1 latency changes had a significant positive association with dose of MK801 (Fig. 2 P < 0.0001, F(4,56) = 9.855). Gamma inter-trial coherence (Fig. 3 P < 0.001, F(4,56) = 5.738) and evoked gamma power (Fig. 3 P < 0.001, F(4,56) = 5.372) had a significant negative relationship with MK801 dose. Gamma baseline power had a significant positive relationship to MK801 dose (Fig. 3 P < 0.0001, F(4,56) = 13.15).
Fig. 2.
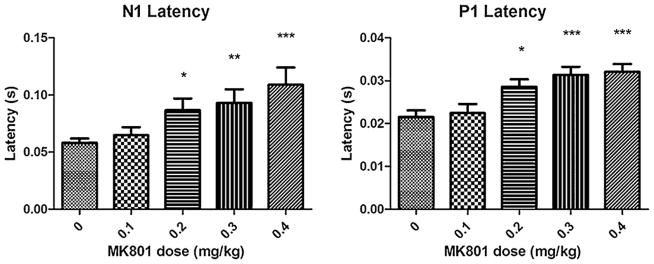
N1 latency was examined following MK801 at 0.1, 0.2, 0.3, 0.4 mg/kg. There was a significant relationship between increasing MK801 dose and longer N1 latency (P < 0.0001, F(4,56) = 8.657). P1 latency was examined following MK801 at 0.1, 0.2, 0.3, 0.4 mg/kg. There was a significant relationship between increasing MK801 dose and longer P1 latency (P < 0.0001, F(4,56) = 9.855). This increase in P1 and N1 latency is similar to the pattern found for the corresponding M50 in children with language impairment using MEG [8,10]. Figure shows mean + SEM (*P < 0.05, **P < 0.01, ***P < 0.001 after Tukey’s test for multiple comparisons between saline and conditions).
Fig. 3.
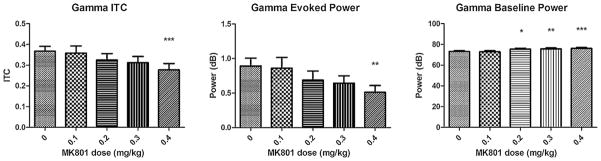
Gamma ITC, evoked, and baseline power were examined following exposure to MK801 at 0.1, 0.2, 0.3, and 0.4 mg/kg. There was a significant relationship between increasing dose of MK801and reduction in gamma ITC and evoked power (gamma ITC: P < 0.001, F(4,56) = 5.738, gamma evoked power: P < 0.001, F(4,56) = 5.372). MK801 dose dependently increased gamma baseline power (P < 0.0001, F(4,56) = 13.15). Thus, MK801 recapitulates the overlapping pattern of reduced gamma ITC and evoked power along with increased gamma baseline power found among patients with either ASD or schizophrenia (Fig. 4). Figure shows mean + SEM (**P < 0.01, ***P < 0.01 after Tukey’s test for multiple comparisons between saline and conditions).
After Tukey’s post-test for multiple comparisons, there were significant differences in N1 latency between saline and 0.2, 0.3, or 0.4 mg/kg MK801 conditions (P < 0.05). There were significant differences in P1 latency between saline and 0.2, 0.3. 0.4 mg/kg MK801 conditions. There were significant differences in gamma inter-trial coherence and evoked power between saline and 0.4 mg/kg MK801 along with 0.1 mg/kg and 0.4 mg/kg MK801 (P < 0.01) conditions. For gamma baseline power, there were significant differences between saline and 0.2, 0.3, 0.4 mg/kg MK801 (P < 0.05). There were no significant associations between dose and P1 or N1 amplitude (P = 0.8553, P = 0.1703).
The current study demonstrates that P1 latency, N1 latency, evoked gamma power, gamma inter-trial coherence, and gamma baseline power changes following MK801 mimic electrophysiological endophenotypes of ASD [20]. N1 latency delays of 10% matched those found in ASD MEG studies which in turn were able to discriminate between patients and controls [3,8]. The ERP alterations following MK801 were specific to P1 and N1 latency, with no amplitude changes observed for P1 or N1 components, also similar to findings in ASD. In addition, while latency changes are small, significant differences existed because measurements had very low variability, indicating tight physiological control of response latency. These electrophysiological data are consistent with previous studies in which MK801 has been used to model the core deficits of ASD in rodents including stereotypies, social deficits and withdrawal [6,7].
One limitation of the current study is that the disruptions in gamma inter-trial coherence, baseline power, and evoked power disruptions match recordings done in ASD, but are also present in schizophrenia. Similarly, stereotypies, social deficits and withdrawal created by MK801 are also found in schizophrenia [21]. Qualitative, but nonsignificant reduction of N1 amplitude was found in the current data set, consistent with the direction but not extent of changes in schizophrenia. Additionally, drugs like MK801 and phencyclidine (PCP) can induce pre-pulse inhibition (PPI) deficits and hyperlocomotion at the doses tested, indicating that deficits are not selectively tied to social function. Thus, MK801 is not specific as a model of ASD and reduced NMDAR-mediated glutamate transmission appears to model the overlap between ASD and schizophrenia. It therefore may be helpful in determining the common, rather than distinguishing aspects of their pathophysiology.
The pattern of endophenotypes that are shared by schizophrenia and ASD illustrate the relative extent of glutamatergic contributions towards the two disorders (Fig. 4). There is evidence to suggest that both disorders have a glutamate component and several studies have shown a connection between autism-like symptoms and glutamate disruption. For example, anti-NMDA receptor encephalitis has been reported to mimic late onset autism [22]. This may indicate that there are localized areas of glutamatergic dysfunction in ASD that give rise to the change in electrophysiological response latencies, gamma coherence, and gamma evoked power without psychosis. In contrast, the glutamatergic dysfunction of schizophrenia may be the driver of psychosis, negative symptoms, and cognitive deficits. Another possible hypothesis is that low levels of glutamatergic disruption may be more ASD like, while greater dysfunction is more schizophrenia like. The qualitative but non-significant reduction in N1 at the MK801 dosages used supports this view because higher doses have previously been reported to reduce N1 amplitude similar to those found in schizophrenia [14]. However, P1 and N1 latency changes are typically not reported in schizophrenia, suggesting the electrophysiological disruptions may be from different domains. Given that patients with schizophrenia have EEG timing deficits, as measured by ITC, and that latency shifts in the current study and ASD are significant because their variability is extremely low, the reduction in consistency of EEG response timing in schizophrenia may mask any changes in latency from being measured.
Fig. 4.
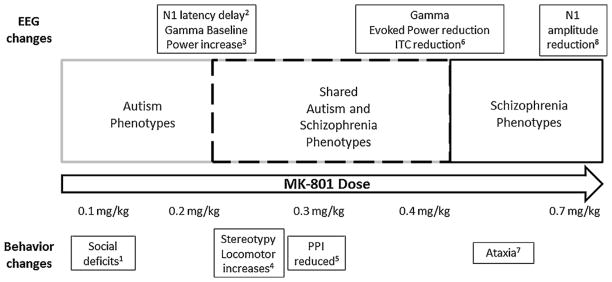
Graphical illustration of the continuum of EEG and behavioral deficits along different doses of MK801. The autism specific and overlapping with schizophrenia phenotypes are elucidated at the doses used in this study (1) [7] (2) [3,8] (3) [20] (4) [23] (5) [24] (6) [3] (7) [25] (8) [14].
In summary, the current study indicates that acute pharmacological disruption of NMDAR signaling recapitulates several aspects of ASD, as well as those of schizophrenia. Furthermore, previous studies suggest that the electrophysiological endophenotypes induced by MK801 are linked to functional measures in both ASD and schizophrenia, this model may be useful for testing novel therapeutics for language impairment among other targets [10]. For example, behavioral effects of NMDA disruption have been reversed with mGluR5 agonist MPEP. Similar findings in future electrophysiological and behavioral studies could suggest that reversal EEG deficits would provide an early indication that novel compounds could be effective in autism and/or schizophrenia.
HIGHLIGHTS.
Low dose MK-801 creates EEG endophenotypes similar to autism.
At the highest doses tested, these changes overlap with schizophrenia findings.
These findings illustrate the relative amount of NMDAR dysfunction in the two disorders.
Acknowledgments
Role of funding agencies
The study was supported by 5R01DA023210-02 (SJS) R01DC008871 (TPR). The funding agencies had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication
Dr. Roberts thanks the Oberkircher Family for the Oberkircher Family Endowed Chair in Pediatric Radiology.
Footnotes
Conflicts of interest
Steven Siegel reports having received grant support from Eli Lilly, AstraZeneca, NuPathe, and Pfizer that is unrelated to the content of this paper and consulting payments from NuPathe, Merck, Sanofi, and Wyeth that are unrelated to this work. Dr Roberts is a consultant for prism clinical imaging. All other authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Edgar JC, Keller J, Heller W, Miller GA. Handbook of psychophysiology. 3. New York: Cambridge University Press; 2007. Psychophysiology in research on psychopathology. [Google Scholar]
- 2.Amann LC, Gandal MJ, Halene TB, Ehrlichman RS, White SL, McCarren HS, et al. Mouse behavioral endophenotypes for schizophrenia. Brain Research Bulletin. 2010;83:147–61. doi: 10.1016/j.brainresbull.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Gandal MJ, Edgar JC, Ehrlichman RS, Mehta M, Roberts TP, Siegel SJ. Validating gamma oscillations and delayed auditory responses as translational biomarkers of autism. Biological Psychiatry. 2010;68:1100–6. doi: 10.1016/j.biopsych.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gandal MJ, Edgar JC, Klook K, Siegel SJ. Gamma synchrony: towards a translational biomarker for the treatment-resistant symptoms of schizophrenia. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bangash MA, Park JM, Melnikova T, Wang D, Jeon SK, Lee D, et al. Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell. 2011;145:758–72. doi: 10.1016/j.cell.2011.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Wu J, Zou H, Strong JA, Yu J, Zhou X, Xie Q, et al. Bimodal effects of MK-801 on locomotion and stereotypy in C57BL/6 mice. Psychopharmacology (Berlin) 2005;177:256–63. doi: 10.1007/s00213-004-1944-1. [DOI] [PubMed] [Google Scholar]
- 7.Zou H, Zhang C, Xie Q, Zhang M, Shi J, Jin M, et al. Low dose MK-801 reduces social investigation in mice. Pharmacology Biochemistry and Behavior. 2008;90:753–7. doi: 10.1016/j.pbb.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts TP, Khan SY, Rey M, Monroe JF, Cannon K, Blaskey L, et al. MEG detection of delayed auditory evoked responses in autism spectrum disorders: towards an imaging biomarker for autism. Autism Research. 2010;3:8–18. doi: 10.1002/aur.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oram Cardy JE, Flagg EJ, Roberts W, Roberts TP. Auditory evoked fields predict language ability and impairment in children. International Journal of Psychophysiology. 2008;68:170–5. doi: 10.1016/j.ijpsycho.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Roberts TP, Cannon KM, Tavabi K, Blaskey L, Khan SY, Monroe JF, et al. Auditory magnetic mismatch field latency: a biomarker for language impairment in autism. Biological Psychiatry. 2011;70:263–9. doi: 10.1016/j.biopsych.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanlon FM, Miller GA, Thoma RJ, Irwin J, Jones A, Moses SN, et al. Distinct M50 and M100 auditory gating deficits in schizophrenia. Psychophysiology. 2005;42:417–27. doi: 10.1111/j.1469-8986.2005.00299.x. [DOI] [PubMed] [Google Scholar]
- 13.Mazhari S, Price G, Waters F, Dragovic M, Jablensky A. Evidence of abnormalities in mid-latency auditory evoked responses (MLAER) in cognitive subtypes of patients with schizophrenia. Psychiatry Research. 2011;187:317–23. doi: 10.1016/j.psychres.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Saunders JA, Gandal MJ, Siegel SJ. NMDA antagonists recreate signal-to-noise ratio and timing perturbations present in schizophrenia. Neurobiology of Disease. 2012;46:93–100. doi: 10.1016/j.nbd.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gandal MJ, Ehrlichman RS, Rudnick ND, Siegel SJ. A novel electrophysiological model of chemotherapy-induced cognitive impairments in mice. Neuroscience. 2008;157:95–104. doi: 10.1016/j.neuroscience.2008.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siegel SJ, Connolly P, Liang Y, Lenox RH, Gur RE, Bilker WB, et al. Effects of strain, novelty, and NMDA blockade on auditory-evoked potentials in mice. Neuropsychopharmacology. 2003;28:675–82. doi: 10.1038/sj.npp.1300087. [DOI] [PubMed] [Google Scholar]
- 17.Ehrlichman RS, Gandal MJ, Maxwell CR, Lazarewicz MT, Finkel LH, Contreras D, et al. N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience. 2009;158:705–12. doi: 10.1016/j.neuroscience.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 18.Umbricht D, Vyssotky D, Latanov A, Nitsch R, Brambilla R, D’Adamo P, et al. Mid-latency auditory event-related potentials in mice: comparison to midlatency auditory ERPs in humans. Brain Research. 2004;1019:189–200. doi: 10.1016/j.brainres.2004.05.097. [DOI] [PubMed] [Google Scholar]
- 19.Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. Journal of Neuro-science Methods. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Lushchekina EA, Podreznaia ED, Lushchekin VS, Strelets VB. Comparative EEG study in normal and autistic children. Zhurnal Vysshei Nervnoi Deiatelnosti Imeni I P Pavlova. 2010;60:657–66. [PubMed] [Google Scholar]
- 21.Burket JA, Cannon WR, Jacome LF, Deutsch SI. MK-801, a noncompetitive NMDA receptor antagonist, elicits circling behavior in the genetically inbred Balb/c mouse strain. Brain Research Bulletin. 2010;83:337–9. doi: 10.1016/j.brainresbull.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 22.Creten C, van der Zwaan S, Blankespoor RJ, Maatkamp A, Nicolai J, van Os J, et al. Late onset autism and anti-NMDA-receptor encephalitis. Lancet. 2011;378:98. doi: 10.1016/S0140-6736(11)60548-5. [DOI] [PubMed] [Google Scholar]
- 23.Ninan I, Kulkarni SK. 5-HT2A receptor antagonists block MK-801-induced stereotypy and hyperlocomotion. European Journal of Pharmacology. 1998;358:111–6. doi: 10.1016/s0014-2999(98)00591-3. [DOI] [PubMed] [Google Scholar]
- 24.Ralph-Williams RJ, Lehmann-Masten V, Otero-Corchon V, Low MJ, Geyer MA. Differential effects of direct and indirect dopamine agonists on prepulse inhibition: a study in D1 and D2 receptor knock-out mice. Journal of Neuroscience. 2002;22:9604–11. doi: 10.1523/JNEUROSCI.22-21-09604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Oliveira RV, Dall’Igna OP, Tort AB, Schuh JF, Neto PF, Santos Gomes MW, et al. Effect of subchronic caffeine treatment on MK-801-induced changes in locomotion, cognition and ataxia in mice. Behavioural Pharmacology. 2005;16:79–84. doi: 10.1097/00008877-200503000-00002. [DOI] [PubMed] [Google Scholar]