

Abstract
The attachment of alkyl and other hydrophobic groups to traditional antibacterial kanamycins and neomycins creates amphiphilic aminoglycosides with altered antimicrobial properties. In this review, we summarize the discovery of amphiphilic kanamycins that are antifungal, but not antibacterial, and that inhibit the growth of fungi by perturbation of plasma membrane functions. With low toxicities against plant and mammalian cells, they appear to specifically target the fungal plasma membrane. These new antifungal agents offer new options for fighting fungal pathogens and are examples of reviving old drugs to confront new therapeutic challenges.
Introduction
Certain natural product aminoglycosides produced by Actinomycetes are among the oldest and most successful antibacterial medications. However, the vast majority of fungi are not affected by these aminoglycosides, and no major class of antifungal aminoglycosides of importance exists. Instead the most widely used antifungals are sterol-binding polyene compounds (such as amphotericin and nystatin) and sterol biosynthesis disruptants (such as imidazoles and triazoles).1,2 The use of the former group is limited due to toxicity issues and fungal resistance to the latter group has become a major public health problem.3 Thus, as with antibacterials, there is an increasing shortage of effective therapeutic antifungal agents. Newer classes of therapeutic antifungals that bypass resistance mechanisms and that possess novel mechanisms of action are needed. Within the last few years, several publications have appeared reporting semi-synthetic modifications of aminoglycosides into cationic amphiphiles by attaching one or more alkyl or aryl groups to alcohol or amine moieties of the parent compounds.4-13 Many of these novel amphiphilic aminoglycoside analogues display improved inhibitory activities against Gram-positive (G+) and Gram-negative (G-) bacteria and, perhaps more importantly, against bacterial strains resistant to the parent aminoglycosides. In addition, antifungal amphiphilic kanamycins with alkyl chains and that concomitantly lack antibacterial activities have been reported.14,15 Several lines of evidence indicate membrane perturbation as the principal mechanism of action for both antibacterial and antifungal amphiphilic aminoglycosides leading to the suggestion that mechanistically they represent a novel group of aminoglycoside antimicrobials.10,11,13-15 This review begins by summarizing findings that reveal the uniqueness and significance of amphiphilic aminoglycosides among antimicrobial agents. It then describes more specific details about kanamycin-based antifungal amphiphilic analogues particularly regarding synthetic strategies, structure-activity analyses, and mechanisms of action.
1 Traditional aminoglycosides: antibacterials and the resistance problem
Traditional aminoglycosides are polycationic di-tri- or tetra-saccharides rich in amino and hydroxyl moieties that impart capabilities for killing a broad spectrum of G+ and G- bacteria. These include streptomycin, neomycin, gentamicin, tobramycin, kanamycin, and kasugamycin.16,17 In addition, newer semi-synthetic versions such as amikacin, dibekacin, and arbekacin are widely used.17 They are imported into growing bacterial cells via membrane-associated ATP-driven transport systems.17 Subsequent binding to the aminoacyl-tRNA decoding A sites of ribosomal 16S rRNAs decreases protein translational fidelity leading to accumulation of defective proteins and eventual cell death.17
Though successful as antibacterials, the long-term and excessive use of traditional aminoglycosides in medicine and agriculture has bred resistance -- rendering some widely used ones ineffective as medically useful antibiotics.3,18, Three major aminoglycoside resistance mechanisms in bacteria are recognized: 1) alteration of the 16S ribosomal RNA A site leading to lower aminoglycoside binding affinities, 2) reduction in the aminoglycoside intracellular concentration by efflux transport systems across the cytoplasmic membrane or by decreasing membrane permeability, and 3) inactivation by enzymatic covalent modification with nucleotidyl, phosphoryl, and acetyl groups.17 With increasing frequency, aminoglycoside-resistant bacterial strains are observed that possess combinations of these resistance mechanisms.11 Inevitably, new combinations and new resistance mechanisms will evolve as new aminoglycosides are developed. Strategies in aminoglycoside synthetic efforts will therefore need to focus on bypassing new resistance mechanisms with systems that are sufficiently flexible to keep up with biological evolution.
Extensive effort has been devoted to structural modifications of aminoglycosides with the goal of reviving their inhibitory activities against drug-resistant bacteria. Synthetic designs based on neomycin have dominated these efforts.4,7,9,12,13,17 Examples of successful modifications include attachment of (S)-4-amino-2-hydroxybutyryl (AHB) group at the N-1 position (neokacin20, butirosin), 3’,4’-dideoxygenation (pyranmycin21), glycosylation at the O-5 position (pyranmycin22,23), glycosylation at the O-5″ position24, and conformationally constrained neomycin derivatives.25-27 Many of these newer synthetic aminoglycosides, albeit showing prominent antibacterial activities in vitro, are not suitable for scale-up production.
2 Amphiphilic aminoglycosides
2.1. Antibacterial
Studies by Hanessian et al.4 revealed that paramomycin derivatives with lipophilic substituents attached at the 2″ position of ring III had unusually strong inhibitory activities against Staphylococcus aureus despite showing altered A-site rRNA binding orientation. Zhang et al. 5,6 observed that addition of stearic and palmitic alkyl chains to the O-5″position of neomycin B imparted ~32-fold greater activity against both methicillin-resistant S. aureus (MRSA) and vancomycin-resistant enterococci (VRE). These initial observations were expanded in subsequent reports revealing enhanced antibacterial activities with attachment of alkyl and other hydrophobic moieties to neamine,8,10,13 paromomycin,7,9,13 and tobramycin.11,12 These new discoveries with antibacterial amphiphilic aminoglycosides immediately raise questions about their mechanisms of action, and (not surprisingly) interactions with cell surface membranes are implicated. Several observations indicate membrane perturbation as the primary antibacterial action of these compounds and not ribosome interaction to cause protein translation misreading.4,10,11,23,28
2.2 Antifungal
Generally, fungi are not susceptible to the traditional aminoglycosides except at high concentrations.29 However, several oomycetous Phytophora and Pythium fungal species are inhibited by paramomycin (in vitro concentrations between 1 and 10 μg mL-1) and to a lesser extent by neomycin, streptomycin, and ribostamycin.29 The mechanisms by which these oomycete species are inhibited by paramomycin and the other aminoglycosides are not known although it may be speculated that mitochondrial ribosomes are targeted to compromise the fidelity of protein translation.30 Chang and colleagues31-33 developed protocols to stereoselectively attach a variety of α-glycosyl donors to neamine to create libraries of kanamycin B analogues altered in the kanosamine moiety. Inspired by the prior work,29 random structure-bioactivity screens of these libraries uncovered a few analogues that inhibited the growth of fungi and yeasts.14 One analogue, FG08, with a C8 alkyl chain at the O-4″ position of ring III displayed strong fungicidal activity but little or no growth inhibition of G+ or G- bacteria.14 As with antibacterial alkyl chain-modified aminoglycosides, FG08 elicits membrane perturbation effects consistent with an antifungal mechanism of action involving plasma membrane permeability changes.15
3 Broader perspectives of antifungal amphiphilic aminoglycosides
The recent discoveries of amphiphilic aminoglycosides with mechanisms of action that differ from those of traditional aminoglycosides could represent a significant advance towards the development of more effective aminoglycosides. In addition, the discovery of alkyl chain-modified kanamycins (e.g. FG08) with antifungal but no antibacterial activities broadens the scope of amphiphilic aminoglycosides as potential therapeutics. It appears that adjusting the length of the alkyl chain, the chemical nature of the hydrophobic moiety, or the attachment site on aminoglycoside core structures can generate novel antimicrobials with taxon and perhaps species specificities. In the following sections of this review, we describe in more detail the synthetic strategies, structure-activity analyses, and mechanism of action studies with kanamycin and neomycin-based aminoglycoside analogues that lead us to suggest these views.
(In our descriptions below, aminoglycoside structures are shown as neutral forms. Aminoglycosides are often prepared or sold as ammonium salts with various anions, such as chloride, sulfate, acetate or trifluoroacetate, which have no apparent effect on the biological activities. Inter-conversion of these anions can be conveniently achieved using ion-exchange resin).
4 Kanamycin-based amphiphilic aminoglycosides and antibacterial and antifungal activities
The kanamycins belong to a group of 4,6-disubstituted 2-deoxystreptamine aminoglycosides which are comprised of several structurally different compounds (Fig.1). There are two common strategies for synthetic production of kanamycin-class aminoglycosides: 1) direct modifications using kanamycin, tobramycin and gentamicin as the starting materials;34,35 and 2) diversification of ring III using glycosylation. 31,33,36 Only amikacin, tobramycin and gentamicin have been used clinically and scale-up production issues have hindered the development of many reported synthetic derivatives of the kanamycins. Earlier attempts at the synthesis of kanamycin derivatives were direct modification approaches that employed kanamycin B as the starting material.37 Following the protection of Boc groups, the 6″-OH, which is the only primary hydroxyl group, were selectively modified using nucleophiic substitution. Derivatives with various functional groups, such as thioether, alkoxyl, alkylamino, alkylamido and esters, were synthesized and evaluated for their antibacterial activities. Antibacterial evaluation of analogues containing linear alkyl chains no longer than C5 (pentyl) showed no significant or novel activities. Related amphiphilic tobramycin derivatives possessing longer alkyl chains have been prepared.11 For example, tobramycin was converted to 6″-modified amphiphilic derivatives with C14 and C16 alkyl chains which displayed relatively strong antibacterial activities against VRE and a tobramycin-resistant E. coli strain whereas a shorter (C8) alkyl chain analogue did not.11 Oxidation of alkylthio derivatives to sulfones or sulfoxides slightly decreased the antibacterial activities. Kanamycin A derivatives synthesized with attached hydrophobic peptides displayed no significantly improved antibacterial activities.38 Other reported designs of amphiphilic aminoglycoside derivatives include 4″,6″-dialkylthio tobramycin derivatives,39 a 6″-cholestolcarbonyl kanamycin A derivative,40 and perbenzylated kanamycin A derivatives.8 The former two showed moderate to excellent broad spectrum antibacterial activities including inhibition of MRSA and a tobramycin resistant S. pyogenes strain. The cholesterol derivative was not evaluated for activity. In the above cases, where direct modification synthetic approaches were used, no antifungal growth inhibitory activities were analyzed or reported.
Fig.1.
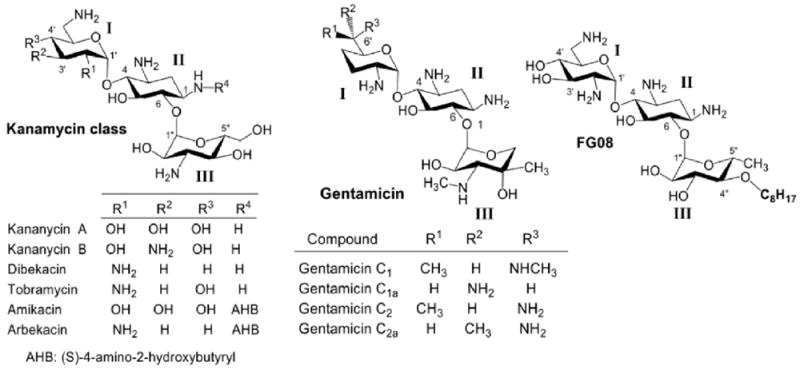
Structure of kanamycin class aminoglycosides
4.1 Glycodiversification for the synthesis of antifungal FG08
In contrast to the above direct modification approaches, a glycodiversification synthetic strategy (Scheme 1) yielded amphiphilic kanamycin derivatives that displayed antifungal activities. Inspired by a report29 that certain aminoglycosides had antifungal activities against Phytophthora and Pythium species, compound libraries derived from kanamycin, neomycin and paramomycin were screened and led eventually to amphiphilic aminoglycoside FG08 possessing antifungal properties.14,31,33 FG08 and its analogues were synthesized through successive regio- and stereoselective glycosylation of an azidoneamine core, deacetylation, Staudinger reduction of azido groups and hydrogenolysis of benzyl groups14 (Scheme 1). It is a C8 alkylated kanamycin analogue modified at the O-4″ position (Fig. 1) and possesses little or no antibacterial activity.14 Instead, FG08 inhibited a variety of fungi including plant pathogens at levels comparable to those observed for widely used crop fungicides (e.g. metconazole) (Table 1) leading to a newly described biological property of amphiphilic aminoglycosides.14,15 FG08’s alkyl chain length at C8 (or perhaps a range somewhere between C5-C11) appears critical for antifungal activity. Analogues FG01 and FG02 (Scheme 1) with C4 and C12 alkyl chains were not antifungal (Table 2).
Scheme 1.
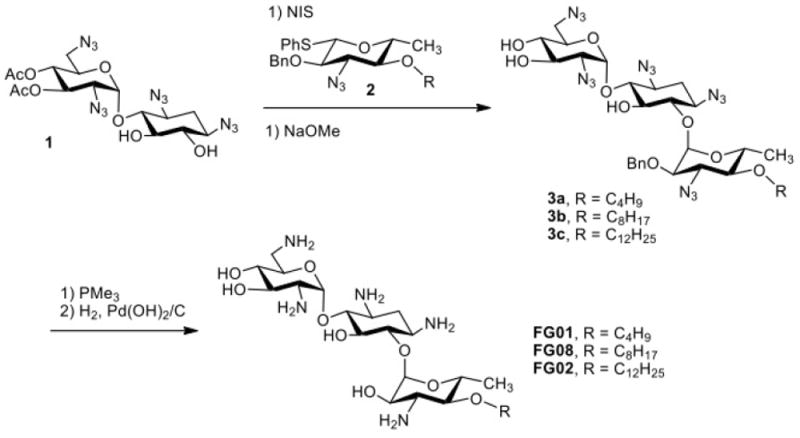
Glycodiversification synthesis of kanamycin B analogues (compound 1: 3’,4’-di-O-acetyl-1,3,2’6,-tetraazidoneamine
Table 1.
aMICs of kanamycin analogues FG08 and K20 against bacteria and fungi
| Organism | MIC(μg mL-1) | |
|---|---|---|
| FG08 | K20 | |
| Bacteria | ||
| Staphylococcus aureus | 62.5 | >250 |
| Escherichia coli ATCC25922 | 250 | 250 |
| Filamentous Fungi | ||
| bBotrytis cinerea | 31.3 | 31.3 |
| Curvularia brachyspora | 31.3 | 62.5 |
| Pythium ultimum | 15.6 | 62.5 |
| Verticillium spp. | 15.2 | cND |
| Microdochium nivale | 3.9 | 3.9-7.8 |
| bFusarium graminearum B4-5A | 7.8 | 7.8 |
| Rhizopus stolonifer | 31.3 | 62.5 |
| Cladosporium cladosporioides | 31.3 | ND |
| Fusarium oxysporum | 7.8 | 31.3 |
| Ulocladium spp. | 7.8 | ND |
| Phoma spp. | 31.3 | ND |
| Yeasts | ||
| Candida albicans 10231 | 31.3 | 15.6 |
| bRhodotorula piliminae | 7.8 | 7.8 |
minimal inhibitory concentration. Experiments were performed with potato dextrose broth as growth medium and diluent.
MICs with metconazole were 7.8, 3.9, and 15.6 μg mL-1 for B. cinera, F. graminearum B4-5A and R. pilimanae, respectively.
ND, not determined
Table 2.
MICs of kanamycin B and (O)-4” alkylated analogues against F. graminearum strain B4-5A
| Compound | Alkyl group | MIC (μg mL-1) |
|---|---|---|
| Kanamycin B | none | >250 |
| FG01 | C4 | >500 |
| FG02 | C12 | >500 |
| FG08 | C8 | 7.8-15.6 |
FG08’s capability as an agrofungicide was shown by its ability to suppress Fusarium head blight (FHB) of wheat caused by F. graminareum.14 At 5 and 30 μg mL-1, FG08 application reduced the rate of spikelet infection by 29% and 49%, respectively, over untreated controls, and FG08 pretreatments at 1080 μg mL-1 reduced disease incidence by 82% with no phytotoxicity (Fig.2).
Fig. 2.
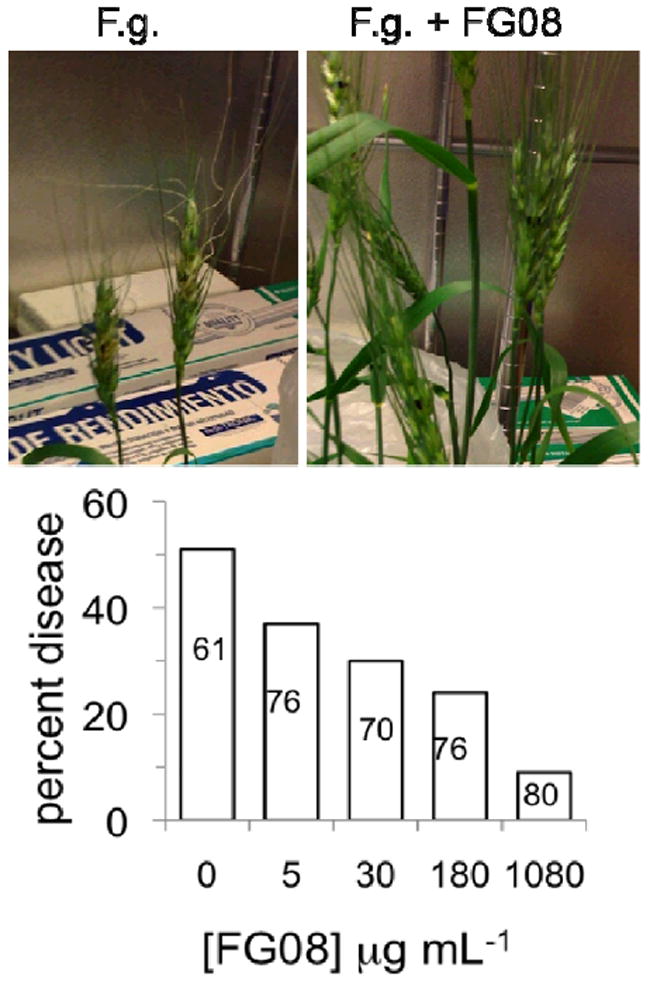
FG08 suppression of FHB disease in wheat spikelet florets. Florets (one per spikelet) of early flowering heads (cultivar Apogee) were pre-treated with FG08 (10 μL, 5 to 1080 μg mL-1 followed by inoculation with F. graminearum (F.g.) B4-5A macroconidia (10 μL, 105 conidia mL-1) 24 h later. (Upper panel) Without FG08 pre-treatment, FHB symptoms (chlorosis and curled spikes) were evident within 4 days, but suppressed with pre-treatment. (Lower panel) Suppression was observed at FG08 concentrations between 5 and 1080 μg mL-1 with reduced disease incidences between 27 and 82%, respectively. Non-inoculated spikelets had visually healthy florets following FG08 treatments at concentrations ranging between 5 and 1080μg mL-1. Numbers in the bars indicate the numbers of florets examined (8 to 12 florets per spikelet), and the florets were scored as diseased or not diseased based on visual inspection.
4.2 Direct modification for the synthesis of antifungal K20
Although FG08 shows prominent antifungal activity, its complex synthesis is not suitable for scale-up production. A relevant quote applies: “Without the ability to make a compound on a large scale, it’s essentially no more than a laboratory curiosity and it’s not going to be of any great benefit to the public” (Jaan Pesti, Bristol Meyers Squibb).41 Thus, synthesizing FG08 does not appear to be practical for purposes of use in medicine and agriculture. Therefore, based on structure-activity relationship investigations of FG08 and related compounds, a more simplified direct modification design was developed for the production of a library of 2nd generation antifungal amphiphilic aminoglycosides from tetra Boc-protected kanamycin A (Scheme 2). Among the compounds generated in this scheme, K20 (6″-O-octanesulfonylkanamycin A) was identified as a particularly active antifungal compound that can be synthesized in the laboratory at the 200-300 g scale per batch.42 Like FG08, K20 possesses strong antifungal activity and lacks antibacterial activity (Table 1). In preliminary field experiments, K20 also suppressed FHB disease of wheat (unpublished). The synthetic route leading to K20 is shorter and chemically more straight forward than for FG08 (Scheme 1) and is scalable to kg amounts making it a feasible lead compound for agricultural as well as therapeutic applications.
Scheme 2.
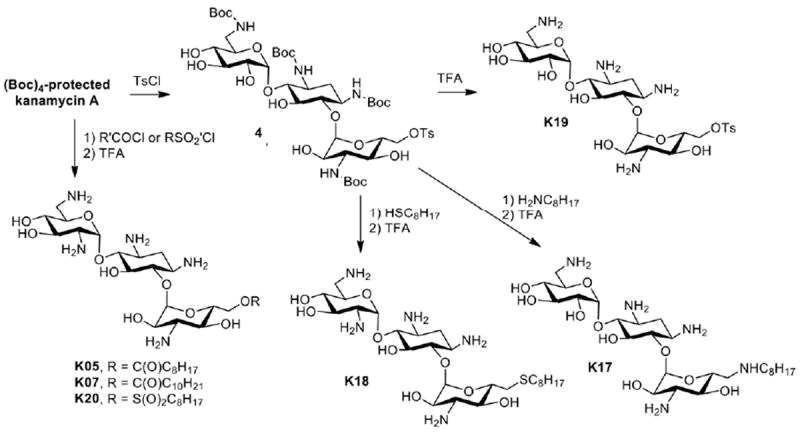
Direct modification synthesis of kanamycin A analogues
5 Comparison of antibacterial and antifungal activities of neomycin-based amphiphilic aminoglycosides
Neomycin belongs to a group of 4,5-disubstituted 2-deoxystreptamine aminoglycosides, which contain several structurally different compounds. Neomycin B is the most cost effective starting material among these aminoglycosides and has been commonly used for the synthesis of amphiphilic aminoglycosides. Neomycin B has only one primary hydroxyl group (5″-OH), offering a convenient site for approaches to incorporate hydrophobic groups. The synthesis of amphiphilic neomycins usually begins with protection of neomycin using t-butoxylcarbonyl (Boc) or carbobenzyloxy (CBz) groups (Scheme 3).
Scheme 3.
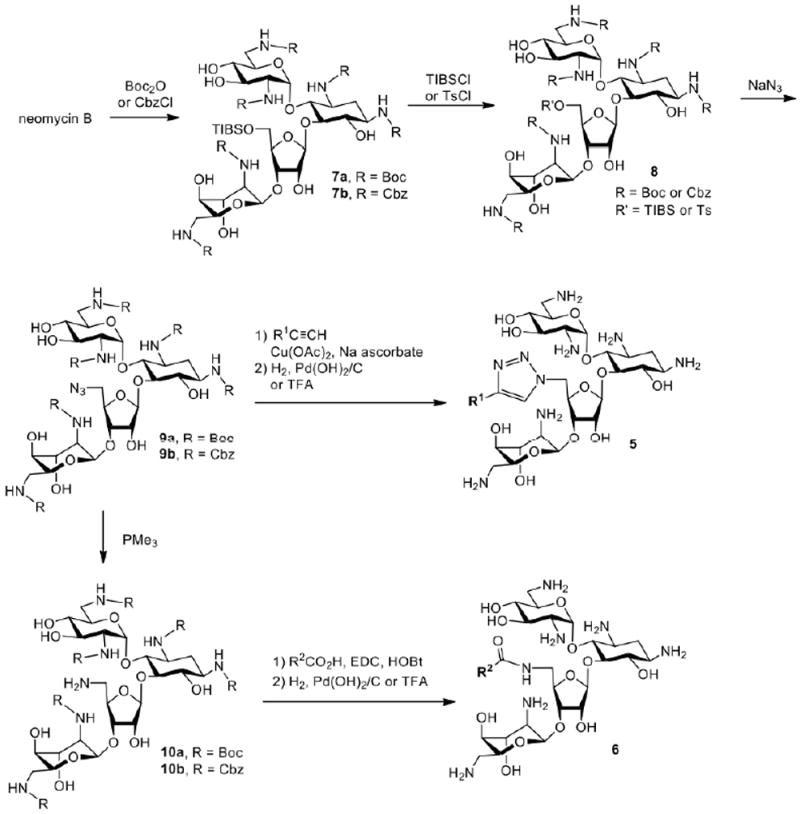
Synthesis of amphiphilic neomycin analogues
Following the regioselective sulfonation using 2,4,6-triisopropylbenzenesulfonyl chloride (TIBSCl) or toluenesulfonyl chloride (TsCl) and a subsequent azide substitution, 5″-azidoneomycin is obtained. The 5″-azido group can undergo 1,3-dipolar cyclization with alkynes to introduce hydrophobic chains. Alternatively, the 5″-azido group of 9 can be reduced to an amino group and coupled with various carboxylic acids for the attachment of hydrophobic groups. Global deprotection of the Boc or Cbz groups of these derivatives from both routes provides amphiphilic neomycin derivatives (Fig. 3). The Schweizer8,43,44 and Chang5,6 groups report similar antibacterial profiles for the variety of amphiphilic neomycin derivatives produced. Shown in Table 3 are antibacterial activities Determined by the Chang group for selected bacterial strains.
Fig. 3.
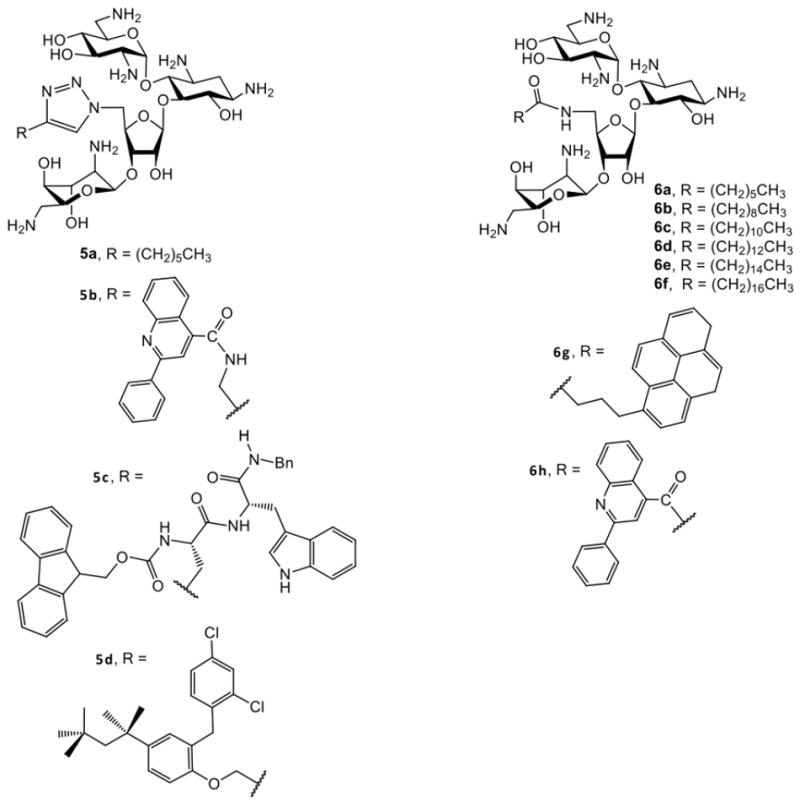
Structures of amphiphilic neomycin analogues
Table 3.
| Compound | MIC, μg mL-1 | ||||
|---|---|---|---|---|---|
| S. aureusa | MRSAb | E. colic | E. faecalisd | P. aeruginosae | |
| neomycin B | 1 | 125 | 4 | ≥ 250 | 64 |
| amikacin | 1 | 8-16 | 1 | ≥ 250 | 0.5-1 |
| vancomycin | 0.5-1 | 1 | 125-250 | 250 | ≥ 250 |
| 5a | 4 | NDf | 32 | ND | ND |
| 5b | 8 | ND | 32 | ND | ND |
| 6a | 2-4 | 125 | 16 | ≥ 250 | ≥ 250 |
| 6b | 16 | 125-250 | 32 | 64-125 | 16-32 |
| 6c | 8-16 | 16-32 | 16-32 | 64-125 | 16 |
| 6d | 2-4 | 4-8 | 8-16 | 8-16 | 8 |
| 6e | 1-2 | 2-4 | 4-8 | 4 | 4 |
| 6f | 2-4 | 2-4 | 4-8 | 8-16 | 8-16 |
| 6h | 4-8 | ND | 8-16 | ND | ND |
ATCC25923,
ATCC33591,
ATCC25922,
ATCC51299 (vancomycin-resistant enterococci),
ATCC27853
ND, not determined
In general, the derivatives are more active against G+ than G- bacteria. For derivatives attached with linear alkyl chains (5a, 6a-f), increasing hydrophobicity enhances activities with 6e (C16, hexadecyl) being the most active. Extending the alkyl chain to C18 (octadecyl), slightly decreases the activity. The most significant activities for 6e are against MRSA and VRE – bacterial pathogens that are generally inactive against traditional aminoglycosides. The surprising activities of 6e suggest a novel antibacterial mode of action for an aminoglycoside. Converting amino groups into guanidine groups did not affect antibacterial activity8 and designs with hydrophobic groups incorporated on all hydroxyl groups of neomycin displayed good activities against MRSA.38 Interestingly, none of the neomycin amphiphilic aminoglycosides synthesized by the Chang group 5,6 including 6e displayed antifungal activities. Fungal susceptibility data were not reported for the neomycin B analogues synthesized by the Schweizer group.8,43,44
6 Imparting antifungal properties with C8 alkyl chains on the kanamycin core
The antifungal amphiphilic aminoglycosides FG08 and K20 uniquely possess ring III C8 alkyl chains. This suggests a role for this hydrophobic feature in imparting antifungal properties as well as the loss of antibacterial capabilities. An examination of the antifungal activities of kanamycin and neomycin-based amphiphilic analogues (Table 4) lends support to this notion.
Table 4.
MICs of aminoglycoside compounds against F. graminearum B4-5A
| Compound | MIC(μg mL-1) |
|---|---|
| neomycin B | 62.5 |
| kanamycin A | >500 |
| 6a | 62.5 |
| 6e | 125 |
| 6f | 125 |
| FG01 | >500 |
| FG02 | >500 |
| FG08 | 31.5 |
| K07 | 62.5 |
| K18 | 15.6 |
| K19 | >500 |
| K20 | 7.8-15.6 |
| aK20 | 1.95-3.9 |
Microbroth dilution MIC assayswere performed with RPMI medium; all others were performed with potato dextrose broth.
As mentioned previously, the O-4″-C8 kanamycin B analog FG08 is antifungal, but the C4 (FG01) and C12 (FG02) isomers are not (Tables 2 and 4). Among analogues derived from tetra-Boc protected kanamycin A (Scheme 2), compound K20 (with 6″-sulfonyl C8) showed antifungal activity against F. graminearum as did K18 (with 6″-sulfo C8) (although with less potency), and non C8 analogues K07 and K19 were not antifungal. Finally, none of the amphiphilic neomycin B derivatives synthesized by the Chang group5,6 possess a C8 alkyl chain (Scheme 3) but rather C7, C16 and C18 alkyl chains as the hydrophobic moiety (e.g. 6a, 6e, and 6f, Table 4), and coincidentally none showed antifungal activities. Altogether, these observations begin to point to a correlation between the occurrence of a C8 alkyl chain and antifungal activity in amphiphilic aminoglycosides. Clearly, further structure –activity analyses are needed, including with additional aminoglycoside core structures, to determine the significance and degree of this possible correlation between C8 alkyl chain and antifungal activity.
7 Hemolytic and cytotoxic properties of antifungal FG08
Several studies reveal that active amphiphilic aminoglycosides have hemolytic and cytoxicity activities, but at concentrations that greatly exceed their effective antibacterial or antifungal concentrations.6,14,15 For example, neomycin derivatives 6e and 6f (Fig. 3) were hemolytic at IC90 values greater than 1 mg mL-1.6 These concentrations are 50 – 500 fold higher than their antibacterial MIC values (ranging from 2 to 16μg mL-1). A similar difference occurs with antifungal FG08.14 FG08 did not hemolyze more than 20% of sheep erythrocytes at 313μg mL-1, a concentration that is more than ten-fold higher than its antifungal MIC, and at 28 μg mL-1 only 2% hemolysis occurred. FG08 showed dose-dependent toxicity to mammalian cell lines C8161.9 (melanoma) and NIH3T3 (mouse fibroblasts).15 However, FG08 IC50s for both C8161.9 and NIH3T3 cells were greater than 250 μg mL-1, or at least 10 to15-fold higher concentrations than the antifungal MICs determined against Saccharomyces cerevisiae. The cell permeabilizing effect of FG08 was also measured on C6181.9 cells.15 Unlike its effect on S. cerevisiae, treatment with 100 μg mL-1 FG08 caused no influx of SYTOX Green dye over 30 min of exposure to the aminoglycoside. Finally, FG08 was minimally phytotoxic when directly applied to wheat leaf tissues (at 30 μg mL-1).15
8 Antifungal mechanism of action of FG08
Several observations indicate that cell membrane perturbation is the primary mechanism of action of FG08 against fungi.14,15 When cells of S. cerevisiae were exposed to FG08 (30 μg mL-1), 8-fold more cells were stained with the membrane impermeable dye fluorescein isothiocyanate, cells had 4 to 6-fold higher K+ efflux rates, and 18-fold more cells were stained with SYTOX Green in comparison to exposure to kanamycin B at 30 μg mL-1 (Fig. 4). Altogether, these results show that FG08 quickly increases the permeability of fungal plasma membrane to impermeant fluorescent dyes and K+ ions. Other observations show the role of membrane lipids in allowing FG08’s antifungal action. Yeast mutants with aberrant membrane sphingolipids were 4 to 8-fold less susceptible to growth inhibition with FG08 and showed 2 to 10-fold lower SYTOX Green uptake rates than did the isogenic wild-type strain.15 FG08 caused leakage of pre-loaded calcein from 50% of small unilamellar vesicles with glycerophospholipid and sterol compositions that mimic the compositions of fungal plasma membranes.15 Less than 5 and 10%of vesicles with glycerophospholipid and sterol compositions that mimic bacterial and mammalian cell plasma membranes, respectively, showed calcein leakage.15 It is suggested that FG08’s growth inhibitory specificity for fungi and lack of inhibitory effects to bacterial and mammalian cells lie in its interactions with plasma membranes by mechanisms that are modulated by membrane lipid composition.
Fig. 4.
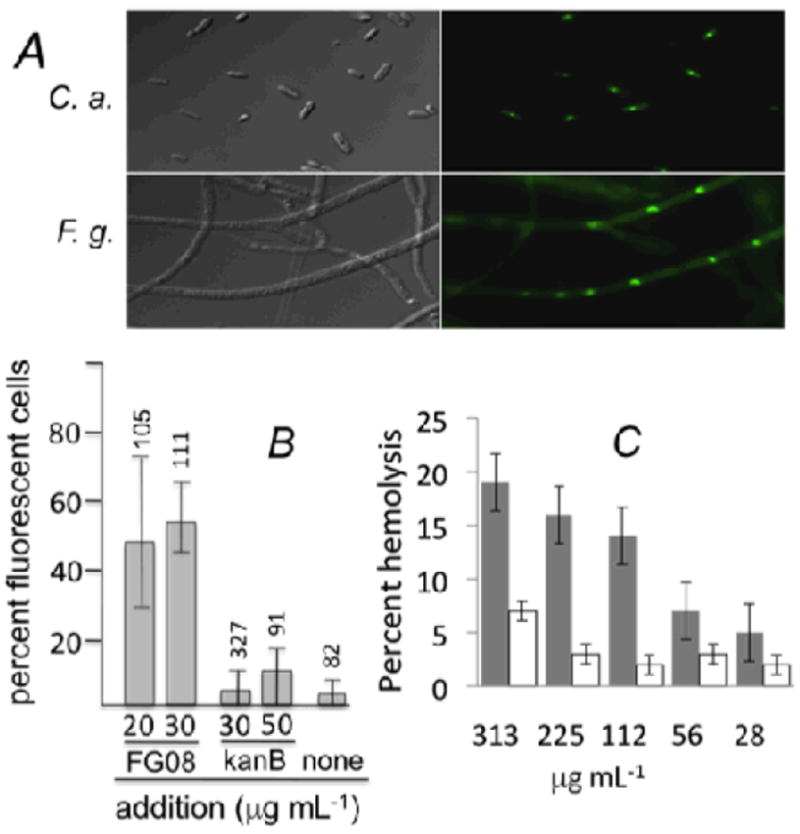
FG08 effects on membrane permeability. (A) Addition of FG08 at 30 and 10 μg mL-1 (10 min) increased the permeability of C. albicans (C. a.) cells and F. graminearum (F. g.) hyphae, respectively, to SYTOX Green (0.2 μM). (B) FG08 was more effective than kanamycin B (kanB) (both with 10 min exposure) in causing SYTOX green dye permeation in C. albicans cells. At 30 μg mL-1, FG08 was ~12 times more effective than kanamycin B. Triton X100 (1%) gave 100% dye permeation (data not shown). Numbers above the range bars = number of cells analyzed. (C) Neither FG08 (grey bars) or kanamycin B (blank bars) caused hemolysis of sheep erythrocytes at concentrations equal to their antifungal or antibacterial MICs, respectively.11
Conclusions and Future Directions
Novel antifungal amphiphilic aminoglycosides FG08 and K20 have been synthesized with attachment of C8 alkyl chains to ring III of core kanamycin B and A structures, respectively. The antibacterial capabilities of the kanamycins are simultaneously lost with C8 alkyl chain attachment, i.e. a “switch“ occurs in growth susceptibility between bacteria and fungi corresponding to non-alkylated and C8 alkylated kanamycin derivatives, respectively.
It is of interest that longer length alkyl chains (e.g C14 - C18) attached to ring III of tobramycin11 and neomycin B5,6 yield strongly antibacterial derivatives that in the case of neomycin B analogues lack antifungal activities. It may be speculated that varying the lengths of the hydrophobic groups attached to aminoglycosides not only expands the spectrum and degree of antibacterial activities, but also alters the preference for growth inhibition of bacteria vs fungi. More studies with more structural variations are needed to determine the validity of this notion, and how generally applicable it may be among the variety of traditional aminoglycosides serving as core structures for amphiphilic derivatives.
As opposed to synthetic designs for most of the described amphiphilic aminoglycosides, the direct modification strategy for synthesizing K20 is simple and efficient.42 In addition and like FG08, K20 appears to be relatively non-toxic to plants and animal cell cultures.42 As a consequence, its development as an antifungal lead compound, particularly for large scale agricultural applications, is attractive. The ready availability of reagents and large stockpiles of kanamycin A (starting material) for K20 synthesis significantly enhance the prospects for its scalable production and use. Also K20’s lack of antibacterial activities helps alleviate concerns about its promotion of bacterial resistance. Nevertheless these factors and well as issues related to stability to abiotic factors (temperature, light), animal toxicities and pharmacokinetics, and adherence to plant surfaces require further investigation and analyses.
Acknowledgments
The authors acknowledge financial support from the Utah Science Technology and Research (USTAR) initiative, Baicor LC, the National Institutes of Health (AI053138 to C.-W.T. Chang) and the National Institute of Food and Agriculture, USDA (Utah Agricultural Experiment Station project UTA 1017 to J.Y. Takemoto)
Notes and references
- 1.Odds FC, Brown AJP, Gow NAR. Trends Microbiol. 2003;11:272–279. doi: 10.1016/s0966-842x(03)00117-3. [DOI] [PubMed] [Google Scholar]
- 2.Ghannoum MA, Rice LB. Clin Microbiol Rev. 1999;12:501–517. doi: 10.1128/cmr.12.4.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ. Nat. 2012;484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanessian S, Pachamuthu K, Szychowski J, Giguere A, Swayze EE, Migawa MT, Francois B, Kondo J, Westhof E. Bioorg Med Chem Lett. 2010;20:7097–7101. doi: 10.1016/j.bmcl.2010.09.084. [DOI] [PubMed] [Google Scholar]
- 5.Zhang L, Chiang F-I, Wu L, Czyryca PG, Li D, Chang C-WT. J Med Chem. 2008;51:7563–7573. doi: 10.1021/jm800997s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Keller K, Takemoto JY, Bensaci M, Litke A, Czyryca PG, Chang C-WT. J Antibiot. 2009;62:539–544. doi: 10.1038/ja.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanessian S, Szychowski J, Adhikari SS, Vasquez G, Kandasamy P, Swayze EE, Migawa MT, Ranken R, François B, Wirmer-Bartoschek J, Kondo J, Westhof E. J Med Chem. 2007;50:2352–2369. doi: 10.1021/jm061200+. [DOI] [PubMed] [Google Scholar]
- 8.Bera S, Zhanel GG, Schweizer F. J Med Chem. 2010;53:3626–3631. doi: 10.1021/jm1000437. [DOI] [PubMed] [Google Scholar]
- 9.Szychowski J, Kondo J, Zahr O, Auclair K, Westhof E, Hanessian S, Keillor JW. ChemMedChem. 2011;6:1961–1966. doi: 10.1002/cmdc.201100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ouberai M, Garch FE, Bussiere A, Riou M, Alsteens D, Lins L, Baussanne I, Dufrêne YF, Brasseur R, Decout J-L, Mingeot-Leclercq M-P. Biochim Biophys Acta. 2011;1808:1716–1727. doi: 10.1016/j.bbamem.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Herzog IM, Green KD, Berkov-Zrihen Y, Feldman M, Vidavski RR, Eldar-Boock A, Satchi-Fainaro R, Eldar A, Garneau-Tsodikova S, Fridman M. Angew Chem Int Ed. 2012;51:5652–5656. doi: 10.1002/anie.201200761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhondikubeer R, Bera S, Zhanel GG, Schweizer F. J Antibiot (Tokyo) 2012;65:495–498. doi: 10.1038/ja.2012.59. [DOI] [PubMed] [Google Scholar]
- 13.Zimmermann L, Bussiere A, Ouberai M, Baussanne I, Jolivalt C, Mingeot-Leclercq M-P, Décout J-L. J Med Chem. 2013;56:7691–7705. doi: 10.1021/jm401148j. [DOI] [PubMed] [Google Scholar]
- 14.Chang C-WT, Fosso M, Kawasaki Y, Shrestha S, Bensaci MF, Wang J, Evans CK, Takemoto JY. J Antibiot. 2010;63:667–672. doi: 10.1038/ja.2010.110. [DOI] [PubMed] [Google Scholar]
- 15.Shrestha S, Grilley M, Fosso MY, Chang C-WT, Takemoto JY. PLoS One. 2013;8:e73843. doi: 10.1371/journal.pone.0073843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Begg EJ, Barclay ML. Brit J Clin Pharmacol. 1995;39:597–603. [PMC free article] [PubMed] [Google Scholar]
- 17.Vakulenko SB, Mobashery S. Clin Microbiol Rev. 2003;16:430–450. doi: 10.1128/CMR.16.3.430-450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy SB, O’Brien TF. Clin Infec Dis. 2005;41(Suppl 4):S219–220. doi: 10.1086/432443. [DOI] [PubMed] [Google Scholar]
- 19.Verweij PE, Snelders E, Kema GH, Mellado E, Melchers WJ. Lancet Infect Dis. 2009;9:789–795. doi: 10.1016/S1473-3099(09)70265-8. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Chiang F-I, Chen H-N, Chang C-WT. J Org Chem. 2007;72:4055–4066. doi: 10.1021/jo062588j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rai R, Chang H, Chen H-N, Chang C-WT. J Carbohydr Chem. 2005;24:131–143. [Google Scholar]
- 22.Chang C-WT, Hui Y, Elchert B, Wang J, Li J, Rai R. Org Lett. 2002;4:4603–4606. doi: 10.1021/ol0269042. [DOI] [PubMed] [Google Scholar]
- 23.Elchert B, Li J, Wang J, Hui Y, Rai R, Ptak R, Ward P, Takemoto JY, Bensaci M, Chang C-WT. J Org Chem. 2004;69:1513–1523. doi: 10.1021/jo035290r. [DOI] [PubMed] [Google Scholar]
- 24.Fridman M, Belakhov V, Yaron S, Baasov T. Org Lett. 2003;5:3575–3578. doi: 10.1021/ol035213i. [DOI] [PubMed] [Google Scholar]
- 25.Asensio JL, Hidalgo A, Bastida A, Torrado M, Corzana F, Chiara JL, Garcia-Junceda E, Canada J, Jimenez-Barbero J. J Am Chem Soc. 2005;127:8278–8279. doi: 10.1021/ja051722z. [DOI] [PubMed] [Google Scholar]
- 26.Blount KF, Zhao F, Hermann T, Tor Y. J Am Chem Soc. 2005;127:9818–9829. doi: 10.1021/ja050918w. [DOI] [PubMed] [Google Scholar]
- 27.Kling D, Hesek D, Shi Q, Mobashery S. J Org Chem. 2007;72:5450–5453. doi: 10.1021/jo0707636. [DOI] [PubMed] [Google Scholar]
- 28.Udumula V, Ham YW, Fosso MY, Chen KY, Rai R, Zhang J, Li J, Chang C-WT. Bioorg Med Chem Lett. 2013;23:1671–1675. doi: 10.1016/j.bmcl.2013.01.073. [DOI] [PubMed] [Google Scholar]
- 29.Lee HB, Kim Y, Kim JC, Choi GJ, Park S-H, Kim C-J, Jung HS. J Appl Microbiol. 2005;99:836–843. doi: 10.1111/j.1365-2672.2005.02684.x. [DOI] [PubMed] [Google Scholar]
- 30.Lynch SR, Puglisi JD. J Mol Biol. 2001;306:1037–1058. doi: 10.1006/jmbi.2000.4420. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Wang J, Czyryca PG, Chang H, Orsak TW, Evanson R, Chang C-WT. Org Lett. 2004;6:1381–1384. doi: 10.1021/ol0497685. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Chen H-N, Chang H, Wang J, Chang C-WT. Org Lett. 2005;7:3061–3064. doi: 10.1021/ol051045d. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Li J, Chen H-N, Chang H, Tanifum CT, Liu H-H, Czyryca PG, Chang C-WT. J Med Chem. 2005;48:6271–6285. doi: 10.1021/jm050368c. [DOI] [PubMed] [Google Scholar]
- 34.Umezawa S, Tsuchiya T. In: Aminoglycoside Antibiotics. Umezawa H, Hooper IR, editors. Springer-Verlag; New York: 1982. pp. 37–110. [Google Scholar]
- 35.Haddad J, Kotra LP, Mobashery S. In: Glycochemistry Principles, Synthesis, and Applications. Wang PG, Bertozzi CR, editors. Marcel Dekker, Inc; New York/Basel: 2001. pp. 353–424. [Google Scholar]
- 36.Chou C-H, Wu C-S, Chen C-H, Lu L-D, Kulkarni S, Wong C-H, Hung S-C. Org Lett. 2004;6:585–588. doi: 10.1021/ol0363927. [DOI] [PubMed] [Google Scholar]
- 37.Schepdael AV, Delcourt J, Mulier M, Busson R, Verbist L, Vanderhaeghe HV, Mingeot-Leclercq MP, Tulkens PM, C1aes PJ. J Med Chem. 1991;34:1468–1475. doi: 10.1021/jm00108a035. [DOI] [PubMed] [Google Scholar]
- 38.Bera S, Zhanel GG, Schweizer F. Bioorg Med Chem Lett. 2010;20:3031–3035. doi: 10.1016/j.bmcl.2010.03.116. [DOI] [PubMed] [Google Scholar]
- 39.Berkov-Zrihen Y, Herzog IM, Feldman M, Fridman M. Org Lett. 2013;15:6144–6147. doi: 10.1021/ol4030138. [DOI] [PubMed] [Google Scholar]
- 40.Sainlos M, Belmont P, Vigneron J-P, Lehn P, Lehn J-M. Eur J Org Chem. 2003;15:2764–2774. [Google Scholar]
- 41.C & EN News. 2010 Sep 6;88:17–28. [Google Scholar]
- 42.US61/422.983, WO2012082650 (A2) U S Pat.
- 43.Bera S, Zhanel GG, Schweizer F. J Med Chem. 2008;51:6160–6164. doi: 10.1021/jm800345u. [DOI] [PubMed] [Google Scholar]
- 44.Bera S, Zhanel GG, Schweizer F. Bioorg Med Chem Lett. 2012;22:1499–1503. doi: 10.1016/j.bmcl.2012.01.025. [DOI] [PubMed] [Google Scholar]