

Abstract
Histone deacetylase inhibitors (HDACi) affect chromatin remodeling and modulate the expression of aberrantly silenced genes. HDACi have single-agent clinical activity in haematological malignancies and have synergistic anti-leukaemia activity when combined with anthracyclines in vitro. We conducted a 2-arm, parallel Phase I trial to investigate 2 schedules of escalating doses of vorinostat (Schedule A: thrice daily (TID) for 14 days; B: TID for 3 days) in combination with a fixed dose of idarubicin in patients with refractory leukaemia. Of the 41 patients enrolled, 90% had acute myeloid leukaemia, with a median of 3 prior therapies. Seven responses (17%) were documented (2 complete response (5%), 1 complete response without platelet recovery (2.5%), and 4 marrow responses). The 3-day schedule of vorinostat was better tolerated than the 14-day schedule. The maximum tolerated dose for vorinostat was defined as 400 mg TID for 3 days. The most common grade 3 and 4 toxicities included mucositis, fatigue, and diarrhoea. Correlative studies demonstrated histone acetylation in patients on therapy and modulation of CDKN1A and TOP2A (topoisomerase II) gene expression. Pharmacokinetic analysis confirmed a dose-related elevation in plasma vorinostat concentrations. The combination of vorinostat and idarubicin is generally tolerable and active in patients with advanced leukaemia and should be studied in the front-line setting.
Keywords: vorinostat, idarubicin, leukaemia, histone, epigenetics
INTRODUCTION
The topology of chromatin is regulated by precise mechanisms that govern consistent packaging and replication of DNA, and also play a critical role in transcriptional regulation. Modulating this process in rapidly dividing cells may be a potentially important anti-neoplastic strategy. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) work in an opposing manner by acetylating and deacetylating histones, respectively. Acetylated histones facilitate an open chromatin configuration, which allows access to transcription factors and ultimately activation of gene transcription. HDACs catalyze the removal of acetyl groups and consequently lead to a compact, “closed” chromatin structure and gene repression.(Bolden, et al 2006, Garcia-Manero and Issa 2005, Marks, et al 2001) Development of small molecule inhibitors of HDACs and demonstration of their antineoplastic activity in haematological malignancies has generated a great deal of interest.(Minucci and Pelicci 2006)
Vorinostat is an HDAC inhibitor (HDACi) that inhibits class I and II HDACs, and causes apoptosis and cell cycle arrest in leukaemia cell lines in vitro.(Marks, et al 2000, Richon, et al 1998, Ruefli, et al 2001) In addition, several Phase I and II trials have confirmed the safety, tolerability, and single-agent activity of vorinostat in patients with haematological malignancies.(Duvic, et al 2007, Garcia-Manero, et al 2008a, Kelly, et al 2005, Kelly, et al 2003, O’Connor, et al 2006, Olsen, et al 2007) In a Phase I dose escalation study of oral vorinostat given twice or thrice daily to patients with advanced leukaemias and myleodysplastic syndrome (MDS), 17% of patients had haematological improvement or response.(Garcia-Manero, et al 2008a) The most common adverse events included diarrhoea, nausea, fatigue, and anorexia. Grade 3/4 drug-related adverse events included fatigue, thrombocytopenia, diarrhoea, nausea, and vomiting. In a Phase II study of vorinostat given on 3 different schedules to patients with refractory cutaneous T cell lymphoma (CTCL), the response rate was 24%.(Duvic, et al 2007) A second Phase II trial in progressive or refractory CTCL reported an objective response rate of 30%.(Olsen, et al 2007) In both studies, the most common adverse events were fatigue, nausea, diarrhoea, and thrombocytopenia. These studies led to the US Food and Drug Administration (FDA) approval of vorinostat in October 2006 for the treatment of cutaneous manifestations in patients with CTCL who have progressive, persistent, or recurrent disease on or following 2 systemic therapies.(Mann, et al 2007)
Although vorinostat has single-agent activity, its optimal use may be in combination with other agents.(Blum, et al 2007, Friedmann, et al 2006, Garcia-Manero, et al 2006, Munshi, et al 2006, Pathil, et al 2006, Soriano, et al 2007, Zhang, et al 2006, Ziauddin, et al 2006) Due to its ability to induce an open chromatin configuration, we hypothesized that vorinostat would allow better access to DNA interactive agents, such as anthracyclines, and thereby enhance their cytotoxic effects.(Kim, et al 2003) We also hypothesized that vorinostat could upregulate the expression of DNA topoisomerase II (topo-II) and thereby sensitize leukaemia cells to anthracyclines, such as idarubicin.(Davies, et al 1988, Kurz, et al 2001) A synergistic effect for the combination of an HDACi with idarubicin was demonstrated in vitro.(Sanchez-Gonzalez, et al 2006) This was accompanied by an increase in histone acetylation, upregulation of CDKN1A, TOP2A, and induction of double-stranded (ds-) DNA breaks. Based on this information, we conducted a CTEP (Cancer Therapy Evaluation Program)-sponsored Phase I clinical trial of the combination of idarubicin and vorinostat in patients with relapsed, refractory acute leukaemia.
The primary objective of this trial was to determine the safety and tolerability of the combination of a fixed dose of idarubicin with escalating doses of vorinostat in patients with relapsed or refractory advanced leukaemia or high-risk MDS. Two different dosing schedules of vorinostat were tested. The maximum tolerated dose (MTD) and dose limiting toxicities (DLTs) of vorinostat in combination with idarubicin were defined for each schedule. Secondary objectives included determining the ability of vorinostat to induce histone acetylation and the expression of TOP2A and CDKN1A in peripheral blood mononuclear cells (PBMCs) as well as the clinical efficacy and pharmacokinetic profiles of vorinostat and idarubicin.
MATERIALS AND METHODS
Eligibility Criteria
This open-label, CTEP-sponsored, randomized Phase 1 study (Protocol 2005-0031) was approved by the Institutional Review Board of the MD Anderson Cancer Center, and all patients provided written, informed consent according to institutional guidelines.
Patients with relapsed or refractory acute myeloid leukaemia (AML), acute lymphoblastic leukaemia (ALL), high-risk myelodysplastic syndrome (MDS), or chronic myeloid leukaemia (CML) in blastic phase that had progressed after imatinib mesylate treatment were eligible for enrollment. Other eligibility criteria included age ≥ 18 years, Eastern Cooperative Oncology Group (ECOG) performance status ≤ 2, adequate hepatic (bilirubin ≤ 34.2 μmol/l; aspartate aminotransferase/alanine aminotransferase ≤ 2.5 times the upper limit of normal), renal (creatinine ≤ 176.8 μmol/l or creatinine clearance > 1 ml/s), and cardiac (ejection fraction ≥ 50%) function. Patients that had received cumulative doses > 290 mg/m2 of idarubicin (or its equivalent in other anthracyclines) were excluded from the study. Patients with central nervous system involvement by leukaemia, human immunodeficiency virus infection, or another clinically significant illness were excluded. Patients who planned to undergo allogeneic bone marrow transplantation within 4 weeks, or who were pregnant, or lactating were also excluded.
Treatment plan and Design
Two different dosing schedules were tested in the study in two parallel arms. If both were open at the time of enrollment, patients were randomly assigned to either arm. In both arms, idarubicin was given at a fixed dose of 12 mg/m2 intravenously (IV) daily over 30 min on days 1 to 3. In arm A, the starting dose of oral vorinostat was 100 mg TID for 14 consecutive days (days 1 to 14). In arm B, the starting dose of vorinostat was 100 mg TID for 3 consecutive days (days 1 to 3). In both arms, a cycle of therapy was defined as 21 days. In both arms, the vorinostat dose was escalated using a “3+3” dose-escalation design with cohorts of n=3 patients. Briefly, if ≥ 2 of 3 or ≥ 2 of 6 patients at a particular dose level experienced a DLT during the first treatment cycle, that dose was considered above the MTD. Dose escalation continued until the MTD was reached. Dose de-escalation was also contemplated for idarubicin if the starting dose proved to be too toxic. Any patient who experienced DLT was to have vorinostat held until the toxicity resolved to ≤ grade 1. When resumed, dosing was reduced by one level in that patient. To confirm the safety of the combination, up to 10 patients were to be enrolled in each schedule at their respective MTD. Continuation of treatment up to 6 cycles or to a cumulative idarubicin dose of 290 mg/m2 (or its equivalent) was permitted at the discretion of the physician if the patient had no excessive toxicity or evidence of disease progression. Then, patients could continue on single-agent vorinostat.
Clinical Evaluation
Pretreatment assessment included: a complete history and physical examination; ECOG performance status; standard clinical laboratory analysis; chest x-ray; electrocardiogram; echocardiogram or multigated acquisition (MUGA) scan; and bone marrow aspiration and biopsy. Patients were reevaluated weekly during the 1st cycle and then at minimum prior to each subsequent cycle. Bone marrow aspiration was performed on days 14 and 21 of cycle 1 and then every 3 weeks, as clinically indicated, to document response as per primary physician criteria. Cardiac ejection fraction was evaluated every other cycle and at the end of study.
Safety and response criteria
The National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events, v3.0 was used to grade adverse events. (NCI-CTEP 1999) DLT was defined as: prolonged myelosuppression with ≤ 5% bone marrow cellularity and no evidence of leukaemia, lasting > 42 days; grade ≥ 3 nausea, vomiting, or diarrhoea despite maximum supportive care; or other grade ≥ 3 non-haematological toxicity related to the study drug. Untreated nausea and vomiting ≥ grade 3, alopecia, study drug-related fever, hyperbilirubinaemia, and electrolyte abnormalities (including K, Na, Cl, HCO3, Mg, Ca) that were ≥ grade 3 did not define the DLT. Because vorinostat in combination with another agent might exacerbate adverse events known to be caused by that other agent, potassium abnormalities (which have been associated with vorinostat) that persisted despite repletion therapy were considered dose-limiting. Clinical responses were evaluated using the revised guidelines of the International Working Group for AML, ALL, and MDS. (Cheson, et al 2003) All responses, except complete remission with incomplete count recovery (Cri), must have lasted at least 4 weeks for AML, ALL, and CML. The duration of response was measured from the time when all response criteria were first met until recurrent or progressive disease was documented.
Quantitation and Pharmacokinetic Analysis of Idarubicin
Blood samples (7 ml) were collected in heparin-containing vacutainers prior to the start of the idarubicin infusion and at 10, 15, 30 min and 1, 2, 4, 8, 10 h following the start of the infusion on day 1. Pre-dose samples were also collected on days 2 and 3, along with samples at 0.5, 1 and 2 h following drug administration. Plasma was generated by centrifugation immediately after sample collection and stored in cryovials at -80°C until analyzed. Just prior to analysis, idarubicin was isolated from plasma using a solid phase extraction process. Briefly, 0.6 ml of plasma was applied to conditioned C18 cartridges (Phenomenex, Torrance, CA) which were then washed with buffer and extracted using chloroform:isopropanol (4:1, v/v) elution buffer. A high performance liquid chromatographic method for the quantitation of idarubicin in human plasma was developed and validated over a dynamic concentration range of 0.5 ng/ml to 1000 ng/ml (r2 > 0.997, linear regression analysis) using a modification of a previously published method. (Cummings, et al 1987) Chromatographic separation was achieved using gradient elution with a mobile phase consisting of 10 mM ammonium formate and acetonitrile, (55%:45% v/v, pH 3.0) and a C8 reverse phase analytical column (Zorbax Rx-C8, 4.6 x 250 mm, 5 μm, Agilent Technologies, Santa Clara, CA). Peak detection was achieved using a fluorescence detector with excitation and emission settings of λ=210 nm and λ=790 nm, respectively (Waters Alliance 2695/2475, Waters Corporation, Milford, MA). The mean inter- and intra-day assay precisions for the method were 5.8% and 4.3%, respectively. The mean extracted recovery from human plasma at idarubicin concentrations of 5 and 500 ng/ml were 74% and 81%, respectively. The pharmacokinetic parameters (maximum plasma concentration [Cmax], half life [t1/2], volume of distribution [Vd], total body clearance [ClT], and area under the plasma concentration time curve [AUC]0-inf) were obtained from each patient dataset using non-compartmental analysis (WinNonlin v5.2, model 202, Pharsight Corp, Mountain View, CA) and summarized using the global 2-stage method as mean values ± SD for dose schedules A and B, respectively.
Quantification and Pharmacokinetic Analysis of Vorinostat
Pharmacokinetic studies of vorinostat were performed during the first cycle of treatment. On day 1, 5 mL of blood were collected in a redtop vacutainer tubes (no anticoagulant; Becton Dickinson, Franklin Lakes, NJ, USA) before and at 0.5, 1, 2, 2.5, 3, 4, 6, and 8 h after ingestion of vorinostat. Blood samples were allowed to coagulate at 4°C for 20 to 30 min and then centrifuged at 2,000 × g for 15 min at 4°C. The resulting serum was stored at -70°C until analyzed for vorinastat content. Concentrations of vorinostat and two metabolites (vorinostat glucuronide and 4-anilino-4-oxobutanoic acid) were quantitated, as previously described, with a validated liquid chromatography-electrospray ionization tandem mass spectrometric method. (Parise, et al 2006)
Isolation of Human PBMCs
Blood and bone marrow aspiration specimens were drawn into heparin (30 u/ml). PBMCs were prepared using Ficoll-Paque PLUS gradient centrifugation (Amersham Biosciences AB, Uppsala, Sweden). The monocyte-enriched cell fraction was collected and washed twice with calcium- and magnesium-free phosphate-buffered saline (PBS). After centrifugation, PBS was removed, and the cell pellets were stored at -80°C.
Analysis of Histone Acetylation
As a molecular marker of exposure to vorinostat, histone-H3 acetylation was measured in PBMCs on days 0, 1, 3, 14, 21 of the first course (for both schedules) as previously described. (Garcia-Manero, et al 2008a) ß-actin (1:5000 dilution; Sigma, St Louis, MO) was used as internal control. HL-60 cells treated with 1 mM valproic acid were used as positive controls.
Real-time polymerase chain reaction (PCR) analysis of DNA TOP2A and CDKN1A mRNA expression
TOP2A is the target enzyme of idarubicin. Because of the potential role of vorinostat in modulating the expression of this enzyme, we measured TOP2A sequentially at the time points during therapy described above for histone acetylation. Because CDKN1A is upregulated in vitro by vorinostat (Huang, et al 2000, Marks 2004, Richon, et al 2000), it was also measured at the same time points described above. TOP2A and CDKN1A mRNA expression was measured by real-time PCR as previously described. (Sanchez-Gonzalez, et al 2006)
RESULTS
Patient characteristics
Forty-one patients were enrolled in this study: 15 on schedule A; and 26 on schedule B. Baseline characteristics are shown in Table 1. The most common diagnosis was relapsed/refractory AML, and the median number of prior therapies was 3 (range, 1-7). The 41 patients received 58 treatment cycles (median 1, range 1-3). In schedule A, the dose regimens evaluated were: (1) idarubicin at 12 mg/m2 with vorinostat at 100 mg TID and (2) idarubicin at 9 mg/m2 (-1 dose level) with vorinostat at 100 mg TID. In schedule B, the dose regimens studied were idarubicin at 12 mg/m2 with vorinostat at 100, 200, 300, 400, and 500 mg TID.
Table 1. Patient Characteristics.
| Characteristic | N=41 |
|---|---|
| Median age, years (range) | 59 (21-80) |
| Male Sex (%) | 21 (51) |
| Diagnosis (%) | |
| AML | 37 (90.2) |
| MDS | 1 (2.4) |
| ALL | 1 (2.4) |
| Biphenotypic | 2 (5) |
| Median WBC (×109/l) | 8.3 (0.2 - 98.9) |
| Median BM Blasts , %, (range) | 63 (0* - 97) |
| Median No. of Prior Therapies (range) | 3 (1 - 7) |
| Cytogenetics (%) | |
| Diploid | 12 (29.3) |
| Poor Risk | 29 (70.7) |
N represents the total number of patients in the study. AML=Acute myeloid leukaemia; MDS=Myelodysplastic syndrome; ALL=Acute lymphoblastic leukaemia; Biphenotypic=Acute biphenotypic leukaemia. WBC=White blood cell count; BM=Bone Marrow; Poor Risk cytogenetics defined as monosomy 5, monosomy 7, 11q23 abnormality, or complex karyotype (3 or more abnormalities).
Included one patient with a rapidly proliferative relapsed MDS/Myeloproliferative neoplasm with 0% bone marrow blasts but 100% marrow infiltration by high-risk myeloid neoplasm.
Safety and tolerability
The number of patients enrolled on the trial and the adverse events (by grade) on each cohort are outlined in Table 2. There were 13 instances of DLTs: 8 in patients on schedule A; and 5 on schedule B. The DLTs included prolonged myelosuppression, mucositis, fatigue, uncontrollable diarrhoea, tumour lysis syndrome, and left ventricular dysfunction. In schedule A, dose level 1 (idarubicin 12 mg/m2/d IV for 3 days with vorinostat 100 mg TID for 14 days) was found to be above the MTD. In this arm, the dose of idarubicin was de-escalated to 9 mg/m2 with vorinostat at 100 mg TID. No excess toxicity was observed at these lower doses when this cohort was expanded to 10 patients. In schedule B, 3 consecutive patients experienced grade 2 toxicities at dose level 5 (idarubicin 12 mg/m2/d IV for 3 days with vorinostat 500 mg TID for 3 days). To better define safety at this dose level, 3 more patients were enrolled. All three experienced grade 3 non-haematological toxicity. We therefore concluded that dose level 4 (idarubicin 12 mg/m2 with vorinostat 400 mg TID) was the MTD of this combination and schedule. Cohort expansion up to 10 patients confirmed the relative tolerability of this dose level.
Table 2. Non-haematological toxicities observed by grade, dose, and schedule.
| Dosing Schedule | Dosage | Grade | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Ida ∣ Vorinostat | N | 1 | 2 | 3 | 4 | |
| Schedule A | 12 mg/m2 ∣ 100 mg | 5 | 25 | 11 | 6 | 1 |
| 9 mg/m2 ∣ 100 mg | 10 | 48 | 20 | 2 | 1 | |
|
| ||||||
| Schedule B | 12 mg/m2 ∣ 100 mg | 3 | 10 | 7 | 3 | 0 |
| 12 mg/m2 ∣ 200 mg | 3 | 13 | 5 | 1 | 0 | |
| 12 mg/m2 ∣ 300 mg | 4 | 17 | 4 | 0 | 0 | |
| 12 mg/m2 ∣ 400 mg | 10 | 62 | 26 | 4 | 0 | |
| 12 mg/m2 ∣ 500 mg | 6 | 33 | 16 | 5 | 0 | |
N represents the number of patients at each dose and schedule. The values under grade represent the numbers of adverse events reported by grade. Ida=Idarubicin. Schedule A is Ida given at 12 mg/m2 IV daily for 3 days with vorinostat given TID for 14 days. Schedule B is Ida given at 12 mg/m2 IV daily for 3 days with vorinostat given TID for 3 days.
Overall, for both schedules, the most common drug-related adverse experiences and laboratory abnormalities observed were fatigue (88%), nausea (76%), liver function test (LFT) abnormalities (76%), diarrhoea (73%), mucositis (61%), cough (61%) and anorexia (46%) (Figure 1a). These events were mostly grades 1 - 2 in severity. Grade 3 and 4 drug-related adverse experiences or laboratory abnormalities included mucositis (5/23; 22%), fatigue (22%), diarrhoea (17%), prolonged myelosuppression (4%), left ventricular systolic dysfunction (4%), confusion (4%), cardiac arrhythmia (4%), pleural effusion (4%), gastrointestinal bleed (4%), dysphagia (4%), tumour lysis syndrome (4%), and musculoskeletal pain (4%) (Figure 1b). Given the patient population studied and the cytotoxic agent used in this trial, treatment-related myelosuppression was expected and universal in all patients. Serious drug-related adverse experiences were observed in two patients (one with grade 4 mucositis and another with grade 4 prolonged myelosuppression of > 42 days duration). Both of these patients were treated on schedule A with vorinostat at 100 mg TID for 14 days. The patient who experienced the prolonged myelosuppression never recovered her counts after having a complete marrow response and complete resolution of leukaemia cutis. She subsequently died of fungal infection and sepsis. Of note, no significant decrease on cardiac ejection fraction related to therapy administration was observed in patients treated on this study (data not shown).
Figure 1. Adverse Events.
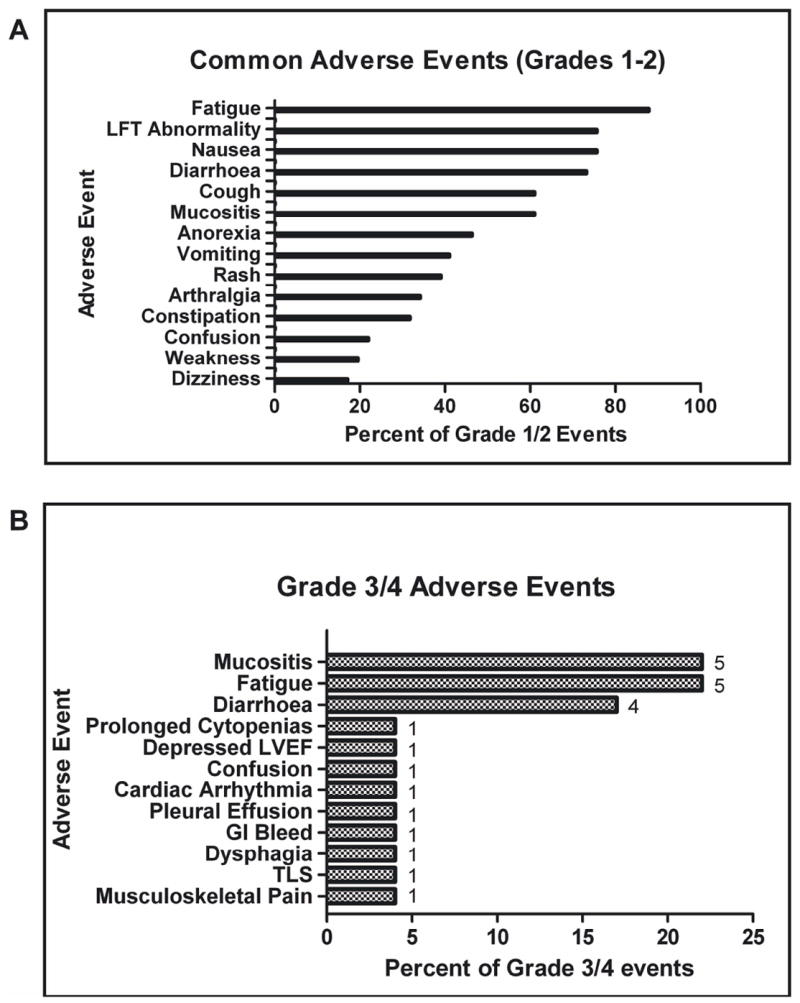
A. Most common grade 1 or 2 non-haematological adverse events seen in patients on the trial. B. Most common grade 3 or 4 adverse events seen in patients on the trial. The bar graphs express a percentage. The numbers next to each bar represent actual numbers of events. LVEF, left ventricular ejection fraction; GI, gastrointestinal; TLS, tumour lysis syndrome.
Clinical Activity
A summary of clinical responses is shown in Table 3. Overall, 7 of 41 patients (17%) achieved a clinical response, including 2 complete responses (CR) and 1 CR without platelet recovery (CRp). There was one CR on schedule A, and one CR and one CRp on schedule B. In addition, there was 1 marrow response on schedule A and 3 on schedule B. In schedule B, each of the responses occurred at doses of vorinostat ≥ 300 mg TID. Six of the 7 responders had a diagnosis of AML, and 1 had a diagnosis of acute biphenotypic leukaemia. Both complete responders and 2 of the patients with marrow response had diploid cytogenetics. The other 2 patients with marrow response had cytogenetic abnormalities including trisomy 8 and –X, respectively. The median age of responding patients was 63 (range, 33-73) years. The median number of prior therapies for the responding patients was 2 (range, 1-3).
Table 3. Responses by dose and schedule.
| Dosing Schedule | Dosage
|
N | CR | CRp | Marrow Response |
|---|---|---|---|---|---|
| Ida ∣ Vorinostat | |||||
|
| |||||
| Schedule A | 12 mg/m2 ∣ 100 mg | 5 | 0 | 0 | 1 |
| 9 mg/m2 ∣ 100 mg | 10 | 1 | 0 | 0 | |
|
| |||||
| Schedule B | 12 mg/m2 ∣ 100 mg | 3 | 0 | 0 | 0 |
| 12 mg/m2 ∣ 200 mg | 3 | 0 | 0 | 0 | |
| 12 mg/m2 ∣ 300 mg | 4 | 1 | 0 | 1 | |
| 12 mg/m2 ∣ 400 mg | 10 | 0 | 1 | 1 | |
| 12 mg/m2 ∣ 500 mg | 6 | 0 | 0 | 1 | |
|
| |||||
| Totals | 41 | 2 | 1 | 4 | |
N represents the total number of patients for each dose and schedule. Ida = Idarubicin. Schedule A is Ida given at 12 mg/m2 IV daily for 3 days with vorinostat given for 14 days. Schedule B is Ida given at 12 mg/m2 IV daily for 3 days with vorinostat given for 3 days. CR = Complete Response. CRp = Complete Response without platelet recovery.
Characteristics of Complete Responses
There were two CRs on the trial, 1 each on schedule A and schedule B. The first patient had a diagnosis of AML with diploid cytogenetics and had been treated with 2 prior therapies, including initial induction with idarubicin and cytarabine. This patient required 3 cycles of therapy on schedule B to achieve CR and had an initial duration of remission of 4 weeks, prior to receiving an allogeneic bone marrow transplant. This patient remained in complete remission at 6-months of follow-up. The second patient had a diagnosis of acute biphenotypic leukaemia with diploid cytogenetics and had 3 prior therapies. This patient achieved CR after the first cycle of idarubicin and vorinostat on schedule A and remained in CR for 7 months on study before relapsing.
Histone Acetylation
Adequate samples for measuring histone acetylation were available from 33 out of the total 41 patients. Increased histone-H3 acetylation was detected in 15 of 33 (46%) available patient samples, distributed equally between schedules A and B (5/11 patients on schedule A and 10/22 patients on B). In some patients, histone acetylation continued to be detectable at day 21 (Figure S1). The relationship between histone acetylation and vorinostat dose could not be evaluated due to the non-quantitative nature of the assay for histone acetylation used here.
Induction of TOP2A and CDKN1A mRNA
Levels of mRNA induction for TOP2A and CDKN1A were calculated relative to pretreatment levels. Overall, there was induction of TOP2A mRNA in some individual patients, but mostly it was unchanged or downregulated (Figure S2). When the data were examined by treatment schedule, there was a statistically significant trend (p=0.016, Wilcoxon signed rank test) towards downregulation of TOP2A mRNA levels observed in patients on schedule A from day 1 to day 21(figure 2). On schedule B, there was a trend towards downregulation of TOP2A mRNA in most patients by day 3 of therapy (p=0.168, Wilcoxon), but TOP2A mRNA returned to levels similar to baseline over the next 2 time points (figure 2). These patterns were consistent with the vorinostat dosing schedule on each arm. In schedule A, with more prolonged vorinostat dosing (14 days), there was a protracted and more pronounced downregulation of TOP2A, whereas in schedule B (3 days dosing), there was a trend towards downregulation of TOP2Aby day 3, but a rebound by day 14.
Figure 2. Expression Levels of Topoisomerase II and p21 by Treatment Arm.
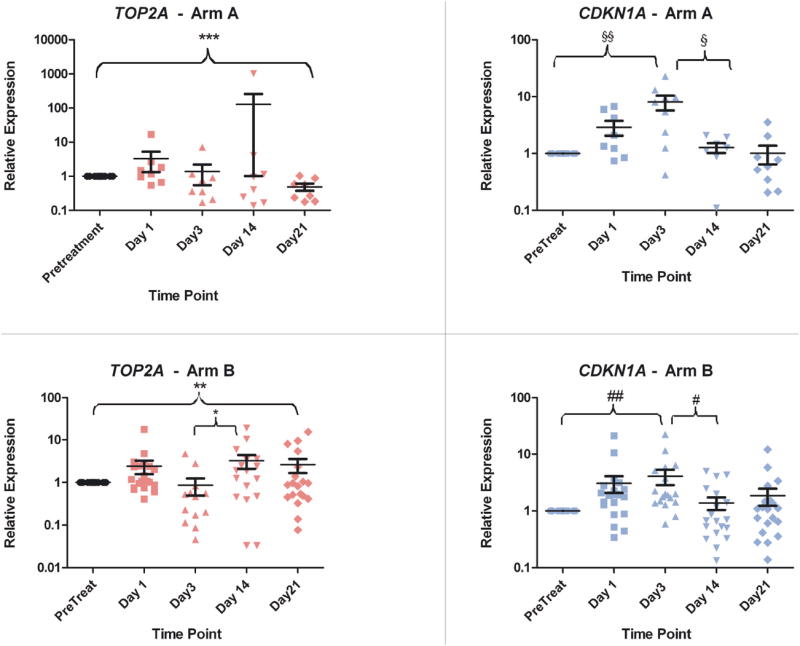
Relative expression of DNA topoisomerase II (topo-II) and p21 by quantitative RT-PCR in patients separated by arm. Time points listed represent the day on study during which the sample was collected. Statistical values ***: p=0.016; **: p=0.984; *: p=0.049; §§: p=0.012; §: p=0.023; ##: p=0.0005; #: p=0.003
There was also a statistically significant trend towards upregulation of CDKN1A mRNA from baseline to day 3 overall (p<0.001, Wilcoxon) (Figure S2). This trend remained significant when the data were analyzed by treatment arm (figure 2). By day 14, these levels dropped and returned to near baseline or below (p= 0.0001, Mann Whitney test). The pattern of CDKN1A mRNA modulation in these patients correlated with the 3-day dosing of idarubicin in both arms. Expression levels of these markers were then examined among responders versus non-responders. There were no significant trends in either TOP2A or CDKN1A expression levels that differentiated responders from nonresponders. There was no significant relationship between CDKN1A or TOP2A mRNA induction and dose of vorinostat.
Pharmacokinetic Profile of Idarubicin
The comparative pharmacokinetic parameters for idarubicin (12 mg/m2) administered by a 30-min intravenous infusion every 24 h for 3 days with two different schedules of vorinostat, schedule A and B, respectively, are shown in Table S1. Peak plasma concentrations of idarubicin were reached at the end of infusion followed by a rapid decline over the first 30 min post-infusion. In schedule A the maximum idarubicin plasma concentration (cmax) ranged from 19.5 to 139.4 ng/ml with a mean value of 66.1 ng/ml. These values were similar to those observed in schedule B where patients were treated with idarubicin in combination with vorinostat given TID for only 3 days; in this group the mean idarubicin peak plasma concentration was 55.4 ng/ml, and peak plasma concentrations ranged from 14.9 and 129.7 ng/ml. The median t1/2 for idarubicin was found to be approximately 11 h in both treatment schedules. The Vd, ClT and AUC revealed no considerable differences between schedule A and B. Noteworthy, Cmax, ClT and Vd showed a high degree of interindividual variability.
Pharmacokinetic Profile of Vorinostat
Thirty-six patients had blood samples drawn to characterize the pharmacokinetics of vorinostat. Vorinostat levels were quantitated in all 36, but due to insufficient authentic standards and appropriate internal standards, concentrations of vorinostat metabolites were only quantitated in the first 19 patients. The pharmacokinetic parameters of vorinostat administered orally TID on a dose escalation schedule for either 14 days or 3 days are shown in Table 4. Peak concentrations of vorinostat (Cmax) were reached between 2 and 6 h. The median Cmax of vorinostat for each patient cohort increased from 0.331 – 0.445 μM at 100 mg TID up to 1.55 μM at 400 mg TID, and 1.62 μM at 500mg TID. The median time at which the peak concentration was reached (Tmax) varied between 1 and 3 h and did not correlate with dose. Because vorinostat was self-administered orally by the patients on a TID schedule, the exact times at which the doses were ingested were not available. This precluded us from modeling the t1/2, Vd, ClT, and AUC of vorinostat. The Cmax of vorinostat and its metabolites in responding and non-responding patients were compared to explore any relationship between peak plasma concentration of vorinostat and response. In this small cohort of patients, the responders had a higher median Cmax for vorinostat (1.4 μM vs. 0.8 μM) and its metabolites than non-responders, but it did not reach statistical significance (p=0.14) (Table S2).
Table 4. Pharmacokinetic parameters of vorinostat (and its metabolites) given orally TID for 14 days or 3 days in combination with idarubicin given at a dose of 9 or 12 mg/m2 IV daily for 3 days.
| Vorinostat Metabolites | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Dose and Schedule of Vorinostat | N | Vorinostat Mean Cmax (μM) | Vorinostat Median Cmax (Range) | Vorinostat Median Tmax (h) | Acid Mean Cmax (SD) | Acid Mean Tmax | Glucuronide Mean Cmax (SD) | Glucuronide Mean Tmax |
|
| ||||||||
| 100 mg TID for 14 days | 11 | 0.554 | 0.445 (0.19 - 1.53) | 2.5 | 2.13 (0.88) | 3.3 | 2.16 (2.1) | 2.8 |
| 100 mg TID for 3 days | 3 | 0.688 | 0.331 (0.23 - 1.51) | 2 | 1.97 (0.9) | 2.3 | 1.21 (0.14) | 2.2 |
| 200 mg TID for 3 days | 3 | 0.522 | 0.443 (0.41 - 0.71) | 1 | 3.08 (0.58) | 2.3 | 1.77 (0.18) | 2.3 |
| 300 mg TID for 3 days | 4 | 0.991 | 0.998 (0.57 - 1.40) | 2.5 | 3.19 (1.35) | 2.8 | 3.33 (1.8) | 2.5 |
| 400 mg TID for 3 days | 10 | 1.66 | 1.55 (0.91 - 2.64) | 2.25 | 4.23 | 1 | 4.52 | 1 |
| 500 mg TID for 3 days | 5 | 2.02 | 1.62 (1.61 - 2.84) | 3 | ||||
N represents the total number of patients at each dose and schedule of vorinostat. Cmax = the maximum plasma concentration reached on each dose and schedule. Cmax is expressed in micromolar (μM). Tmax = the time point at which the maximum plasma concentration was reached. Tmax is expressed in hours. SD = standard deviation from the mean value of the samples. Two metabolites of vorinostat were measured in only the first 19 patients (due to limited reagents) – the ‘acid’ metabolite and the ‘glucuronide’ metabolite. The acid metabolite is 4-Anilino-4-oxobutanoic acid. The glucuronide metabolite is vorinostat-glucuronide. The columns labeled ‘Acid’ and ‘Glucuronide’ refer to these metabolites, respectively. At the 400 mg dose, vorinostat metabolites were measured in only 1 patient. At the 500 mg dose, metabolites were not measured in any patients.
DISCUSSION
In this phase I study of idarubicin and vorinostat in patients with relapsed/refractory acute leukaemia, we sought to evaluate whether the combination was tolerable and feasible in this patient population. We designed a randomized phase I study that evaluated 2 different schedules of vorinostat. In schedule A, vorinostat was given TID for 14 days, similar to a prior single-agent phase I trial of vorinostat in relapsed/refractory leukaemia.(Garcia-Manero, et al 2008a) In schedule B, vorinostat was given TID for only 3 days. This schedule mirrored our preclinical experience, where we combined these agents and showed synergy over 3 days of treatment.(Sanchez-Gonzalez, et al 2006) Also, given the addition of a cytotoxic agent, this shorter schedule could potentially have been better tolerated and allow the administration of higher doses of vorinostat. Our observations on the trial confirmed this. On schedule A, the 14-day dosing proved to be above the MTD, requiring a dose de-escalation of idarubicin. On schedule B, the shorter 3-day dosing allowed us to escalate vorinostat up to 500 mg TID before encountering DLTs. The shorter schedule allowed for a major increase in vorinostat dose. Because the study was not powered to find differences in terms of response among different dose levels, it is possible that lower doses of vorinostat could be effective in combination.
The most common grade 3 and 4 toxicities on the study were mucositis in 5 patients, fatigue in 5 patients, and diarrhoea in 4 patients. One patient had grade 4 cardiac dysfunction. No clinically significant QT interval prolongation was detected in the patients while on the study. The most common grade 1 and 2 toxicities included fatigue, nausea, liver function test abnormalities, diarrhoea, mucositis, and cough. Although fatigue has been a prominent side effect of HDACi’s, many of the other common toxicities on this trial are proably a result of the addition of idarubicin. Mucositis, diarrhoea, and cough, for example, were more prominent in this trial compared to single-agent trials of HDACi.(Byrd, et al 2005, Garcia-Manero, et al 2008a, Garcia-Manero, et al 2008b, Giles, et al 2006)
A secondary objective of the trial was to describe the clinical activity of this combination. In this heavily pretreated population, many of whom had previously received anthracycline-based therapy, we observed a 17% response rate. Complete responders had significant durations of remission, and one was able to proceed to an allogeneic stem cell transplant. These responses, in a population where the expectation of response is very low, may indicate a signal of activity that should be evaluated further in a cohort of untreated patients. Alternatively, this combination may represent a new paradigm, by which anthracycline resistance can be modulated with HDACi. Further experience with this combination in more homogeneous groups of patients is needed to understand fully the clinical significance of HDACi-based combination therapy. Recently, the combination of etoposide (VP-16), cytarabine, and vorinostat has been shown in vitro to be synergisitic in a sequence-dependant manner.(Shiozawa, et al 2009) Based on this preclinical work, we are currently conducting a phase 2 study of idarubicin, cytarabine, and vorinostat in untreated patients with AML.(Jabbour, et al 2009)
In the preclinical model of idarubicin and vorinostat(Sanchez-Gonzalez, et al 2006), we analyzed a number of biomarkers to understand the mechanism of action of the combination. As part of this study, we developed assays to analyze mRNA expression of CDKN1A, a cyclin dependent kinase inhibitor whose expression is associated with HDACi exposure(Marks 2004, Richon, et al 2000), and TOP2A, the target of idarubicin. As mechanisms for synergy, we hypothesized that an HDACi could sensitize malignant cells to the effects of an anthracycline in 2 possible ways. First, by inducing an open chromatin configuration, vorinostat could allow for more efficient intercalation of the anthracycline into DNA, to exert its cytotoxic effect.(Kim, et al 2003) Second, an HDACi could upregulate the expression of TOP2A and thereby increase the potency of idarubicin within the cell.(Davies, et al 1988, Kurz, et al 2001) To test this, we measured the induction of TOP2A mRNA and CDKN1A mRNA after treatment with vorinostat during treatment. Contrary to our hypothesis, we did not observe an overall increase in TOP2A mRNA after treatment with vorinostat. In fact, when we analyzed the group by vorinostat schedule (14 days versus 3 days), there was a trend towards downregulation of TOP2A mRNA during treatment. In the 3-day schedule, there was a downward trend during vorinostat treatment, followed by a rebound back to baseline or higher at day 14. In view of this, in the clinical setting, it may be more advantageous to pretreat patients with vorinostat for 3 days, followed by the anthracycline in a sequenced fashion to exploit the rebound in TOP2A levels. This should be examined further in future studies. Notably, there was no significant difference in TOP2A modulation between responders and nonresponders in our small cohort of patients.
We observed a statistically significant trend towards CDKN1A mRNA induction in the overall cohort of patients by day 3. This trend remained significant when each treatment arm was examined separately. By day 14, however, the CDKN1A mRNA levels had returned to baseline or lower in both cohorts. In that idarubicin, which was administered over days 1 to 3, is also known to be a potent inducer of CDKN1A, it is not clear how much of the day 3 induction could be attributed to it versus vorinostat. The effect of vorinostat may be evident when examining the day 14 samples from schedule A (14 day) versus B (3 day), when both cohorts have long completed idarubicin. There was a suggestion that levels of CDKN1A mRNA fell below baseline in many patients on schedule B at day 14, but not in schedule A. Presumably, there may be continued CDKN1A mRNA induction by vorinostat in patients on schedule A at day-14 as reflected by higher maintained levels compared to patients on schedule B. Given the small numbers of patients that are being compared on this study, this is not a statistically significant observation. As with TOP2A, there was no significant difference in CDKN1A modulation between responders and nonresponders. These observations suggest that the utility of CDKN1A as a biomarker of HDACi exposure may be confounded when used in combination with anthracyclines. However, as CDKN1A induction by HDACi has been shown in some studies to attenuate apoptosis in malignant cell lines, strategies to interfere with this induction with agents, such as flavopiridol, heat shock protein 90 inhibitors, or phosphatidyl inositol-3 kinase inhibitors, can be monitored clinically using similar CDKN1A assays.(Almenara, et al 2002, Rahmani, et al 2003a, Rahmani, et al 2003b, Vrana, et al 1999)
Western blot analysis was used as a biomarker of vorinostat exposure in this study. Histone-H3 acetylation was detected in about half the patients. In principle, the half-life of histone acetylation is approximately 4-10 h (Kelly, et al 2005, Kelly, et al 2003). Therefore, we did not anticipate the long-lasting histone acetylation (up to day 21) that we observed in several of the patients on the trial. The 14-day schedule more consistently showed day 21 histone acetylation, as might be expected. However, 50% of the patients on the 3-day schedule of vorinostat also had detectable histone acetylation at day 21. We did not find a correlation between histone acetylation duration and clinical response.
The pharmacokinetics for idarubicin presented in this study were similar to those previously reported by several investigators(Crivellari, et al 2004, Danesi, et al 2002) and not significantly different between the 2 arms. The pharmacokinetics of vorinostat showed a dose-related increase in the mean maximum plasma concentration of vorinostat with each escalation of the dose, but it reached a plateau between 400 mg TID and 500 mg TID. Although there was a trend, there was no significant difference in peak plasma vorinostat concentration between responders and nonresponders. With the TID dosing for short durations (3-day), dose levels of 400 mg and 500 mg were able to achieve plasma concentrations greater 2 μM.
In conclusion, the combination of idarubicin 12 mg/m2/d IV for 3 days combined with vorinostat for 3 days was generally well tolerated, with the MTD of vorinostat being 400 mg TID. The alternate schedule tested, with vorinostat given TID for 14 days, was not tolerated and was associated with excessive DLTs, including severe mucositis and prolonged myelosuppression. The combination of idarubicin and vorinostat had a signal of activity in a heavily pretreated, refractory population and should be evaluated further in the frontline setting.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Cancer Institute 2 UO1 CA062461-15, NCI contract N01-CO-124001, subcontract 25XS115-Task Order 2; and NCI grant P30 CA47904. TMK is supported in part by K12-CA088084. GG-M is supported in part by the Physician Scientist Program – MD Anderson Cancer Center (Houston, TX), and the Leukemia & Lymphoma Society of America (New York, NY). MJE is an ASCO Cancer Foundation Translational Research Professor.
Footnotes
AUTHORSHIP
Contribution: TMK helped perform correlative studies, analyzed the data, and wrote the manuscript. HY, SM performed correlative studies and analyzed the data. CS, TLM, JLH, MJE performed pharmacokinetic analyses and analyzed the data. MJE also helped edit the paper. BS-G performed preclinical studies. AF, FR, DAT, and HK enrolled patients on the study. WN followed the patients and collected the data. I E-D, JZ helped in designing the study, analyzing the data, and writing the manuscript. GG-M designed the study, analyzed the data and wrote the manuscript.
CONFLICT OF INTEREST
Conflict of Interest: The authors declare no competing financial interests or conflict of interest.
References
- Almenara J, Rosato R, Grant S. Synergistic induction of mitochondrial damage and apoptosis in human leukemia cells by flavopiridol and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) Leukemia. 2002;16:1331–1343. doi: 10.1038/sj.leu.2402535. [DOI] [PubMed] [Google Scholar]
- Blum W, Klisovic RB, Hackanson B, Liu Z, Liu S, Devine H, Vukosavljevic T, Huynh L, Lozanski G, Kefauver C, Plass C, Devine SM, Heerema NA, Murgo A, Chan KK, Grever MR, Byrd JC, Marcucci G. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol. 2007;25:3884–3891. doi: 10.1200/JCO.2006.09.4169. [DOI] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, Lucas DM, Fischer B, Shank R, Tejaswi SL, Binkley P, Wright J, Chan KK, Grever MR. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–967. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, Schiffer CA, Doehner H, Tallman MS, Lister TA, Lo-Coco F, Willemze R, Biondi A, Hiddemann W, Larson RA, Lowenberg B, Sanz MA, Head DR, Ohno R, Bloomfield CD. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21:4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- Crivellari D, Lombardi D, Spazzapan S, Veronesi A, Toffoli G. New oral drugs in older patients: a review of idarubicin in elderly patients. Crit Rev Oncol Hematol. 2004;49:153–163. doi: 10.1016/S1040-8428(03)00120-3. [DOI] [PubMed] [Google Scholar]
- Cummings J, Milroy R, Banham SW, Kaye SB. Method for the determination of 4-demethoxydaunorubicin, its quinone and hydroquinone metabolites in human plasma and urine by high-performance liquid chromatography. Cancer Chemother Pharmacol. 1987;19:296–300. doi: 10.1007/BF00261476. [DOI] [PubMed] [Google Scholar]
- Danesi R, Fogli S, Gennari A, Conte P, Del Tacca M. Pharmacokinetic-pharmacodynamic relationships of the anthracycline anticancer drugs. Clin Pharmacokinet. 2002;41:431–444. doi: 10.2165/00003088-200241060-00004. [DOI] [PubMed] [Google Scholar]
- Davies SM, Robson CN, Davies SL, Hickson ID. Nuclear topoisomerase II levels correlate with the sensitivity of mammalian cells to intercalating agents and epipodophyllotoxins. J Biol Chem. 1988;263:17724–17729. [PubMed] [Google Scholar]
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann I, Atmaca A, Chow KU, Jager E, Weidmann E. Synergistic effects of valproic acid and mitomycin C in adenocarcinoma cell lines and fresh tumor cells of patients with colon cancer. J Chemother. 2006;18:415–420. doi: 10.1179/joc.2006.18.4.415. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G, Issa JP. Histone deacetylase inhibitors: a review of their clinical status as antineoplastic agents. Cancer Invest. 2005;23:635–642. doi: 10.1080/07357900500283119. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes J, O’Brien S, Estey E, Bueso-Ramos C, Fiorentino J, Jabbour E, Issa JP. Phase I/II study of the combination of 5-aza-2’ -deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G, Rosner G, Loboda A, Fantin VR, Randolph SS, Hardwick JS, Reilly JF, Chen C, Ricker JL, Secrist JP, Richon VM, Frankel SR, Kantarjian HM. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008a;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, Newsome WM, Miller WH, Jr, Rousseau C, Kalita A, Bonfils C, Dubay M, Patterson TA, Li Z, Besterman JM, Reid G, Laille E, Martell RE, Minden M. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008b;112:981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, Kantarjian H, Dugan M, Albitar M, Bhalla K. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006;12:4628–4635. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- Huang L, Sowa Y, Sakai T, Pardee AB. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene. 2000;19:5712–5719. doi: 10.1038/sj.onc.1203963. [DOI] [PubMed] [Google Scholar]
- Jabbour E, Faderl S, Ravandi F, Konopleva M, Verstovsek S, Cortes J, Wierda W, Newsome WM, Yang H, Kantarjian H, Garcia-Manero G. Phase II study of vorinostat (V) in combination with idarubicin and high-dose cytarabine (IA) as front-line therapy in patients (pts) with high-risk myelodyplsatic syndrome (MDS) or acute myeloid leukemia (AML) J Clin Oncol (Meeting Abstracts) 2009;27:7004. [Google Scholar]
- Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9:3578–3588. [PubMed] [Google Scholar]
- Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003;63:7291–7300. [PubMed] [Google Scholar]
- Kurz EU, Wilson SE, Leader KB, Sampey BP, Allan WP, Yalowich JC, Kroll DJ. The histone deacetylase inhibitor sodium butyrate induces DNA topoisomerase II alpha expression and confers hypersensitivity to etoposide in human leukemic cell lines. Mol Cancer Ther. 2001;1:121–131. [PubMed] [Google Scholar]
- Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Marks PA. The mechanism of the anti-tumor activity of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA) Cell Cycle. 2004;3:534–535. doi: 10.4161/cc.3.5.827. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther. 2006;5:1967–1974. doi: 10.1158/1535-7163.MCT-06-0022. [DOI] [PubMed] [Google Scholar]
- NCI-CTEP. Common Toxicity Criteria. 1999 Available at : http://ctep.cancer.gov/forms/CTCv20_4-30-992.pdf.
- O’Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C, Portlock C, Horwitz S, Zelenetz AD, Frankel S, Richon V, Marks P, Kelly WK. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24:166–173. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, Duvic M. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- Parise RA, Holleran JL, Beumer JH, Ramalingam S, Egorin MJ. A liquid chromatography-electrospray ionization tandem mass spectrometric assay for quantitation of the histone deacetylase inhibitor, vorinostat (suberoylanilide hydroxamicacid, SAHA), and its metabolites in human serum. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;840:108–115. doi: 10.1016/j.jchromb.2006.04.044. [DOI] [PubMed] [Google Scholar]
- Pathil A, Armeanu S, Venturelli S, Mascagni P, Weiss TS, Gregor M, Lauer UM, Bitzer M. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology. 2006;43:425–434. doi: 10.1002/hep.21054. [DOI] [PubMed] [Google Scholar]
- Rahmani M, Yu C, Dai Y, Reese E, Ahmed W, Dent P, Grant S. Coadministration of the heat shock protein 90 antagonist 17-allylamino- 17-demethoxygeldanamycin with suberoylanilide hydroxamic acid or sodium butyrate synergistically induces apoptosis in human leukemia cells. Cancer Res. 2003a;63:8420–8427. [PubMed] [Google Scholar]
- Rahmani M, Yu C, Reese E, Ahmed W, Hirsch K, Dent P, Grant S. Inhibition of PI-3 kinase sensitizes human leukemic cells to histone deacetylase inhibitor-mediated apoptosis through p44/42 MAP kinase inactivation and abrogation of p21(CIP1/WAF1) induction rather than AKT inhibition. Oncogene. 2003b;22:6231–6242. doi: 10.1038/sj.onc.1206646. [DOI] [PubMed] [Google Scholar]
- Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci U S A. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, Smyth MJ, Johnstone RW. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001;98:10833–10838. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gonzalez B, Yang H, Bueso-Ramos C, Hoshino K, Quintas-Cardama A, Richon VM, Garcia-Manero G. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood. 2006;108:1174–1182. doi: 10.1182/blood-2005-09-008086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozawa K, Nakanishi T, Tan M, Fang HB, Wang WC, Edelman MJ, Carlton D, Gojo I, Sausville EA, Ross DD. Preclinical studies of vorinostat (suberoylanilide hydroxamic acid) combined with cytosine arabinoside and etoposide for treatment of acute leukemias. Clin Cancer Res. 2009;15:1698–1707. doi: 10.1158/1078-0432.CCR-08-1587. [DOI] [PubMed] [Google Scholar]
- Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A, Pierce S, Estey EH, Issa JP, Kantarjian HM, Garcia-Manero G. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM, Ehinger M, Fisher PB, Grant S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18:7016–7025. doi: 10.1038/sj.onc.1203176. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yashiro M, Ren J, Hirakawa K. Histone deacetylase inhibitor, trichostatin A, increases the chemosensitivity of anticancer drugs in gastric cancer cell lines. Oncol Rep. 2006;16:563–568. [PubMed] [Google Scholar]
- Ziauddin MF, Yeow WS, Maxhimer JB, Baras A, Chua A, Reddy RM, Tsai W, Cole GW, Jr, Schrump DS, Nguyen DM. Valproic acid, an antiepileptic drug with histone deacetylase inhibitory activity, potentiates the cytotoxic effect of Apo2L/TRAIL on cultured thoracic cancer cells through mitochondria-dependent caspase activation. Neoplasia. 2006;8:446–457. doi: 10.1593/neo.05823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.