

Abstract
We synthesized two series of imatinib mesylate (STI-571) analogs to develop a Bcr-Abl and c-KIT receptor-specific labeling agent for positron emission tomography (PET) imaging to measure Bcr-Abl and c-KIT expression levels in a mouse model. The methods of molecular modeling, synthesis of STI-571 and its analogs, in vitro kinase assays, and radiolabeling are described. Molecular modeling revealed that these analogs bind the same Bcr-Abl and c-KIT binding sites as those bound by STI-571. The analogs potently inhibit the tyrosine kinase activity of Bcr-Abl and c-KIT, similarly to STI-571. [18F]-labeled STI-571 was prepared with high specific activity (75 GBq/μmol) by nucleophilic displacement and an average radiochemical yield of 12%. [131I]-labeled STI-571 was prepared with high purity (>95%) and an average radiochemical yield of 23%. The uptake rates of [18F]-STI-571 in K562 cells expressing Abl and in U87WT cells overexpressing c-KIT were significantly higher than those in the U87 cell and could be inhibited by STI-71 (confirming the specificity of uptake). PET scans of K562 and U87WT tumor-bearing mice with [18F]-STI-571 as a contrast agent showed visible tumor uptake and tumor-to-non-target contrast.
1. Introduction
Activating mutations of KIT are associated with certain human neoplasms, including those in the majority of patients with systemic mast cell disorders as well as those in patients with seminoma, acute myelogenous leukemia (AML), gastrointestinal stromal tumors (GISTs), and hypopigmentary disorders1 Improved knowledge of the mechanisms causing pathological mast cell growth will lead to the discovery of novel treatment options including drugs targeting the mutated KIT protein. The small-molecule tyrosine kinase inhibitor imatinib mesylate (Gleevec®, STI-571) is a potent inhibitor of wild-type (WT) KIT and certain mutant KIT isoforms and has become the standard of care for treating patients with metastatic GIST.2 However, distinct forms of tyrosine kinase domain (TKD), juxtamembrane domain, exon 8, and internal tandem duplication (ITD) mutations of c-KIT were observed in about 46% of core binding factor leukemia (CBFL) patients, and acquired KIT activation loop mutations can be associated with imatinib mesylate resistance in GIST.3
The current lack of diagnostic assays and markers predictive of sensitivity to c-KIT inhibitors limits the possibility of proper selection of patients for clinical trials to assess drugs targeting the c-KIT kinase. The serious clinical challenges for selection of optimal setting(s) in which to test and monitor both the biological efficacy and the therapeutic efficacy of these novel drugs include a) identification of patients whose disease would likely respond well to therapy with c-KIT kinase inhibitors, b) individualization of the therapeutic dose(s) and therapeutic regimens, c) proper integration of these novel drugs into established cytotoxic therapeutic protocols, and d) development of effective markers and methods for noninvasive monitoring of the early efficacy of therapy.
Noninvasive positron emission tomography (PET) imaging with c-KIT kinase-specific radiolabeled agents could provide a better assessment of the levels and heterogeneity of c-KIT expression and activity in tumors in individual patients and could provide selection criteria for inclusion of patients in redesigned clinical trials. Also, the ability to monitor the levels of expression and activity of c-KIT at the kinase level should provide a direct measure of biological drug efficacy (kinase inhibition) in tumors as opposed to surrogate tissues (e.g., hair, skin, etc.) before any therapeutic effect is expected.
The use of [18F]-fluoro-2-deoxy-D-glucose ([18F]-FDG) in the staging of and early prediction of response to STI-571 therapy in patients with recurrent GISTs has been extensively studied by Gayed et al.,4 who demonstrated that [18F]-FDG is superior to CT in predicting early response to therapy. In addition, more recently, Jager et al.5 as well as other investigators confirmed this finding. Noteworthy is the observation that GIST shares immunohistochemical, ultrastructural, and histogenetic similarities with the interstitial cells of Cajal. Both GIST and the interstitial cells of Cajal express c-KIT, the receptor tyrosine kinase that is the protein product of the c-KIT proto-oncogene. It has also been found that c-KIT from GIST cells has a high frequency of mutations that lead to constitutive activation of the c-KIT tyrosine kinase in the absence of stimulation by the kinase’s physiologic ligand (stem cell factor), which in turn causes uncontrolled stimulation of downstream signaling cascades with aberrant cellular proliferation and resistance to apoptosis.3b While these studies are of the great clinical value, they cannot truly identify before treatment those patients who will be responsive to imatinib mesylate chemotherapy. Our long term aim is to use kinase-specific [131I]- and [18F]-labeled STI-571 analogs for PET imaging to help identify patients with GISTs that overexpress c-KIT and that would respond favorably to therapy with STI-571. On PET, overexpression would correlate with high tumor uptake and retention of labeled STI-571 analogs, and favorable tumor response would be indicated by early decreases in tumor uptake and retention of labeled analogs. Similarly, we aim to identify GISTs with low c-KIT expression or activity as having low [131I]- or [18F]STI-571 analog uptake and retention and thus as being potentially poor responders to STI-571 therapy.
As part of an ongoing program to develop new potent inhibitors of c-KIT tyrosine kinases,6,7 we have developed a class of STI-571 analogs (4-(3-pyridyl)-2-pyrimidines) with significant c-KIT inhibition activities, and we aim to synthesize analogs of STI-571 radiolabeled with 131I or 18F. The plan to modify imatinib with radioactive fluorine was also pursued by Larson and colleagues,8 who used an 18F-radiolabeled derivative of imatinib to measure Bcr-Abl expression. In this study, we aimed to measure both Bcr-Abl and c-KIT expression levels in a mouse model.
2. Results and Discussions
2.1. Molecular modeling
Our FlexX docking results for the highest ranked configuration of each STI analog/c-KIT combination showed that STI-571 (11a), STI-F (11b), and STI-I (11e) (Scheme 1) had similar scores (−41.47, −38.54, −37.70, respectively) and nearly identical binding configurations. Scores for the same structures in Abl were −37.09, −38.39, and −40.58. As the scores are an estimate of the free energy of binding, more negative values are better. There is no evidence to suggest that these modifications would cause any unfavorable steric overlap. Therefore, the modified molecules should have activities for c-KIT and Abl kinase similar to those of STI-571.
Scheme 1.
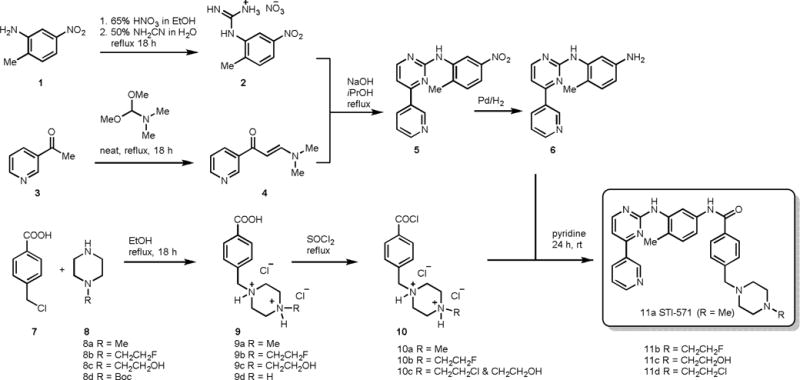
Synthesis of STI-571 and its analogs
2.2. Synthesis of STI-571 and its analogs
The synthesis of STI-571 and its analogs began with treatment of 2-methyl-5-nitroaniline 1 with 65% nitric acid in ethanol followed by the addition of cyanamide to give the corresponding 2-methyl-5-nitroaniline-guanidine nitrate salt 2 (Scheme 1). Pyridine derivative 4 was synthesized by refluxing 3-acetylpyridine 3 with N,N-dimethylformamide dimethyl acetal under neat conditions for about 18 h. The resulted compound 4 was subjected to condensation reaction with nitrate salt 2 in presence of sodium hydroxide in refluxing isopropanol to produce pyrimidineamine derivative 5, which was subsequently hydrogenated with 10% palladium on carbon to obtain N-(2-Methyl-5-aminophenyl)-4-(3-pyridyl)-2-pyrimidineamine 6 in high yield. In order to synthesize the STI-571 and its analogs, the coupling acid chloride derivatives 10 were prepared in two simple steps. First, alkylation of piperizine derivatives 8 with α-chloro-p-toluic acid 7 in refluxing ethanol to produce the corresponding acid 9, which was subsequently activated with thionyl chloride to give the corresponding acid chloride 10. It was noted that, during the acid chloride formation most of the primary alcohol in 10c was converted into chloride and obtained as mixture. The acid chlorides were immediately used for the next reaction without further purification as they found to be less stable. Condensation of N-(2-Methyl-5-aminophenyl)-4-(3-pyridyl)-2-pyrimidine-amine 6 and acid chloride derivatives 10 in pyridine yielded the STI-571 and its analogs (Scheme 1) in good yields.
Furthermore, 4-iodobenzyl substituted STI derivative 11e and 4-n-tributyltinbenzyl substituted STI derivative 11f (Scheme 2) were also synthesized. The salt 9e was prepared by alkylation of 9d with 4-iodobenzyl bromide 12, and in other hand, palladium catalyzed transmetallation of 13 with tributyltin metal, then reductive amination of the resultant stannyl benzaldehyde 14 with piperazine benzoic acid 9d gave 9f as a salt.9 The salts 9e and 9f were treated with thionyl chloride to obtain 10e and 10f respectively, which were then subjected to condensation with 6 in pyridine to produce 11e and 11f respectively in good yields (Scheme 2).
Scheme 2.
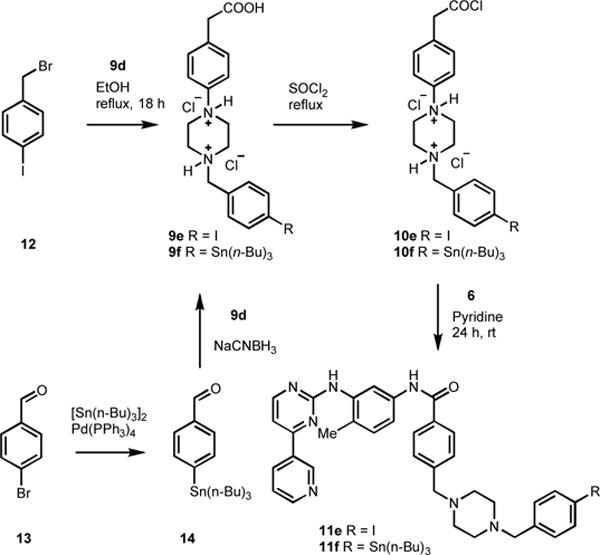
Synthesis of STI-I and STI-Sn analogs.
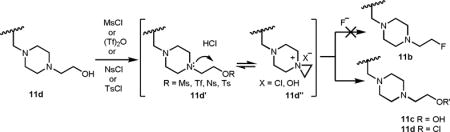
In additions to the synthesis of STI-571 and its analogs, we chose to make radiolabeled STI analogs. Converting the primary alcohol of 11c into as a good leaving group then knocking it off with radiolabeled nucleophiles would lead to the radiolabeled STI analogs. However, all our efforts to synthesize the mesylate, tosylate or triflate derivative of 11c were unsuccessful due the internal attack of the piperazine amine as a nucleophile onto the leaving group to form highly unstable aziridinium salt which was then decomposed to give back 11c (STI-OH) or 11d (STI-Cl).10 Since the conversion of primary alcohol of 11c (STI-OH) into a good leaving was ineffective, we chose 11d (STI-Cl) as a precursor for the synthesis of radiolabeled STI analog.
2.3. Radiolabeling
STI-[18F] 11g was prepared by fluorination of the STI-Cl 11d with n-Bu4N[18F] followed by HPLC purification (scheme 3). n-Bu4N[18F] was prepared in situ from n-Bu4NHCO3 and aqueous H18F using 0.40 mL (1% solution, ~7 mmol) of n-Bu4NHCO3 to elute the activity from the ion exchange cartridge.11 After radiofluorination of the precursor 11d, the crude reaction mixture was passed through a silica gel cartridge (900 mg; Alltech), and the crude product was eluted with 30% MeOH in dichloromethane. Silica gel holds the unreacted [18F] fluoride from the product and helps to avoid overloading the HPLC column with high levels of free fluoride during purification. The recovered 18F-labeled STI was purified by semi preparative HPLC and was eluted at 11.6 min. using a 50% MeCN and 50% water-containing 0.1% ammonium formate solvent system with a flow of 4 mL/min. Ammonium formate (0.1%) in water was used to improve the peak shape. Quality control analysis of the final product STI-[18F] 11g was performed by analytical HPLC using the same solvent system at a flow rate of 1 mL/min. Co-injection of the final product 11g with an authentic sample of STI-F 11b showed that the radiolabeled compound and standard STI-F were co-eluted at 6.7 min. The radiochemical yield of this synthesis was 8–16% with an average of 12% in 12 runs (corrected for decay). The radiochemical purity was >99%, with specific activity >74 GBq/mmol. The synthesis time was 65–75 min from the end of bombardment.
Scheme 3.
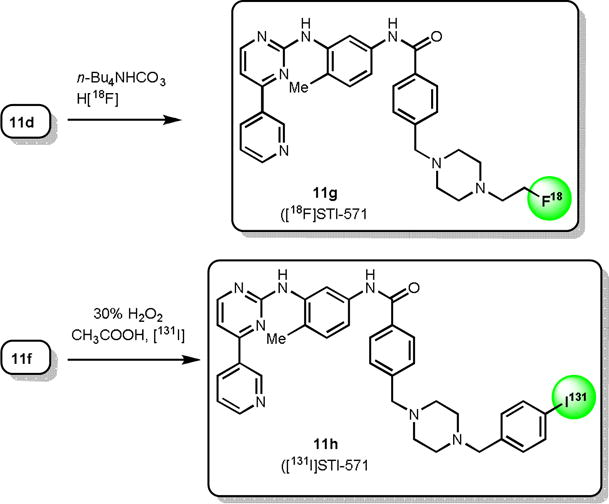
Synthesis of radiolabeled [18F]STI-571 and [131I]STI-571 analogs.
STI-[131I] 11h was prepared by treating STI-Sn 11f precursor with a mixture of 30% hydrogen peroxide/glacial acetic acid (1:3) in the presence of [131I]iodide for 3 min at room temperature (scheme 3).12 The reaction was quenched with an aqueous solution of saturated sodium metabisulfite and loaded onto a Sep-Pak C18 column. The column was washed with water, and the product was eluted with ethanol. Crude STI-[131I] was purified by preparatory HPLC, concentrated on a Sep-Pak C18 column, and isolated by elution with ethanol. Radiochemical purity was determined by radio-thin-layer chromatography using chloroform/MeOH (85:15) on silica gel plates. STI-[131I] 11h was also characterized by radio-HPLC on an analytical C8 column using acetonitrile/0.1% ammonium formate in water (1:1) at a 1 mL/min flow rate, with UV detection set at 265 nm. A mixture of standard STI-I 11e and STI-[131I] 11h was found to co-elute. The radiochemical yield of STI-[131I] in crude form averaged 23.3% (19–27%, n = 3). The radiochemical purity of the final product was >95%.
3. Bcr-Abl kinase assay
The kinase assay was performed on STI-F (11b), and STI-I (11e), STI-OH (11c) and STI-571 (11a). STI-571 inhibits Bcr-Abl with an IC50 of 0.11 μM; STI-F and STI-I IC50 values were 1.3 μM and 0.73 μM respectively. STI-OH was the least potent, with an IC50 of 7.08 μM (Figure 1).
Figure 1.
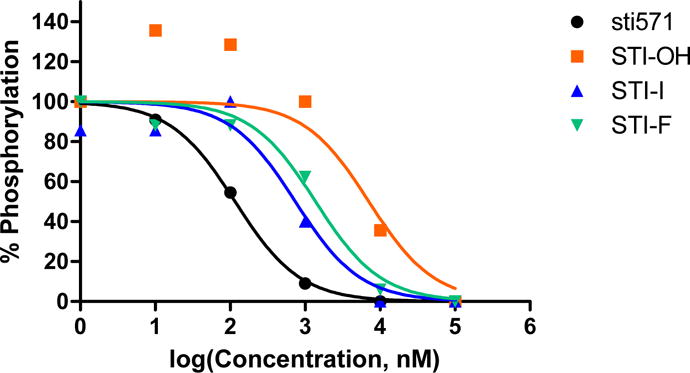
Kinetic inhibitory effect rates of STI-571 and its analogs (determined by measuring phosphorylation rates in spectrophotometric assays of Bcr-Abl kinase activity).
4. Free energy perturbation (FEP) Calculation results
In order to better understand the observed experimental difference in inhibitory effect of the compounds versus STI-571 (11a), we ran Monte Carlo/free energy perturbation (MC/FEP) calculations for 11a, 11b, and 11c. These calculations were performed in stages starting from the docked STI-571 structure. For set 1, perturbations involved Ethyl->Methyl, Propyl->2-hydroxyethyl and Propyl->Ethyl. In set 2, perturbations were 2-hydroxyethyl->2-fluoroethyl and Propyl->2-hydroxyethyl. Shown in Figure 2 is the structure with lowest average total system energy out of those stored at the end of each 250K configurations. The majority of interactions were identical to the crystal structure. One exception was the piperazine unit, which is nearby the backbone carbonyls of His 95/Arg 96 in the crystal, but directly hydrogen bonds with Asp115 in the simulation. Extending STI-571 by one or two carbons on the piperazine was increasingly disfavored as shown in Table 1. This is expected, since the additional methyl groups are not interacting with a hydrophobic area in the protein. Changing to 2-hydroxyethyl was favorable compared to the propyl group by 1.62 ± 0.11 kcal/mol, but overall unfavorable by 3.42 ± 0.20 kcal/mol in comparison to 11a. This was most likely due to the hydrogen bonding capability of the OH group, which accepts a hydrogen bond from Arg 96 in the lowest energy structure (not shown). The experimental ΔΔGbind value for this change was 2.44 kcal/mol. The calculated ΔΔGbind for 2-hydroxyethyl to 2-fluoroethyl was 0.67 ± 0.09 kcal/mol, but the experimental determined value was −0.97. With 2-fluoroethyl, there is a water bridging the interaction to Asp115, which is also observed for 2-hydroxyethyl. Error in the theoretical values could be due to incomplete sampling and another estimate may be determined from comparing the set 1 and set 2 perturbations, which repeat the Propyl->2-hydroxyethyl calculations from a different starting point. In that calculation, the ΔΔGbind was −2.51 ± 0.12 kcal/mol as compared with −1.62 ± 0.11 kcal/mol from set 1. Using that value, the ΔΔGbind would give 2.53 ± 0.21 kcal/mol and 3.20 ± 0.23 kcal/mol for 2-hydroxyethyl and 2-fluoroethyl, respectively. As shown in the IC50 plots, there is also error associated with the biological experiments, which could also explain the differences observed between the theoretical and experimental values.
Figure 2.
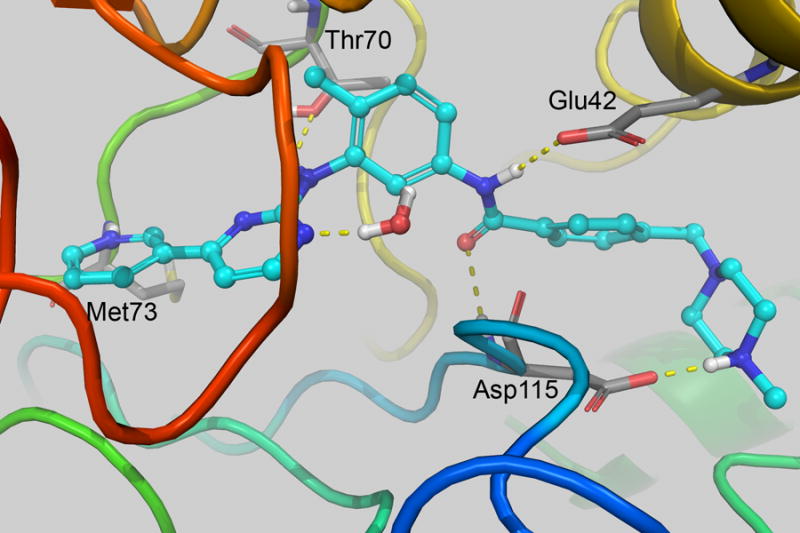
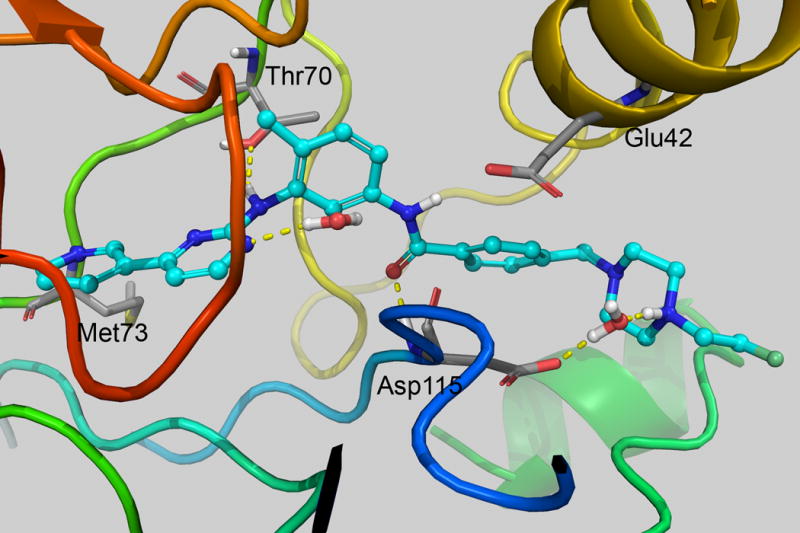
(left) Interaction of STI-571 and (right) 11b from MCPRO+ calculations. Only nearby residues and water with H-bonding interactions with the ligand are shown. Images were created with PyMOL v1.5.0.4.26
Table 1.
Comparison of experimental and theoretically calculated ΔΔGbind for STI-571 and analogs.
| Compound | ΔΔGbind (calc) | ΔΔGbind (exptl)a | IC50 (nM)b |
|---|---|---|---|
| 11a | 0 | 0 | 116.0 |
| 11b | 4.09 ± 0.22 | 1.47 | 1378 |
| 11c | 3.42 ± 0.20 | 2.44 | 7083 |
| 11e | – | 1.10 | 739.5 |
| R = Ethyl | 2.58 ± 0.12 | – | |
| R = Propyl | 5.04 ± 0.17 | – |
Calculated from ΔΔGbind = RT ln [IC50(cmpd)/IC50(11a)]. RT = 0.59219 at 298 K; values are listed in kcal/mol.
Determined by fit of IC50 curve to log(conc) versus normalized %Phosphorylation with GraphPad Prism v5.04.
5. In vitro studies
U87WT cells were produced by retroviral transduction of U87 cells with a wild-type human c-KIT receptor. Western blotting of protein extracts from K562, U87, and U87WT cells was performed to assess Bcr-Abl and c-KIT expression levels (Figure 3). K562 cells expressed Bcr-Abl, and U87WT cells expressed c-KIT.
Figure 3.
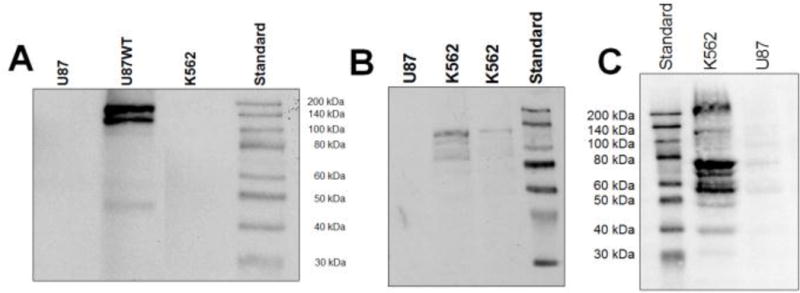
(A) Western blot of c-KIT expression in U87WT cells. (B and C) K562 cells expressed Bcr-Abl, Bcr, and Abl but not c-KIT.
In vitro radiotracer accumulation, washout, and blocking studies performed in our laboratory showed that the binding of [18F]STI-571 diminished as excess STI-571 was added to the cell cultures (A & B, Figure 4). The uptake and washout of [18F]STI-571 at 1 and 2 h in K562 cells revealed an increase in residual intracellular radioactivity (C, Figure 4), which may be explained, at least in part, by intracellular re-compartmentalization and entrapment of the Abl kinase domain-[18F]STI-571 complex in the compartment with poor exchange (i.e., proteasome). The uptake and washout at 1 and 2 h in wild-type U87 cells showed no increase in residual radioactivity (D, Figure 4). The results of uptake and washout studies indicated that [18F]STI-571 and STI-571 have the same binding target.
Figure 4.

(A) [18F]STI-571 rapidly accumulates in K562 cells and then reaches a dynamic at 30 min. (B) The [18F]STI-571 equilibrium in K562 cells was progressively decreased by exposure to increasing concentrations of STI-571. (C) Uptake and washout of [18F]STI-571 at 1 and 2 h in K562 cells. (D) Uptake and washout of [18F]STI-571 at 1 and 2 h in wild-type U87 cells.
6. Small-animal PET studies
The rates of [18F]STI-571 uptake in K562 cells expressing Abl and in U87WT cells overexpressing c-KIT were significantly higher than that in wild-type U87 cells and were susceptible to inhibition by cold STI-571 (confirming the specificity of uptake). A PET scan imaging study of K562 and U87WT-bearing mice was undertaken to compare [18F]FDG levels and the results were shown in Figure 5. Tumors implanted in the right shoulder were visible in all mice; the uptake of the tracer in the digestive tract was quite high. The tumors were visualized at 60 and 120 min after injection. The large amounts of radioactivity in the liver and the intestines were not unexpected, because imatinib is eliminated mainly through biliary excretion or hepatic metabolism, presumably as a result of catabolism and defluorination of [18F]STI-571 in mice, as evidenced by extensive accumulation of [18F] in the bone. In contrast, [18F]FDG was clearly visible in the mice at 45 min. [18F]STI-571 PET images showed higher uptake of the radiotracer in c-KIT-overexpressing U87WT tumors (right shoulder) than in U87 tumors (left shoulder), which don’t express c-KIT. In both studies, tumor-to-muscle and c-KIT+ tumor-to-control ratios were the highest at 60 min after [18F]STI-571 administration.
Figure 5.
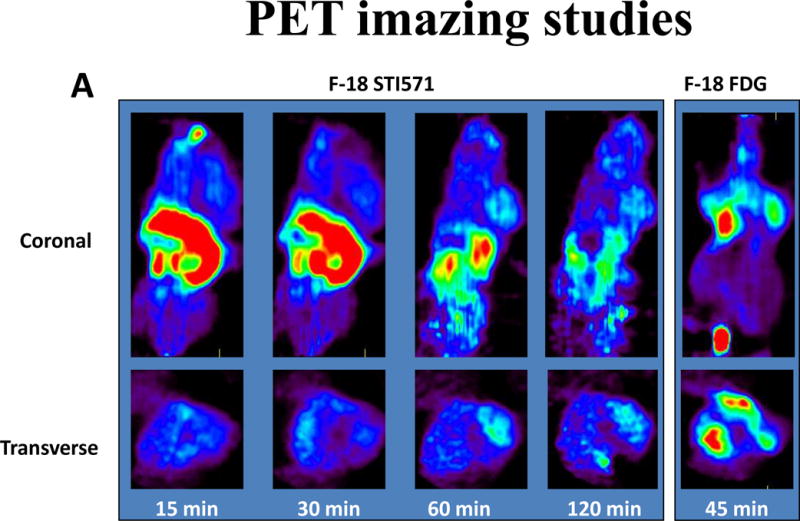
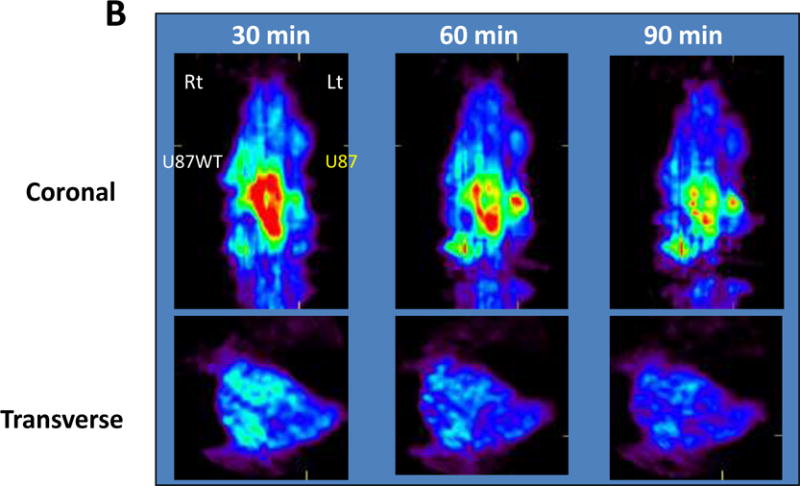
(A) [18F]STI-571 PET images of Bcr-Abl expression in K562 tumors (right shoulder). PET images of [18F]FDG accumulation are provided for comparison. (B) [18F]STI-571 PET images showing the higher uptake of the radiotracer in c-KIT overexpressing U87WT tumors (right shoulder) than in wild-type U87 tumors (left shoulder), which do not express c-KIT.
7. Conclusion
In summary, we have synthesized a series of non-labeled STI-571 analogs along with [18F] and [131I] radiolabeled versions of two of them. The goal was to develop a labeling agent for positron emission tomography (PET) imaging that could measure Bcr-Abl and c-KIT kinase expression levels. We have shown that these analogs inhibit the tyrosine kinase activity of Bcr-Abl and c-KIT, albeit to a lesser degree than STI-571, which is further supported by our FEP calculations. The uptake rates of [18F]-STI-571 in K562 cells expressing Abl and in U87WT cells overexpressing c-KIT were significantly higher than those in the wild type U87 and diminished with excess of STI-571 confirming the specificity of uptake. PET scans of K562 and U87WT tumor–bearing mice with [18F]-STI-571 as a contrast agent showed visible tumor uptake and tumor-to-non-target contrast. Thus, [18F]-STI-571 could be utilized as a marker of sensitivity to Bcr-Abl and c-KIT inhibitors and assist with the selection of patients that could most likely benefit from drugs targeting the Bcr-Abl and c-KIT kinases. Further studies with 131I-labeled STI-571 are under way.
8. Materials and Methods
8.1. Modeling
8.1.1 Docking of STI-571 and F- or I-labeled analogs
The docking calculations were completed on a 4-processer R16000 SGI Tezro running Sybyl 7.214 and FlexX15 docking software. The crystal structures of STI-571 bound to c-KIT (PDB code 1T46) and Abl (PDB code 1IEP) were obtained from the Protein Data Bank.13 The structures were brought into Sybyl and all additional chains, waters, and ions were removed before docking. The STI-571 ligand from 1IEP was extracted and modified with correct atom types for the Tripos force field and protonated at the piperazine nitrogen furthest from the phenyl group. A minimization was completed using the Powell method and a gradient cutoff of 0.05 kcal/(mol Å). This structure was the basis for creating STI-F and STI-I, which were also minimized in the same manner. The primary docking region selected for FlexX included all residues within 6.5 Å of the STI-571. Ten configurations were requested for each ligand, and formal charge assignment for the ligand was template based.
8.1.2 Free energy perturbation (FEP) calculations
The free energy perturbation (FEP) calculations were completed on a 4-quad core (16-core) 2.4 Ghz AMD Opteron system using the RHEL 5.x OS and a 2-eight core (16-core) 3.0Ghz AMD Opteron system. The Suite 2012 from Schrödinger was utilized for all docking and Monte Carlo/free-energy perturbation (MC/FEP) calculations. The PDB structure 2hyy was downloaded from the Protein Data Bank16 and read into Maestro17 with alternative positions turned off, and all chains except chain A were removed. A basic protein preparation was performed using program defaults, with the addition of filling in missing side chains and deleting water molecules beyond 5 Å from heteroatoms. The complete preparation of the protein portion of the model involved the following: 1) basic protein preparation; 2) assignment of heteroatom states; 3) H-bond assignment, including PROPKA; 4) deletion of waters with less than three H-bonds to non-waters; 5) Impref “H-only” minimization; and 6) Impref “Minimize All” to root mean square deviation of 0.5. The structure of STI-571 was drawn in ChemDrawUltra 12.0 on a PC, transferred to the Linux box and converted to an SDF formatted file. A Ligprep18 calculation was performed on the structure by using default criteria and Epik19 ionization. The Glide Grid for docking was created with crystal ligand as basis and no constraints were selected during the calculation. The STI-571 structure was docked using the virtual screening workflow,20 which involved docking with Glide XP mode starting with three extra conformations and rescoring with the Prime MMGBSA ΔGbind.21 Variations were selected in the Ligand Functional Group Mutation by FEP panel for MCPRO+.22 Perturbations for set 1 involved Ethyl->Methyl, Propyl->2-hydroxyethyl and Propyl->Ethyl. For set 2, the starting structure was modified to contain a propyl group instead of a methyl group. No optimization of the structure was done and the residue was renumbered and renamed. Perturbations for set 2 were 2-hydroxyethyl->2-fluroethyl and Propyl->2-hydroxyethyl. Simulations were completed with default parameters. Briefly, this allowed for full ligand flexibility and protein flexibility for residue side chains within 8 Å of ligand and those beyond 11 Å were chopped. Solvation involved a 22 Å TIP3P water cap around the isolated ligand and complex. Atom type parameters came from the OPLS2005 force field. The MC/FEP protocol for MCPRO+ involved running 10 M configurations of solvent only, 5 M of system equilibration, and 10 M of system averaging at 25 °C.
8.2. Chemistry
All chemicals and solvents were obtained from Sigma-Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA) and used without further purification. Analytical HPLC was performed on a Varian Prostar system, with a Varian Microsorb-MW C18 column (250 × 4.6 mm; 5μ) using the following solvent system: A = H2O/0.1% TFA, B = acetonitrile/0.1% TFA. The Varian PrepStar preparative system equipped with a Prep Microsorb–MW C18 column (250 × 41.4 mm; 6μ; 60 Å) was used for preparative HPLC with the same solvent system. Low resulution Mass spectra (ion spray, a variation of electrospray) were acquired on an Applied Biosystems Q-Trap 2000 LC-MS-MS system. High resolution Mss spectra (ion spray) were acquired on an Agilent Time of Flight Accurate Mass 6220A system with Agilent LC1200 HPLC at the front end. UV was measured on a PerkinElmer Lambda 25 UV/Vis spectrometer. IR was measured on a PerkinElmer Spectrum One FT-IR spectrometer. 1H-NMR, and 13C-NMR spectra were recorded on a Bruker Biospin spectrometer with a B-ACS 60 autosampler (600.13 MHz for 1H-NMR and 150.92 MHz for 13C-NMR). Chemical shifts (δ) were determined relative to d4-MeOH (referenced to 3.34 ppm (δ) for 1H-NMR and 49.86 ppm for 13C-NMR). Proton-proton coupling constants (J) are given in hertz, and spectral splitting patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet or overlapped (m), and broad (br). Flash chromatography was performed using Merck silica gel 60 (mesh size 230–400 ASTM) or a Teledyne Isco (Lincoln, NE) CombiFlash Companion and SQ16x flash chromatography system with RediSep columns (normal phase silica gel; mesh size 230–400 ASTM) and Fisher Optima TM grade solvents. Thin-layer chromatography (TLC) was performed on E. Merck (Darmstadt, Germany) silica gel F-254 aluminum-backed plates with visualization under UV (254 nm) and by staining with potassium permanganate or ceric ammonium molybdate.
8.2.1 2-Methyl-5-nitrophenyl-guanidine nitrate (2)23
2-Methyl-5-nitroaniline 1 (100 g, 0.657 mol) was dissolved in ethanol (250 mL), and a 65% aqueous nitric acid solution (48 mL, 0.65 mol) was added thereto. When the exothermic reaction was stopped, cyanamide (41.4 g) dissolved in water (41.4 g) was added. The brown mixture was reacted under reflux for 24 h. The reaction mixture was cooled to 0 °C, filtered, and washed with ethanol-diethyl ether (1:1, v/v) to give 2-methyl-5-nitrophenyl-guanidine nitrate 2 (98 g). RF = 0.1 (DCM/MeOH/25% aq. ammonia, 150:10:1). MS: 195.2 (M+H); 1H-NMR (DMSO-d6) 8.63–8.64 (br, 1H), 7.21–7.27 (m, 1H), 6.72–6.76 (d, 1H), 6.59 (s, 3H), 1.43 (s, 3H).
8.2.2 3-Dimethylamino-1-(3-(6-Methyl-pyridyl))-2-propen-1-one (4)23
3-Acetylpyridine 3 (100 g, 0.19 mol) was added to dimethylformamide dimethyl acetal (156.5 g, 1.27 mol), and the mixture was reacted under reflux for 23 h. After the reaction mixture was cooled to 0 °C, a mixture of diethyl ether and hexane (3:2, v/v) (500 mL) was added, and the whole mixture was stirred for 4 h. The resulting solid was filtered and washed with a mixture of diethyl ether and hexane (500 mL, 3/2, v/v) to give 3-dimethylamino-1-(3-pyridyl)-2-propen-1-one 4 (120 g, 85%). RF = 0.46 (DCM/MeOH, 9:1). 1H and 13C NMR were in agreement with the reported data.
8.2.3 N-(2-methyl-5-nitrophenyl)-4-(3-pyridyl)-2-pyrimidine-amine (5)23
Dimethylamino-1-(3-pyridyl)-2-propen-1-one 4 (25 g, 0.14 mol), 2-methyl-5-nitrophenyl-guanidine nitrate 2 (36 g, 0.14 mol), and sodium hydroxide powder (6.5 g, 0.163 mol) were dissolved in isopropanol and reacted under reflux for 18 h. The reaction solution was cooled to 0 °C, filtered, washed with isopropanol and MeOH, and dried to give N-(2-methyl-5-nitrophenyl)-4-(3-pyridyl)-2-pyrimidine-amine 5 (20 g). RF = 0.6 (DCM/MeOH, 9:1). NMR was in agreement with the reference.
8.2.4 N-(5-amino-2-methylphenyl)-4-(3-pyridyl)-2-pyrimidine-amine (6)23
N-(2-methyl-5-nitrophenyl)-4-(3-pyridyl)-2-pyrimidine-amine 5 (35 g, 0.114 mol) and stannous chloride dihydrate (128.5 g, 0.569 mol) were dissolved in a solvent mixture of ethyl acetate and ethanol (250 mL, 10/1, v/v), and the reaction solution was refluxed for 4 h. The solution was cooled to room temperature, washed with 10% aqueous sodium hydroxide solution, and concentrated to give N-(5-amino-2-methylphenyl)-4-(3-pyridyl)-2-pyrimidine-amine 6 (35 g). RF = 0.45 (DCM/MeOH, 9:1). NMR was in agreement with the reference.
8.2.5 4-(4-Methylpiperazinomethyl)benzoic acid dihydrochloride (9a)24
To a well-stirred suspension consisting of 17.1 g (0.10 mol) of α-chloro-p-toluic acid 7 in 150 mL of absolute ethanol under a nitrogen atmosphere at room temperature (~20 °C), a solution consisting of 44.1 g (0.44 mol) of N-methylpiperazine 8a dissolved in 50 mL of ethanol was added drop wise. The resulting reaction mixture was refluxed for 16 h and then cooled to room temperature. The cooled reaction mixture was concentrated in vacuo, and the residue was partitioned between 100 mL of diethyl ether and 100 mL of 3 N NaOH. The separated aqueous layer was then washed three times with 100 mL of diethyl ether, cooled in an ice-water bath and subsequently acidified with concentrated hydrochloric acid. The resulting solids were filtered, air-dried, triturated with 150 mL of boiling isopropyl alcohol, and stirred for 2 min. After the product was filtered while hot and dried, 9.4 g (35%) of pure 4-(6-methylpiperazinomethyl)benzoic acid dihydrochloride 9a was obtained as the hemihydrate (M.P. 310–312 °C). 1H NMR (D2O) δ 8.04 (d, J = 8.21 Hz, 2H), 7.59 (d, J = 8.21 Hz, 2H), 3.50 (s, 2H), 3.63 (br, 8H), 2.97 (s, 3H); 13C NMR δ 170.18, 133.13, 131.91, 130.90, 60.22, 50.61, 48.77, 43.25.
8.2.6 4-(4-(2-Fluoro-ethyl)-piperazinomethyl)benzoic acid dihydrochloride (9b)
Synthesis of 9b followed as described for synthesis of 9a by using N-(2-fluoro-ethyl)-piperazine 8b, compound 9b was obtained as a solid. 1H NMR (D2O) δ 8.05 (d, J = 8.21 Hz, 2H), 7.63 (d, J = 8.21 Hz, 2H), 4.85 (t, J = 48.0 Hz H–F coupling, H–H 1–3 coupling 4.9 Hz, 2H), 4.54 (s, 2H), 3.71 (b, 10H). 13C NMR (151 MHz, D2O) δ 169.88, 132.84, 131.85, 131.64, 130.59, 78.57, 77.47, 59.93, 56.64, (JC–F = 22 Hz, 1–3 coupling), 49.17, 48.80. (JC–F = 112 Hz, 1–2 coupling). 19F NMR (D2O) δ −221.51 (1H decoupled), −221.41 (1H coupled), −221.46, −221.49, −221.51, −221.54, −221.58, −221.59, −221.63, −221.67. HRMS (C14H19FN2O2) calcd. 267.1503 found 267.1504 [M+H].
4-(4-(2-Hydroxy-ethyl-piperazinomethyl)benzoic acid dihydrochloride (9c)
Synthesis of 9c followed as described for synthesis of 9a by using N-(2-hydroxy-ethyl)-piperazine 8c, compound 9c was obtained as a solid. 1H NMR (D2O) δ 8.02 (d, J = 8.21 Hz, 2H), 7.57 (d, J = 8.21 Hz, 2H), 4.48 (s, 2H), 3.86 (m, 2H), 3.63 (br, 8H), 3.37 (m, 2H); 13C NMR δ 169.85, 132.77, 131.84, 131.50, 130.49, 59.86, 54.85, 48.78, 48.17, 40.52. HRMS (C14H20N2O3) calcd. 265.1547 found [M+H]
8.2.7 4-(Piperazinomethyl)benzoic acid dihydrochloride (9d)24
Synthesis of 9d followed as described for synthesis of 9a by using N-Boc-piperazine 8d, compound 9d was obtained as a solid. 1H NMR (D2O) δ 8.02 (d, J = 7.96 Hz, 2H), 7.51 (d, J = 8.21 Hz, 2H), 4.40 (s, 2H), 3.46 (br, 8H), 3.37 (m, 2H).
8.2.8 4-(4-(4-Iodobenzyl)-piperazinomethyl)benzoic acid dihydrochloride (9e)
To a well-stirred suspension consisting of 5 g (17 mmol) of 4-iodobenzyl bromide 12 in 50 mL of anhydrous ethanol under an argon atmosphere at room temperature, a solution of 7 g (17 mmol) of compound 9d in 50 mL of ethanol was added drop wise; then, 0.7 mL triethylamine was added. The resulting reaction was refluxed for 16 h and then cooled to room temperature. The cooled reaction mixture was concentrated in vacuo and the resulting residue was partitioned between 50 mL of diethyl ether and 50 mL of 3 N NaOH. The separated aqueous layer was then washed three times with 50 mL of diethyl ether, cooled in an ice-water bath, and acidified with concentrated hydrochloric acid. The resulting solids were filtered and air-dried, titrated with 100 mL of boiling isopropyl alcohol, and stirred for 2 min. After filtering and drying, the material was used for the next reaction without characterization. 1H NMR (600 MHz, D2O) δ 8.02 (d, 2H), 7.83 (d, J = 8.1, 2H), 7.55 (d, J = 8.0, 2H), 7.19 (d, J = 8.1, 2H), 4.41 (s, 2H), 4.31 (s, 2H), 3.53 (s, 8H). 13C NMR (151 MHz, D2O) δ 138.60, 132.88, 131.52, 131.39, 130.49, 130.45, 60.23, 59.93, 48.44, 48.19, 40.77. HRMS (C19H21IN2O2) calcd. 437.0720 found 437.0719 [M+H].
8.2.9 4-(4-(4-Tributylstannnylbenzyl)-piperazinomethyl)benzoic acid dihydrochloride (9f)
Compound 9d (446 mg, 1 mmol) was dissolved in 20 mL of DMF and cooled at 0 °C. 4-Tributylstannylbenzaldehyde 14 (395 mg, 1 mmol) dissolved in 2.5 mL of acetic acid was added drop wise to the solution, followed by sodium cyanoborohydride (42 mg). The reaction mixture was kept at room temperature for 18 h. The solvent was dried, and the residue was subjected to silica gel chromatography using 10% MeOH (7 M ammonia)/DCM; 400 mg was obtained 9f (70%). 1H NMR (DMSO) δ 7.786 (t, J = 7.29 Hz, 4H), 7.16 (d, J = 8.21 Hz, 2H), 3.41 (s, 2H), 3.34 (s, 2H), 2.31 (br, 8H), 1.48 (m, 6H), 1.27 (m, 6H), 0.86 (t, 6H), 0.81 (t, 9H). 13C NMR (75 MHz, CDCl3) δ 142.75, 139.79, 136.60, 133.02, 129.96, 129.26, 129.08, 62.92, 62.58, 55.66, 53.35, 29.39, 27.52, 14.14, 9.82. HRMS (C31H48N2O2Sn) calcd 601.2811, found 601.2821 [M+H]
8.2.10 4-(4-Methylpiperazinomethyl)benzoyl chloride dihydrochloride (10a)24
To 20 g (0.065 mol) of 4-(4-methylpiperazinomethyl)benzoic acid dihydrochloride 9a under a nitrogen atmosphere, 119 mL of thionyl chloride (194 g, 1.625 mol) was added to form a beige-white suspension. The reaction mixture was refluxed for 24 h and then cooled to room temperature (~20 °C). The resulting suspension was filtered, and the recovered solids were washed with diethyl ether and dried to 17.0 g (81%) of pure 4-(4-methylpiperazinomethyl)benzoyl chloride dihydrochloride 10a. The material was used for the next reaction without characterization.
8.2.11 N-4-[3-(4-methyl)-pyridyl]-2-pyrimidine amine (11a)(STI-571)4
A mixture of N-(2-methyl-5-aminophenyl)-4-pyridyl))-2-pyrimidine-amine 6 (5 g, 18 mmol) and 4-(4-methylpiperazinomethyl)benzoyl chloride dihydrochloride 10a (5 g, 20 mmol) was stirred in 50 mL of anhydrous pyridine at 20 °C for 18 h. The reaction mixture was concentrated in a vacuum. The residue was subjected to silica gel chromatography using 5% MeOH (7 M ammonia) in DCM to produce 11a in 5 g (60% yield). HRMS (C29H31N7O) calcd. 494.2663, found 494.2653 [M+H].
8.2.12 N-4-[3-(4-methyl)-pyridyl]-2-pyrimidine amine (STI-571, mesylate salt)25
The free base STI-571 11a was dissolved in 15 mL of ethanol and 1 equivalent of methyl sulfonic acid was added. The solution was reacted at 45 °C for 2 h, and the solvent was evaporated to give the mesylate salt. 1H NMR (D2O) δ 8.63 (s, 1H), 8.20 (d, J = 5 Hz, 1H), 8.04 (d, J = 5 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.83 (s, 1H), 7.49 (d, J = 7.5 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H), 7.05 (dd, 1H), 6.96 (d, J = 8.0 Hz, 1H), 6.85 (m, 2H), 3.58 (s, 2H), 3.0 (br, 8H), 2.80 (s, 3H), 2.75 (s, 3H). 13C NMR (151 MHz, D2O) δ 167.18, 159.65, 158.69, 158.16, 144.00, 142.91, 140.61, 136.11, 135.68, 135.10, 132.07, 131.53, 131.07, 128.28, 127.22, 117.62, 108.47, 59.84, 50.45, 48.48, 42.89, 38.57, 16.61. HRMS (C29H31N7O) calcd. 494.2663, found 494.2653 [M+H], 590.2542 [M+CH3SO3H].
8.2.13 STI-F (11b)
Synthesis of 11b followed as described for synthesis of 11a. STI-F was obtained as a solid. 1H NMR (CD3OD) δ 10.17 (s, 1H), 9.28 (s, 1H), 8.98 (s, 1H), 8.69 (d, J = 3 Hz, 1H), 8.51 (dd, J = 4.8, 1.8 Hz, 1H), 8.48 (dd, J = 7.8, 1.8 Hz, 1H), 8.10 (s, 1H), 7.91 (d, J = 6.6, 2H), 7.52 (t, J = 7.8 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.431 (m, 3H), 7.21 (d, J = 5.0 Hz, 1H), 4.52 (dt, JH–F = 48.0 Hz, JH–H = 4.9 Hz, 2H), 3.55 (s, 2H), 3.34 (b, 4H), 2.65 (dt, JH–F = 28.0 Hz, JH–H = 5 Hz, 2H), 2.50 (b, 4H), 2.23 (s, 3H); 13C NMR (CD3OD) δ 165.71, 162.10, 161.68, 159.92, 151.84, 148.68, 138.28, 137.68, 134.88, 134.32, 132.71, 130.48, 129.13, 128.05, 124.23, 117.72, 117.24, 107.99, 82.72, 81.63, 61.98, 57.97, 57.85 (JC–F = 18 Hz, 1–3 coupling), 53.32, 52.97 (JC–F = 52.82 Hz, 1–2 coupling). 19F NMR (CD3OD) δ −217.20 (1H decoupled), δ (1H coupled), −217.05, −217.10, −217.14, −217.15, −217.19, −217.22, −217.27, −217.32. HRMS (C30H32FN7O) calcd. 526.2725, found 526.2719 [M+H].
8.2.14 STI-OH (11c) and STI-Cl (11d)
Syntheses of 11c and 11d were followed as described for synthesis of 11a. STI-OH and STI-Cl were separated via silica gel chromatography using 5% MeOH (7 M ammonia) in DCM.
8.2.14.1 STI-OH (11c)
1H NMR (CD3OD) δ 9.29 (d, 1H), 8.65 (dd, J = 6.5, 1.5 Hz, 1H), 8.60 (d, J = 8.0 Hz, 1H), 8.48 (d, J = 5 Hz, 1H), 8.22 (s, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.56 (dd, J = 8.0, 1.5 Hz, 1H), 7.51 (t, 3H), 7.43 (t, J = 8.0 Hz, 2H), 7.37 (d, J = 5.0 Hz, 1H), 7.28 (d, J = 8.5 Hz, 1H), 3.71 (t, J = 8.0 Hz, 2H), 3.63 (s, 2H), 3.58 (s, 2H), 2.70 (br, 8H), 2.57 (t, J = 8.0 Hz, 2H), 2.32 (s, 3H). 13C NMR (151 MHz, DMSO) δ 165.28, 161.58, 161.15, 159.44, 151.33, 148.13, 142.03, 137.76, 137.15, 134.42, 133.71, 132.19, 130.01, 128.62, 127.58, 127.52, 123.77, 117.19, 116.74, 107.50, 61.62, 60.20, 58.45, 53.12, 52.62, 17.60. HRMS (C30H33N7O2) calcd.524.2768, found 524.2761[M+H].
8.2.14.2 STI-Cl (11d)
1H NMR (CD3OD) δ 9.28 (s, 1H), 8.68 (dd, 1H), 8.51 (dd, J = 4.8, 1.8 Hz, 1H), 8.48 (dd, J = 7.8, 1.8 Hz, 1H), 8.11 (s, 1H), 7.91 (d, J = 6.6, 2H), 7.52 (t, J = 7.8 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.431 (m, 3H), 7.21 (d, J =5.0 Hz, 1H), 3.67 (t, J = 8.0 Hz, 2H), 3.63 (s, 2H), 3.58 (s, 2H), 2.50 (br, 8H), 2.63 (t, J = 8.0 Hz, 2H), 2.23 (s, 3H). 13C NMR (126 MHz, DMSO) δ 165.78, 162.07, 161.64, 159.96, 151.85, 148.65, 138.26, 137.65, 134.92, 134.25, 132.68, 130.52, 129.17, 128.05, 128.02, 124.28, 117.67, 117.22, 108.00, 61.99, 59.58, 52.95, 41.91, 40.41, 18.11. HRMS (C30H32ClN7O) calcd. 542.2419, found 542.2426 [M+H].
8.2.15 STI-I (11e)
1H NMR (DMSO) δ 10.16 (S, 1H), 9.29 (d, 1H), 8.698 (S, 1H), 8.67 (dd, J = 6.5, 1.5 Hz, 1H), 8.51 (d, J = 8.0 Hz, 1H), 8.48 (d, J = 5 Hz, 1H), 8.07 (s, 1H), 7.89 (d, J = 8.5 Hz, 2H), 7.66 (dd, J = 8.0, 1.5 Hz, 2H), 7.52 (dd, J = 8, 1.5 Hz, 1H), 7.47 (d, J = 5 Hz, 1H), 7.43 (m, 3H), 7.20 (d, J = 8.50 Hz, 1H), 7.10 (d, J = 8.0 Hz, 2H), 3.53 (s, 2H), 2.24 (s, 2H), 2.70 (br, 8H), 2.22 (s, 3H). 13C NMR (126 MHz, DMSO) δ 165.76, 162.06, 161.63, 159.96, 151.85, 148.64, 142.46, 138.60, 138.24, 137.64, 137.38, 134.92, 134.21, 132.68, 131.61, 130.51, 129.12, 128.08, 128.03, 124.28, 117.66, 117.21, 107.99, 93.12, 62.03, 61.73, 53.06, 52.97, 18.11. HRMS (C35H34IN7O) calcd. 696.1942, found 696.1929 [M+H].
8.2.16 STI-Sn (11f)
1H NMR (DMSO) δ 10.16 (S, 1H), 9.29 (d, 1H), 8.97 (S, 1H), 8.67 (dd, J = 6.5, 1.5 Hz, 1H), 8.58 (d, 1H), 8.51 (d, J = 8.0 Hz, 1H), 8.48 (d, J = 5 Hz, 3H), 8.10 (s, 1H), 7.89 (d, J = 8.5 Hz, 2H), 7.45 (m, 6H), 7.22 (d, 2H), 3.53 (s, 2H), 3.42 (s, 2H), 2.70 (br, 8H), 2.22 (s, 3H), 1.48 (m, 6H), 1.27 (m, 6H), 0.86 (t, 6H), 0.81 (t, 9H). HRMS (C47H61N7OSn) calcd. 860.4032, found 860.4058 [M+H].
9. Kinase assay
Anti-phospho-Abltide (rabbit, polyclonal), anti-phosphotyrosine (rabbit, polyclonal), anti-rabbit (HRP conjugate) antibodies, Abl, Abltide (biotin conjugate), and c-KIT, Poly(Glu4-Tyr) (biotin conjugate) were obtained from Upstate Biotechnology, Lake Placid, NY. All chemicals were obtained from Sigma-Aldrich, St. Louis, MO. TMB (tetramethylbenzidine) was obtained from Pierce Biotechnology, Rockford, IL. Ninety-six-well plates (streptavidin coated) were obtained from Nunc, Roskilde, Denmark. A plate reader was obtained from Tecan, Salzburg, Austria.
Phosphorylation levels and anti-enzymatic activities of STI-571 and its derivatives were assayed as follows. Biotinylated peptides (25 μM Abltide or 150 nM Poly(Glu4-Tyr)) in 100 mM sodium bicarbonate buffer (pH 8.0) were incubated in 96-well plates for 2 h at room temperature. After that, nonspecific binding sites were blocked by incubation with 3% BSA for 1 h. Phosphorylation was initiated by adding of 10 ng of Abl or 25 ng of c-KIT in an assay buffer (20 mM TRIS (pH 7.4), 10 mM MgCl2, 1 mM MnCl2, 0.2 mM ATP, 1 mM dithiothreitol, 25 mM glycerol phosphate, 1 mM sodium orthovanadate, 5 mM EGTA). Different concentrations of drugs were added simultaneously, and the mixtures were incubated for 30 min at 37 °C. Anti-phospho-Abltide or antiphosphotyrosine and anti-rabbit antibodies (dilution 1:1000) were incubated, in consecutive order, for 1 h at room temperature. A color reaction was launched by adding TMB, and absorbance was measured 10 min later at 450 nm. The working volume was 100 μl (300 μl for BSA). A wash (5 × 100 μl 20 mM TRIS(pH 7.4) was performed after each step.
10. Western blots
U87 cells were plated at 2.5 × 105/well in 6-well plates in RPMI-1640 medium supplemented with 10% FBS. After the cells were allowed to attach for 24 h, they were treated with 1 μM STI-F or imatinib for 6 h. For Western blotting, 50 μg of total protein was separated by SDS-PAGE (Bio-Rad) and blotted onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). The antibodies were anti-phospho-c-KIT polyclonal antibody (Tyr703 and Tyr721) (Zymed Laboratories Inc., South San Francisco, CA), anti-c-KIT monoclonal antibody E-1 (sc-17806) (Santa Cruz Biotechnology, Dallas, TX); and anti-β-actin monoclonal antibody (Sigma, St. Louis, MO).
K562 cells were plated at 5 × 105/well. Two hours later, the cells were treated for 6 h with 1 μM Sti-F. The antibodies were anti-phospho-c-Abl (Tyr 412) monoclonal antibody 247C7 (Cell Signaling Technology, Inc., Danvers, MA) and anti-Abl monoclonal antibody (Sigma).
11. Small-animal PET studies
A dedicated small-animal PET scanner (microPET Focus 120; Siemens Medical Solutions, Malvern, PA) was used for mouse imaging. K562 human CML cells and U87 human glioblastoma cells were obtained from the ATCC. U87WT cells were produced by retroviral transduction of U87 cells with a wild-type human c-KIT receptor. STI-571 was labeled with F-18 by nucleophilic substitution. Tumor cells were injected into dorsal shoulder areas of nude mice (107 cells/200 μl in Matrigel). PET imaging was performed at 30, 60, and 90 min after the [18F]STI-571 injection (100 μCi/mouse i.v.). Some animals were sacrificed at 60 or 90 min after injection, for tissue sampling and autoradiography.
Acknowledgments
The authors would like to acknowledge the Cancer Center Support grant CA016672 for the support of the Translational Chemistry Core Facility (TCCF) and NMR facility at the University of Texas M. D. Anderson Cancer Center.
References and notes
- 1.Schittenhelm MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, Bokemeyer C, Deininger MWN, Druker BJ, Heinrich MC. Cancer Res. 2006;66:473–481. doi: 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- 2.Crossman LC, O’Brien SG. Hematol Oncol Clin N. 2004;18:605–617. doi: 10.1016/j.hoc.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wilkins R, Janes S. Clin Lab Haematol. 2004;26:73–75. doi: 10.1111/j.0141-9854.2003.00582.x. [DOI] [PubMed] [Google Scholar]; (b) Silva Cd, Reid R. Pathol Oncol Res. 2003;9:13–19. doi: 10.1007/BF03033708. [DOI] [PubMed] [Google Scholar]; (c) Potenza L, Luppi M, Riva G, Morselli M, Ferrari A, Imovilli A, Giacobbi F, Temperani P, Donelli A, Narni F, Torelli G. Am J Hematol. 2006;81:45–50. doi: 10.1002/ajh.20421. [DOI] [PubMed] [Google Scholar]; (d) Finnegan D, Jones F, McMullin M. Hematol Oncol. 2005;23:133–135. doi: 10.1002/hon.759. [DOI] [PubMed] [Google Scholar]; (e) Xavier S, Fagundes E, Hassan R, Bacchi C, Conchon M, Tabak D, Spector N, Zalcberg I. Leuk Res. 2003;27:1063–1066. doi: 10.1016/s0145-2126(03)00070-5. [DOI] [PubMed] [Google Scholar]
- 4.Gayed I, Vu T, Iyer R, Johnson M, Macapinlac H, Swanston N, Podoloff D. J Nucl Med. 2004;45:17–21. [PubMed] [Google Scholar]
- 5.Jager P, Gietema J, Graaf VD. Nucl Med Commun. 2004;25:433–438. doi: 10.1097/00006231-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Wisniewski D, Lambek C, Liu C, Strife A, Veach D, Nagar B, Young M, Schindler T, Bornmann WG, Bertino J, Kuriyan J, Clarkson B. Cancer Res. 2002;62:4244–4255. [PubMed] [Google Scholar]
- 7.Nagar BB, Bornmann WG, Pellicena P. Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- 8.Glekas AP, Pillarsetty NK, Punzalan B, Khan N, Smith-Jones P, Larson SM. J Nucl Med. 2011;52:1301–1307. doi: 10.2967/jnumed.110.085050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sessler JL, Wangt B, Harriman A. J Am Chem Soc. 1995;217:704–714. [Google Scholar]
-
10.The highly unstable aziridinium ion 11d″ was readily underwent ring-opening reaction with the readily available counter ions such as Cl− or OH− before the attack of the external F− ion and hence resulted 11c or 11d as major products.
- 11.(a) Kim DW, Jeong HJ, Lim ST, Sohn M-H. Nucl Med Mol Imaging. 2010;44:25–32. doi: 10.1007/s13139-009-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alauddin MM, Conti PS, Fissekis JD, Labelled J. J Labelled Comp Rad. 2002;45:583–590. [Google Scholar]; (c) Alauddin MM, Conti PS, Fissekis JD. J Labelled Comp Rad. 2003;46:285–289. [Google Scholar]
- 12.Zhang M-R, Kumata K, Maeda J, Haradahira T, Noguchi J, Suhara T, Halldin C, Suzuki K. J Med Chem. 2007;50:848–855. doi: 10.1021/jm061127n. [DOI] [PubMed] [Google Scholar]
- 13.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Sybyl Software, version 7.2. Tripos Inc; St Louis, MO, USA: 2006. [Google Scholar]
- 15.(a) Kramer B, Rarey M, Lengauer T. Proteins. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (b) Rarey M, Wefing S, Lengauer T. J Comput Aided Mol Des. 1996;10:41–54. doi: 10.1007/BF00124464. [DOI] [PubMed] [Google Scholar]
- 16.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maestro, Version 9.3. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 18.LigPrep, Version 2.5. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 19.Epik, Version 2.3. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 20.Schrödinger Suite 2012 Virtual Screening Workflow; Glide version 5.7. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]; LigPrep version 2.5. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]; QikProp version 3.4. Schrödinger, LLC; New York, NY: 2012. Schrödinger, LLC, New York, NY, 2012. [Google Scholar]
- 21.Prime, Version 3.1. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 22.MCPRO+, Version 2.9. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 23.Kim DY, Kim JG, Cho DJ, Lee GY, Kim HY, Woo SH, Kim YS, Bae WC, Lee SA, Han BC. US20040248918A1. 2004. [Google Scholar]
- 24.Lombardino JG, Niantic C. US4623486. 1986. [Google Scholar]
- 25.(a) Anli H, Xing L, Zelikovitch L, Kaspi J. Patent: US2006/149061 A1. 2006; (b) Fernández A, Sanguino A, Peng Z, Ozturk E, Chen J, Crespo A, Wulf S, Shavrin A, Qin C, Ma J, Trent J, Lin Y, Han H-D, Mangala LS, Bankson JA, Gelovani J, Samarel A, Bornmann WG, Sood AK, Lopez-Berestein G. J Clin Invest. 2007;117:4044–4054. doi: 10.1172/JCI32373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; [Google Scholar]