

Abstract
Background
Intraperitoneal accumulation of mucinous ascites in pseudomyxoma peritonei (PMP) promotes an inflammatory/fibrotic reaction that progresses to bowel obstruction and eventual patient demise. Cytokines and inflammation-associated transcription factor binding sites, such as glucocorticoid response elements and COX-2, regulate secretory mucin, specifically MUC2, production. We hypothesized that anti-inflammatory drugs targeting inflammation-associated pathways may reduce mucin production and subsequent disease morbidity in PMP.
Methods
The effects of dexamethasone and Celebrex were assessed in mucin-secreting human colon cancer LS174T cells in vitro and murine xenograft models of LS174T and human appendiceal PMP in vivo by serial parametric measurements, MUC2 transcripts via real-time RT-PCR, and MUC2 protein expression via immunofluorescence assays.
Results
Dexamethasone significantly inhibited basal MUC2 mRNA levels in LS174T cells, inhibited mucinous tumor accumulation in an intraperitoneal PMP xenograft model, and prolonged survival in a subcutaneous LS174T xenograft model. Celebrex significantly inhibited sodium butyrate-stimulated MUC2 mRNA levels in LS174T cells and demonstrated a statistically nonsignificant trend toward reduced mucinous tumor growth and prolonged survival in the xenograft models. MUC2 protein analysis by immunofluorescence demonstrated a dual effect of dexamethasone on mucin production and tumor cell count.
Conclusions
Inflammatory mediators are known to regulate mucin production and may promote overexpression of MUC2 by neoplastic cells with goblet cell phenotype in PMP. Anti-inflammatory drugs, dexamethasone and Celebrex, could inhibit extracellular mucin production in PMP by targeting inflammatory cascades and, therefore, may decrease compressive symptoms, increase the disease-free interval, and reduce the extent or frequency of morbid cytoreductive surgeries.
Pseudomyxoma peritonei (PMP) is a rare malignancy characterized by intraperitoneal accumulation of mucinous ascites and predominantly occurs in the setting of appendiceal neoplasms.1 The tumor biology of mucinous appendiceal neoplasms varies from a low-grade, noninvasive malignancy to a high-grade invasive form of the disease.2–5 The clinical course is determined by the degree of epithelial differentiation and extent of extracellular mucin production. Progressive accumulation mucin compresses intra-abdominal organs, leading to organ dysfunction, morbidity, and eventual patient demise. Cytoreductive surgery with perioperative intraperitoneal chemotherapy is considered the standard of care in managing these tumors.6,7
The epithelial cells responsible for extracellular mucin secretion are the intestinal goblet cells.8 The tightly regulated expression of secreted mucins (MUC2, MUC5AC, MUC5B, MUC6, and MUC19) may be compromised by inflammation and cancer.9–11 In fact, the inflammatory milieu of the intestinal tract is highly conducive to mucin promoter upregulation. Inflammatory cytokines (IL-1β, IL-6, TNF-α, INF-γ), bacterial products (LPS, LTA), and growth factors (retinoids, EGF, TGF-α) have been implicated in stimulating mucin secretion. Inflammation-associated transcription factor binding sites in the MUC2 promoter include those for activator protein (AP-1, AP-2), NF-κB, glucocorticoid response elements (GRE), and cAMP response elements. The only known downstream signaling cascade involved in MUC2 promoter regulation, Src/Ras/MAPK/pp90rsk pathway, also is intimately involved in inflammatory signaling. The inflammatory mediator COX-2 also has been found to be highly overexpressed in PMP.12,13
We hypothesized that targeted reduction of mucin production may minimize disease-associated symptoms, tumor recurrence, and the need for repeated surgical interventions. The purpose of our current study was to investigate the role of anti-inflammatory agents in targeted inhibition of mucin in appendiceal neoplasm with PMP.
MATERIALS AND METHODS
LS174T Human Colon Cancer Cell Line
LS174T is a human colon adenocarcinoma cell line, with “characteristics of goblet cells,” that produces secretory mucin. This cell line was purchased from American Type Culture Collection (Manassas, VA, USA) and cultured according to recommended conditions [Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g/l of glucose, 20% fetal bovine serum, 2 mM of L-glutamine, 20 mM of HEPES, 100 IU/ml of penicillin, and 100 μg/ml of streptomycin] at 37°C and 5% CO2.
Human Patient PMP Samples
Primary and metastatic tumor samples were obtained from preoperatively consented patients who were diagnosed with mucinous appendiceal neoplasms with PMP. Specimens were flash-frozen within 30 minutes of removal from the patients and stored in our tissue bank at −80°C. Experimental procedures were approved by the University of Pittsburgh Institutional Review Board (UPCI IRB# 02-077).
Murine Xenograft Model of LS174T Human Colon Cancer Cells
Murine studies were approved by the University of Pittsburgh Animal Care and Use Committee (IACUC Protocol #0901696B-3). LS174T cells (106) were injected subcutaneously in the right flank of mice (athymic nude mice; Taconic; Tarrytown, NY USA) to develop the LS174T murine xenograft model. Tumor cells were allowed to grow for 1 week until a subcutaneous nodule was seen. Animals were then randomized to different treatment groups to ensure comparable tumor volumes at the start of the experiment.
Murine Xenograft Model of Human Appendiceal PMP
The development of the murine xenograft model of human appendiceal PMP has been published.14 Fresh tumor samples were immersed in phosphate-buffered saline (PBS) containing 100 U/ml of penicillin and 100 μg/ml of streptomycin for 3 hr, and then implanted in the peritoneal cavity of nude mice. Tumor sample PMP-754 continued to be successfully passaged to subsequent generations in nude mice with 100% reliability and retained the clinical and pathologic characteristics of the original human tumor.
Small Molecule Drugs to Inhibit Mucin Production In Vitro
Preconfluent LS174T cells, grown in 6-well cell-culture plates (Costar, Cambridge, MA, USA) were treated with supplemented medium (DMEM) containing dexamethasone sodium phosphate (Dex) (APP Pharmaceuticals, Schaumburg, IL, USA) (10−6, 10−7, 10−8 M) or Celebrex (Pfizer, KS) (10−6, 10−5, 5 × 10−5 M) for periods of 24–72 h. Fresh DMEM containing the drug of interest was replaced every 24 h. We performed in vitro experiments at both basal and MUC2-stimulated conditions to simulate the inflammatory milieu of the intestinal tract. Low-dose sodium butyrate (NaB; 2.5 mM; Cayman Chemical Company, Ann Arbor, MI, USA) was used to stimulate MUC2 production.15 Viability of cells (>95%) was confirmed by using trypan blue stain. Harvested cells were snap-frozen for subsequent assays.
Small Molecule Drugs to Inhibit Mucin Production In Vivo
Treatment by Dex (2 mg/kg/mouse, IP, daily) or Celebrex (20 mg/kg/mouse, oral, daily) was commenced 7 days after subcutaneous tumor inoculation (LS174T xenograft model) or 4 days after intraperitoneal tumor inoculation (human appendiceal PMP murine xenograft model). Control mice received daily sham IP injection or oral gavage with PBS respectively. In the LS174T xenograft model, weekly measurements of gross body weight and subcutaneous tumor dimensions (width and height) were recorded. Tumor volumes were calculated from these dimensions (volume = [length × (width)2]/2). Treatments were continued until death (natural or once maximum tumor diameter was ≥2 cm per IACUC guidelines). In the PMP xenograft model, weekly measurements of gross body weight and abdominal girth were recorded. At 5–6 weeks after tumor inoculation, once significant tumor burden had developed, control and treated mice were euthanized. Abdominal contents (abdominal organs + mucinous tumor deposits) were harvested en bloc and weighed. Intra-abdominal mucinous tumor deposits were snap-frozen for further analysis.
Quantification of MUC2 mRNAs by Real-Time Reverse Transcription-PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA) and quantified using a spectrophotometer. Reverse transcription (RT) was performed in a Peltier Thermal Cycler (MJ Research, Waltham, MA, USA) using random hexamers. Real-time PCR was then performed in an ABI Cycler System (ABI Prism SDS 7000; Applied Biosystems, Foster City, CA, USA), using commercially available primers and probes, specific for MUC2, MUC5AC, MUC5B, MUC6, and MUC19 cDNAs (ABI). Relative amounts of mRNAs for gel-forming mucins were determined after normalization of individual mucin transcripts to that of β-actin (ABI), using software supplied by the manufacturer (ABI).
Quantification of MUC2 Protein by Immunofluorescence
Mucinous tumor specimens from xenografts were placed in Tissue Path Disposable Base Molds (Fisher Scientific, Pittsburgh, PA) and snap-frozen in Tissue-Tek O.C.T. compound (Fisher Scientific). Using a cryostat microtome, 6-micron frozen sections of tumor tissue were mounted on Superfrost Plus microscope slides (Fisher Scientific) and maintained at −20°C. Cryosections were permeabilized with 0.1% Triton-X. Slides were blocked for 45 minutes with 2% BSA (Sigma Aldrich, St. Louis, MO, USA), incubated for 60 min with MUC2 rabbit polyclonal primary antibody (MUC2 H-300; Santa Cruz Biotechnology, Santa Cruz, CA), and diluted 1:100 in 0.5% BSA. The slides were then washed and incubated with CY-3-conjugated goat-anti-rabbit secondary antibody (Santa Cruz Biotech) and diluted 1:10,000 in 0.5% BSA for 60 min. Dapi nuclear stain was applied to slides for 30 seconds. Slides were cover-slipped with gel-vatol (Fisher Scientific) and stored at 4°C. Nikon 90i Motorized Upright Microscope at ×20 magnification was used to acquire multiple sequential images of preset pixels for each slide using cy-3 channel (mucin) and DAPI (nuclei). NIS-Elements 3.2 64-bit program was used to stitch together the small sequential images to generate a single large picture image of each slide. Images of each slide were then analyzed using MetaMorph to determine the number of nuclei (DAPI:blue) and the amount of mucin (Cy-3:red).
Statistical Analysis
Experimental means were reported ± standard error of the mean (SEM). Data were analyzed with paired Student’s t test. Survival times were estimated by using the Kaplan–Meier method and compared by using the log-rank test. Values were considered significantly different if P < 0.05.
RESULTS
Relative Contribution of Secreted Mucins in Experimental Models of PMP
Using real-time RT-PCR, we found in LS174T cells that MUC2 mRNA accounted for approximately 98% of the overall message encoding for gel-forming mucins, with a relatively small contribution from other known gel-forming mucins (MUC5AC, MUC6, and MUC19). Similar results were seen in the murine xenograft model. We therefore focused our subsequent studies on targeted inhibition of MUC2.
In Vitro and In Vivo Inhibition of MUC2 Production by Dex
The basal (unstimulated) MUC2 mRNA expression in LS174T cells was inhibited by various concentrations of Dex, with persistent inhibition up to 72 h (DMEM: 0.04 ± 0.002 vs. Dex 10−6 M: 0.03 ± 0.001; 48 h; P = 0.0004; DMEM: 0.05 ± 0.008 vs. Dex 10−6 M: 0.03 ± 0.001; 72 h; P = 0.04; Fig. 1a). There was a statistically nonsignificant trend toward reduced NaB-stimulated MUC2 mRNA production by Dex at various dose levels, most effectively at 48 h (NaB 2.5 mM: 0.054 ± 0.003 vs. Dex 10−6 M + NaB 2.5 mM: 0.045 ± 0.004; 48 h; P = 0.07; Fig. 1b).
FIG. 1.
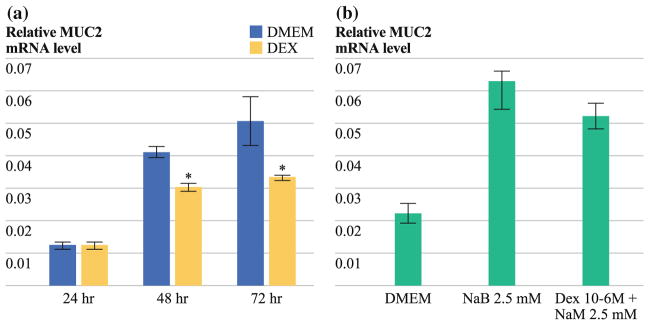
a Dose- and time-dependent reduction in basal MUC2 mRNA expression in LS174T cells as demonstrated by real-time RT-PCR (DMEM: 0.04 ± 0.002 vs. Dex 10−6 M, 48 h: 0.03 ± 0.001; P = 0.0004; DMEM: 0.05 ± 0.008 vs. Dex 10−6 M: 0.03 ± 0.001; 72 h; P = 0.04); b Trend toward reduction in sodium butyrate-stimulated MUC2 mRNA expression by Dex in LS174T cells by real-time RT-PCR (NaB 2.5 mM: 0.054 ± 0.003 vs. Dex 10−6 M + NaB 2.5 mM: 0.045 ± 0.004; 48 h; P = 0.07). Preconfluent cell layers were treated with Dex at various doses for periods of up to 72 h. For stimulated mucin expression experiments, LS174T cells were treated with NaB (2.5 mM) alone or in combination with Dex. Real-time PCR was then performed by using commercially available primers and probe specific for MUC2 cDNA. Relative MUC2 mRNA levels were normalized to that of β-actin. Maximal MUC2 reduction was demonstrated with continuous exposure to 10−6 M Dex for up to 72 h. Experiments were performed in triplicate (n = 3). Error bars depict SEM; means were compared by using paired Student’s t test, * P < 0.05
In the subcutaneous LS174T murine xenograft model, we demonstrated a significant reduction in tumor volume at days 14, 21, and 28 in animals treated with Dex (at day 28: PBS-IP: 2,998 ± 496 mm3 vs. Dex-IP: 1,533 ± 403 mm3; P = 0.03; Fig. 2a), which translated into a significant improvement in median overall survival (PBS-IP: 25 days vs. Dex-IP: 43 days; P = 0.0172; Fig. 2b).
FIG. 2.
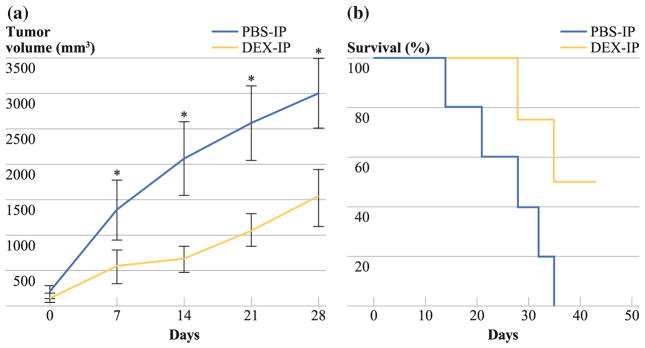
Treatment of a subcutaneous LS174T murine xenograft model with Dex. a Significant reduction in tumor volume at days 14, 21, and 28 in Dex-treated mice compared with PBS-treated mice (day 28: PBS-IP: 2,998 ± 496 mm3 vs. Dex-IP: 1,533 ± 403 mm3; P = 0.03). b Survival analysis of mice treated with Dex compared with PBS-treated controls. Data refer to five PBS-treated and eight Dex-treated mice from day 1 of therapy until death. Dex-treated mice had a significantly longer median survival (PBS-IP: 28 days vs. Dex-IP >40 days; log-rank test, P = 0.0172)
In the intraperitoneal PMP xenograft model, we demonstrated significant reduction in mucinous tumor mass (Figs. 3a–d) at days 13 and 36 after chronic treatment with Dex, as determined by serial measurements of xenograft weight (day 36: PBS-IP: 29.2 ± 1.09 g vs. Dex-IP: 21.7 ± 0.34 g; P = 0.002; Fig. 3e), abdominal girth (day 36: PBS-IP: 30.1 ± 1.2 cm vs. Dex-IP: 23.4 ± 0.63 cm; P = 0.003; Fig. 3f), and the weight of en bloc abdominal contents (day 36: PBS-IP: 13.02 ± 0.96 g vs. Dex-IP: 6.72 ± 0.17 g; P = 0.002; Fig. 3g).
FIG. 3.

Intraperitoneal PMP xenograft model showing decreased abdominal distention and mucinous tumor at days 13 and 36 after Dex treatment. a PBS-IP, before laparotomy; b PBS-IP, after laparotomy; c Dex-IP, before laparotomy; and d Dex-IP, after laparotomy; all mice were euthanized on day 36. Chronic treatment with Dex (2 mg/kg/day, IP) led to a serial reduction in (e) gross body weight (day 36: PBS-IP: 29.2 ± 1.09 g vs. Dex-IP: 21.7 ± 0.34 g; P = 0.002), f abdominal girth (day 36: PBS-IP: 30.1 ± 1.2 cm vs. Dex-IP: 23.4 ± 0.63 cm; P = 0.003), and g weight of en bloc abdominal contents (day 36: PBS-IP: 13.02 ± 0.96 g vs. Dex-IP: 6.72 ± 0.17 g; P = 0.002). Error bars denote SEM (n = 5); means were compared using paired Student’s t test; *P < 0.05
Immunofluorescence analysis of samples from PBS- and Dex-treated animals at day 36 demonstrated a nonsignificant gross reduction in MUC2 protein (PBS-IP: 9,677,261 ± 2,397,075 μm2 vs. Dex-IP: 5,752,493 ± 1,924,477 μm2; P > 0.05; Fig. 4a, b) and a significant reduction in tumor cell count (PBS-IP: 20,248 ± 5,196 cells vs. Dex-IP: 4,264 ± 827 cells; P = 0.04) in Dex-treated animals compared with controls (Fig. 4c). This suggests a direct inhibitory effect of Dex on tumor cells, in addition to reduction in MUC2 accumulation.
FIG. 4.
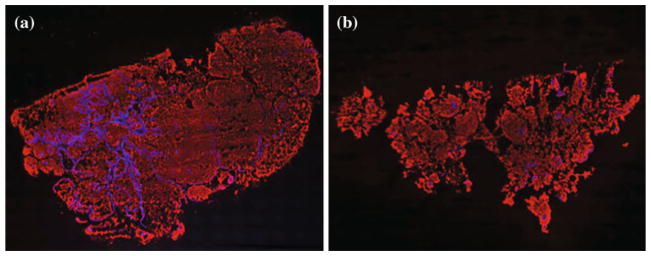
Immunofluorescence imaging of representative tumor cryosections from a PBS-treated and b dexamethasone-treated PMP xenograft mice at day 36. CY-3 (red) stain represents mucin and DAPI (blue) stain represents cell nuclei within the tumor mass. c Nonsignificant overall gross reduction in mucin staining in Dex-treated mice (PBS-IP: 9,677,261 ± 2,397,075 μm2 vs. Dex-IP 5,752,493 ± 1,924,477 μm2; P = 0.2). Simultaneous reduction in tumor cells (PBS-IP 20,248 ± 5,196 cells vs. Dex-IP 4,264 ± 827 cells; P = 0.04)
In Vitro and In Vivo Inhibition of MUC2 Production by Celebrex
In LS174T cells, basal MUC2 mRNA expression was not affected by Celebrex treatment (Fig. 5a). However, we demonstrated a significant reduction in NaB-stimulated MUC2 mRNA production by Celebrex, most effectively at 48 h (NaB: 0.11 ± 0.004 vs. Celebrex 10−5 M + NaB 2.5 mM: 0.06 ± 0.01; P < 0.0001; Fig. 5b).
FIG. 5.
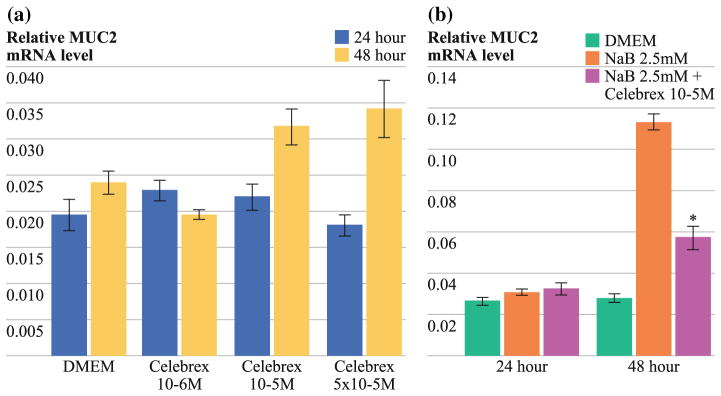
a No dose- or time-dependent effect of Celebrex on basal MUC2 mRNA synthesis in LS174T cell line as demonstrated by real-time RT-PCR. b Significant reduction in sodium butyrate-stimulated MUC2 mRNA synthesis by Celebrex at 48 h (NaB 2.5 mM: 0.11 ± 0.004 vs. Celebrex 10−5 M + NaB 2.5 mM: 0.06 ± 0.01; P < 0.0001). Preconfluent cell layers were treated with Celebrex at various doses for periods of up to 72 h. For stimulated mucin experiments, LS174T cells were treated with NaB (2.5 mM) alone or in combination with anti-inflammatory drugs. Real-time PCR was then performed using commercially available primers and probe specific for MUC2 cDNA. Relative amounts of MUC2 were normalized to β-actin. Experiments were performed in triplicate (n = 3); error bars depict SEM; means were compared using paired Student’s t test; *P < 0.05
In vivo studies using the subcutaneous LS174T murine xenograft model demonstrated a statistically nonsignificant trend toward reduced tumor growth in animals treated with Celebrex (day 14: PBS-PO: 2,550 ± 1,129 mm3 vs. Celebrex-PO: 1,264 ± 532 mm3; P > 0.05; Fig. 6a) and a corresponding trend toward improved median overall survival (PBS-PO: 18 days vs. Celebrex-PO: 34 days; P > 0.05; Fig. 6b).
FIG. 6.

Treatment of subcutaneous LS174T murine xenograft model with Celebrex. a Statistically nonsignificant trend toward reduced subcutaneous tumor volume over time in Celebrex-treated mice compared with PBS-treated mice (day 14: PBS-PO: 2,550 ± 1,129 mm3 vs. Celebrex-PO: 1,264 ± 532 mm3; P > 0.05). b The survival curve reports the number of mice still alive on different days. Data refer to five PBS-treated and eight Celebrex-treated mice from day 1 of therapy until death. There was a trend toward longer median survival in Celebrex-treated mice (PBS-PO: 18 days vs. Celebrex-PO: 34 days; log-rank test, P > 0.05). Intraperitoneal PMP xenograft model treated with Celebrex (20 mg/kg/day, PO) demonstrated a statistically nonsignificant serial reduction in (c) gross body weight (PBS-PO: 30.5 ± 2.5 g vs. Celebrex-PO: 26.6 ± 1.7 g; P >0.05), d abdominal girth (PBS-PO: 32.2 ± 2.1 cm vs. Celebrex-PO: 28.1 ± 1.2 cm; P > 0.05), and (e) weight of en bloc abdominal contents (organ block + mucinous tumor; PBS-PO: 17.4 ± 1.9 g vs. Celebrex-PO: 13.1 ± 1.6 g; P > 0.05) at the time they were euthanized. Error bars denote SEM (n = 5); means were compared using paired Student’s t test
In vivo studies using the intraperitoneal PMP xenograft model demonstrated a statistically nonsignificant trend toward reduced mucinous tumor mass at days 37 and 49 after chronic treatment with oral Celebrex therapy as measured by serial measurements of xenograft weight (day 49: PBS-PO: 30.5 ± 2.5 g vs. Celebrex-PO: 26.6 ± 1.7 g; P > 0.05; Fig. 6c), abdominal girth (day 49: PBS-PO: 32.2 ± 2.1 cm vs. Celebrex-PO: 28.1 ± 1.2 cm; P > 0.05; Fig. 6d), and weight of en bloc abdominal contents (day 49: PBS-PO: 17.4 ± 1.9 g vs. Celebrex-PO: 13.1 ± 1.6 g; P > 0.05; Fig. 6e).
DISCUSSION
Secretory mucins, specifically MUC2, play a significant role in the clinical course of PMP. Patient demise predominantly occurs as a result of progressive mucin accumulation, with its associated inflammatory and fibrotic reaction, leading to intestinal obstruction.1 Intestinal epithelial goblet cells are the predominant source of MUC2 and the inflammatory milieu of the intestinal tract is highly conducive to MUC2 promoter upregulation.12,13 We investigated the role of anti-inflammatory agents as a novel approach to disease control in PMP.
Our experimental models included a mucin-secreting human colon cancer cell line (LS174T) and murine xenograft models of LS174T (subcutaneous) and human PMP tissue (intraperitoneal). NaB was used to stimulate MUC2 production in LS174T cells, to simulate the inflammatory milieu of the intestinal tract. In LS174T cells, Dex treatment significantly inhibited basal MUC2 mRNA, whereas Celebrex had a significant MUC2-inhibitory effect under NaB-stimulated conditions. Whether MUC2 inhibition is a function of altered transcription at the promoter level or RNA stability is currently unclear. In the LS174T xenograft model, chronic Dex therapy significantly prolonged survival. This corresponded to a statistically nonsignificant trend toward less tumor growth by parametric measurements. In addition, tumor growth was significantly reduced by dexamethasone in the PMP xenograft model. Given the gross reduction in overall tumor volume, by parametric measurements, and inhibition of MUC2 mRNA levels in vitro, we had expected to see a corresponding reduction in MUC2 protein staining with an increase in relative numbers of tumor cells. Paradoxically, immunofluorescence analysis revealed a reduction in tumor cell count, in combination with gross overall reduction in MUC2 protein volume after chronic Dex therapy. This suggests a dual inhibitory effect of Dex on mucin production and cell proliferation.
Cytokines have been implicated in goblet cell hyperplasia/metaplasia and mucin hypersecretion in inflammatory diseases of the lung and gastrointestinal tract.16–18 Glucocorticoids may directly inhibit MUC2 production via GRE in the MUC2 promoter region or indirectly via transrepression of inflammation-associated transcription factors, including NF-κB or AP-1.19,20 Our research suggests that the anti-inflammatory effects of Dex may be applicable to the inflammatory environment of the gastrointestinal tract for MUC2 regulation in mucinous malignancies. COX-2 is involved in inflammation, is overexpressed in a number of tumors, and correlates with poor clinical outcome. In addition, COX-2 protein levels increase during inflammation, as a result of transcript stabilization by inflammatory cytokines.21 Gray and colleagues demonstrated elevated mucin production in normal human tracheobronchial epithelium in response to IL-1β stimulation, as a result of COX-2 overexpression.22 Our experiments demonstrated a reduction in NaB-stimulated MUC2 production but no inhibition of basal MUC2 synthesis after chronic Celebrex therapy. This suggests that Celebrex may have a negative regulatory role only under inflammatory states, thereby countering the inflammation-induced COX-2 upregulation.
Normally, secreted mucins form a protective layer covering epithelial-lined surfaces. However, mucinous appendiceal neoplasms with PMP are characterized by the excessive intraperitoneal accumulation of extracellular secreted gel-forming mucins. This paradoxical increase in mucin, in the setting of malignancy, may have a number of theoretical explanations. Mucinous appendiceal tumors may represent a goblet cell malignancy, with early goblet cell lineage switch during carcinogenesis, leading to goblet cell hyperplasia and, correspondingly, increased MUC2 accumulation. On the other hand, overexpression of MUC2 by existing goblet cells due to the inflammatory milieu of the intestinal tract or epigenetic activation of goblet cells during malignant transformation may play a role in the mucinous phenotype of this malignancy. Inflammatory mediators have been shown to play a significant role in the transcriptional regulation of mucin production. Our data suggest a potential role for anti-inflammatory drugs in targeted reduction of mucin production in PMP. Further studies are needed to determine the exact mechanism by which these drugs exert their effects and their potential for therapeutic intervention in this disease.
Acknowledgments
Supported by a research grant from the National Organization for Rare Diseases (NORD) (1/2011–1/2012), Division of Surgical Oncology, University of Pittsburgh (Proposal #22812); Pseudomyxoma peritonei Philanthropic Research Fund, Division of Surgical Oncology, University of Pittsburgh, and the Koch fund, University of Pittsburgh.
Footnotes
CONFLICT OF INTERESTS None.
Contributor Information
Haroon Asif Choudry, Email: choudrymh@upmc.edu.
David L. Bartlett, Email: bartlettdl@upmc.edu.
References
- 1.Sugarbaker PH. New standard of care for appendiceal epithelial neoplasms and pseudomyxoma peritonei syndrome? Lancet Oncol. 2006;7(1):69–76. doi: 10.1016/S1470-2045(05)70539-8. [DOI] [PubMed] [Google Scholar]
- 2.Misdraji J. Appendiceal mucinous neoplasms: controversial issues. Arch Pathol Lab Med. 2010;134(6):864–70. doi: 10.5858/134.6.864. [DOI] [PubMed] [Google Scholar]
- 3.Pai RK, Longacre TA. Appendiceal mucinous tumors and pseudomyxoma peritonei: histologic features, diagnostic problems, and proposed classification. Adv Anat Pathol. 2005;12(6):291–311. doi: 10.1097/01.pap.0000194625.05137.51. [DOI] [PubMed] [Google Scholar]
- 4.Ronnett BM, Zahn CM, Kurman RJ, Kass ME, Sugarbaker PH, Shmookler BM. Disseminated peritoneal adenomucinosis and peritoneal mucinous carcinomatosis. A clinicopathologic analysis of 109 cases with emphasis on distinguishing pathologic features, site of origin, prognosis, and relationship to “pseudomyxoma peritonei”. Am J Surg Pathol. 1995;19(12):1390–408. doi: 10.1097/00000478-199512000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Bradley RF, Stewart JH, Russell GB, Levine EA, Geisinger KR. Pseudomyxoma peritonei of appendiceal origin: a clinicopathologic analysis of 101 patients uniformly treated at a single institution, with literature review. Am J Surg Pathol. 2006;30(5):551–9. doi: 10.1097/01.pas.0000202039.74837.7d. [DOI] [PubMed] [Google Scholar]
- 6.Yan TD, Black D, Savady R, Sugarbaker PH. A systematic review on the efficacy of cytoreductive surgery and perioperative intraperitoneal chemotherapy for pseudomyxoma peritonei. Ann Surg Oncol. 2007;14(2):484–92. doi: 10.1245/s10434-006-9182-x. [DOI] [PubMed] [Google Scholar]
- 7.Glehen O, Mohamed F, Gilly FN. Peritoneal carcinomatosis from digestive tract cancer: new management by cytoreductive surgery and intraperitoneal chemohyperthermia. Lancet Oncol. 2004;5(4):219–28. doi: 10.1016/S1470-2045(04)01425-1. [DOI] [PubMed] [Google Scholar]
- 8.O’Connell JT, Tomlinson JS, Roberts AA, McGonigle KF, Barsky SH. Pseudomyxoma peritonei is a disease of MUC2-expressing goblet cells. Am J Pathol. 2002;161(2):551–64. doi: 10.1016/S0002-9440(10)64211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9(12):874–85. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4(1):45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- 11.Thai P, Loukoianov A, Wachi S, Wu R. Regulation of airway mucin gene expression. Annu Rev Physiol. 2008;70:405–29. doi: 10.1146/annurev.physiol.70.113006.100441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Seuningen I, Pigny P, Perrais M, Porchet N, Aubert JP. Transcriptional regulation of the 11p15 mucin genes. Towards new biological tools in human therapy, in inflammatory diseases and cancer? Front Biosci. 2001;6:D1216–34. doi: 10.2741/seuning. [DOI] [PubMed] [Google Scholar]
- 13.Vincent A, Perrais M, Desseyn JL, Aubert JP, Pigny P, Van Seuningen I. Epigenetic regulation (DNA methylation, histone modifications) of the 11p15 mucin genes (MUC2, MUC5AC, MUC5B, MUC6) in epithelial cancer cells. Oncogene. 2007;26(45):6566–76. doi: 10.1038/sj.onc.1210479. [DOI] [PubMed] [Google Scholar]
- 14.Mavanur AA, Parimi V, O’Malley M, Nikiforova M, Bartlett DL, Davison JM. Establishment and characterization of a murine xenograft model of appendiceal mucinous adenocarcinoma. Int J Exp Pathol. 2010;91(4):357–67. doi: 10.1111/j.1365-2613.2010.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatayama H, Iwashita J, Kuwajima A, Abe T. The short chain fatty acid, butyrate, stimulates MUC2 mucin production in the human colon cancer cell line, LS174T. Biochem Biophys Res Commun. 2007;356(3):599–603. doi: 10.1016/j.bbrc.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 16.Kim YD, Kwon EJ, Kwon TK, Baek SH, Song SY, Suh JS. Regulation of IL-1beta-mediated MUC2 gene in NCI-H292 human airway epithelial cells. Biochem Biophys Res Commun. 2000;274(1):112–6. doi: 10.1006/bbrc.2000.3107. [DOI] [PubMed] [Google Scholar]
- 17.Enss ML, Cornberg M, Wagner S, Gebert A, Henrich M, Eisenblätter R, et al. Proinflammatory cytokines trigger MUC gene expression and mucin release in the intestinal cancer cell line LS180. Inflamm Res. 2000;49(4):162–9. doi: 10.1007/s000110050576. [DOI] [PubMed] [Google Scholar]
- 18.Iwashita J, Sato Y, Sugaya H, Takahashi N, Sasaki H, Abe T. mRNA of MUC2 is stimulated by IL-4, IL-13 or TNF-alpha through a mitogen-activated protein kinase pathway in human colon cancer cells. Immunol Cell Biol. 2003;81(4):275–82. doi: 10.1046/j.1440-1711.2003.t01-1-01163.x. [DOI] [PubMed] [Google Scholar]
- 19.Schoneveld OJ, Gaemers IC, Lamers WH. Mechanisms of glucocorticoid signalling. Biochim Biophys Acta. 2004;1680(2):114–28. doi: 10.1016/j.bbaexp.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Kai H, Yoshitake K, Hisatsune A, Kido T, Isohama Y, Takahama K, et al. Dexamethasone suppresses mucus production and MUC-2 and MUC-5AC gene expression by NCI-H292 cells. Am J Physiol. 1996;271(3 Pt 1):L484–8. doi: 10.1152/ajplung.1996.271.3.L484. [DOI] [PubMed] [Google Scholar]
- 21.Brown JR, DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23(12):2840–55. doi: 10.1200/JCO.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 22.Gray T, Nettesheim P, Loftin C, Koo JS, Bonner J, Peddada S, et al. Interleukin-1beta-induced mucin production in human airway epithelium is mediated by cyclooxygenase-2, prostaglandin E2 receptors, and cyclic AMP-protein kinase A signaling. Mol Pharmacol. 2004;66(2):337–46. doi: 10.1124/mol.66.2.337. [DOI] [PubMed] [Google Scholar]