

Abstract
Recent studies have suggested that antithrombin (AT) could act as a significant physiologic regulator of FVIIa. However, in vitro studies showed that AT could inhibit FVIIa effectively only when it was bound to tissue factor (TF). Circulating blood is known to contain only traces of TF, at best. FVIIa also binds endothelial cell protein C receptor (EPCR), but the role of EPCR on FVIIa inactivation by AT is unknown. The present study was designed to investigate the role of TF and EPCR in inactivation of FVIIa by AT in vivo. Low human TF mice (low TF, ∼1% expression of the mouse TF level) and high human TF mice (HTF, ∼100% of the mouse TF level) were injected with human rFVIIa (120 µg kg−1 body weight) via the tail vein. At varying time intervals following rFVIIa administration, blood was collected to measure FVIIa-AT complex and rFVIIa antigen levels in the plasma. Despite the large difference in TF expression in the mice, HTF mice generated only 40–50% more of FVIIa-AT complex as compared to low TF mice. Increasing the concentration of TF in vivo in HTF mice by LPS injection increased the levels of FVIIa-AT complexes by about 25%. No significant differences were found in FVIIa-AT levels among wild-type, EPCR-deficient, and EPCR-overexpressing mice. The levels of FVIIa-AT complex formed in vitro and ex vivo were much lower than that was found in vivo. In summary, our results suggest that traces of TF that may be present in circulating blood or extravascular TF that is transiently exposed during normal vessel damage contributes to inactivation of FVIIa by AT in circulation. However, TF’s role in AT inactivation of FVIIa appears to be minor and other factor(s) present in plasma, on blood cells or vascular endothelium may play a predominant role in this process.
Introduction
Tissue factor pathway inhibitor (TFPI) is the primary physiological regulator of factor VIIa (FVIIa)-tissue factor (TF)-induced blood coagulation [1], [2]. Although antithrombin III (AT) was shown to inhibit FVIIa [3]–[6], the physiological significance of this inhibition was debatable [7], [8]. AT could effectively inhibit FVIIa only when it was bound to TF and not free FVIIa [3], [4]. Still, compared to TFPI, AT was a poor inhibitor of FVIIa-TF [5], [8], [9]. Interestingly, Smith et al. [10] showed that levels of FVIIa-AT complex were surprisingly abundant in plasma (2% of plasma FVII antigen), and suggested that AT could be a significant regulator of FVIIa function and turnover in plasma. Recently, Agerso et al. [11] showed that rFVIIa-AT complex formation was responsible for 65% of the total rFVIIa clotting activity clearance following intravenous administration of rFVIIa in hemophilia patients. This is somewhat surprising as in vitro studies showed little inhibition of FVIIa by AT in the absence of TF, even in the presence of saturating concentrations of heparin [3], [4]. Furthermore, circulating blood contains either no detectable TF, or at best, traces of TF [12]–[14]. The above studies raise an interesting question that whether TF, either circulating or intravascular, or some other factors in blood are responsible for relatively rapid inactivation of FVIIa by AT in vivo.
Since AT preferentially inhibits FVIIa bound to TF, and FVIIa-AT complex rapidly disassociates from TF [9], the circulating levels of FVIIa-AT complex is thought to be an important indirect indicator of intravascular TF exposure in vivo [10]. Therefore, a number of recent studies measured plasma FVIIa-AT levels in various patient groups to investigate whether FVIIa-AT levels in plasma could predict hypercoagulable state and thrombotic risk [15]–[18]. Although these studies indicate that the FVIIa-AT levels may be useful in identifying hypercoagulable state in specific patient groups, they strongly suggest that large prospective cohort studies were needed to consider the clinical application of FVIIa-AT complex determination [17]. More importantly, to date, there is no empirical data in the literature showing that exposure of blood to intravascular TF is primarily responsible for the generation of FVIIa-AT complex in vivo.
Studies from our laboratory [19] and others [20], [21] have established that FVIIa binds endothelial protein C receptor (EPCR) in a true ligand manner. The interaction between FVIIa and EPCR is capable of not only eliciting protease activated receptor-1 (PAR1)-mediated barrier protective signaling [22], [23], but also promotes internalization of the receptor–ligand complex [24]. At present, it is unknown whether FVIIa binding to EPCR influences AT inactivation of FVIIa. Recently, we have shown that rFVIIa administered to mice intravenously (i.v) associates with EPCR, and EPCR facilitates the entry of FVIIa from circulation into perivascular tissues [25]. Once entered into perivascular tissues, FVIIa was retained there in functionally active state for extended time periods (24 h to 7 days) [25], which sharply contrasts to the short circulating half-life (<30 min) of FVIIa [26]. Since EPCR is present primarily on the endothelium whereas TF is mainly localized in extravascular cells, it raises the possibility that TF may be involved in FVIIa retention in extravascular tissues.
Development of transgenic mice that express either low levels of human TF (low TF) or high levels of human TF (HTF) in place of murine TF were useful in obtaining valuable insights into TF’s role in hemostasis, thrombosis and vascular development [27], [28]. Compared to wild-type TF, low TF mice express ∼1% of TF [29] whereas HTF mice express ∼100% of TF, except in the heart [30]. Similarly, development of EPCR-deficient and EPCR-overexpressing mice helped in elucidating the role of EPCR in hemostasis and inflammation [31], [32]. In the present study, we used the above mice to investigate the role of TF and EPCR in generation of FVIIa-AT complex in vivo. In order to compare the rates of AT inactivation of FVIIa in vitro, ex vivo and in vivo, human rFVIIa was administered to mice, or added to whole blood or plasma. Evaluation of the role of TF and EPCR in AT inactivation of exogenously administered rFVIIa is clinically relevant as AT was believed to be primarily responsible for rapid inactivation of therapeutically administered rFVIIa to hemophilic patients [11]. In addition, we also measured endogenous levels of FVIIa-AT complex in wild-type, TF and EPCR transgenic mice.
Materials and Methods
Ethics statement
Human participants: Blood from healthy donors was obtained following a written consent. Human subject research was approved by the Institutional Review Board at The University of Texas Health Science Center at Tyler.
Animals: All studies involving animals were conducted in accordance with the animal welfare guidelines set forth in the Guide for the Care and Use of Laboratory Animals and Department of Health and Human Services, and approved by the Institutional Animal Use and Care Committee of The University of Texas Health Science Center at Tyler, Tyler, TX (Animal Welfare Assurance Number A3589-01; Protocol Number: 530).
Reagents
Human rFVIIa was obtained from Novo Nordisk A/S (Maaloev, Denmark). Mouse rFVIIa was provided by Mirella Ezban/Lars Petersen, Novo Nordisk (Denmark). Affinity purified polyclonal antibodies against human FVIIa were provided by the late Walter Kisiel (University of New Mexico, Albuquerque, NM, USA). Murine FVIIa antibodies were raised in-house by immunizing rabbits with recombinant mouse FVIIa. Antithrombin and sheep anti-AT antibodies, for both human and murine, were purchased from Haematologic Technologies, Inc (Essex Junction VT, USA). Human factor X was from Enzyme Research Laboratories (South Bend, IN, USA). Chromogenic substrate Chromogenix S-2765 was from DiaPharma (West Chester, OH, USA). Rat anti-mouse TF mAb (1H1) antibodies were provided by Daniel Kirchhofer, Genentech, CA, USA. Donkey anti-sheep biotinylated IgG was obtained from Thermo Scientific (Rockford, IL, USA). Lipopolysaccharide (LPS) from Escherichia coli 0111:B4 were from Sigma (St. Louis. MO, USA). Streptavidin alkaline phosphatase (ALP) and BluePhos microwell phosphatase substrate system were from KPL (Gaithersburg, MD, USA).
Cells
Primary human umbilical vein endothelial cells (HUVEC), EBM-2 basal medium, and growth supplements were purchased from Lonza (Walkersville, MD, USA). Endothelial cells were cultured in EBM-2 basal medium supplemented with growth supplements, 1% penicillin/streptomycin, and 2% fetal bovine serum.
Mice
The generation of low TF and HTF mice was described earlier [29], [30]. Breeding pairs of EPCR-deficient (EPCR-def) and EPCR-overexpressing (EPCR-OE) mice were obtained from Chuck Esmon (Oklahoma Medical Research Foundation, Oklahoma City, OK, USA), and their generation was described in earlier reports [31], [32]. Where available, littermate controls were used as wild-type mice. Otherwise, wild-type mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA) or in-house breeding program. All mice were in C57BL/6J genetic background.
Administration of rFVIIa to mice, collection of blood and tissues
Human rFVIIa was administered to mice via the tail vein at a dose of 120 µg kg−1 body weight in 100 µL of Tris-buffered saline (TBS, 50 mM Tris–HCl, 0.15 M NaCl, pH 7.5). At various time intervals, ranging from 5 min to 7 days after rFVIIa administration, blood was collected either via submandibular vein or by cardiac puncture at right ventricle if it was a terminal time point. Only two or three blood samples were obtained from each mouse. Blood was collected into 1/10 volume of 0.13 M sodium citrate anticoagulant unless otherwise specified. Mice were subsequently exsanguinated by severing the renal artery and perfused by flushing ice-cold saline containing 5 mM CaCl2+1 mM MgCl2 through the heart. Knee bone joints were excised, rinsed briefly with ice-cold saline containing 5 mM CaCl2+1 mM MgCl2, and then processed for measuring FVIIa activity and antigen levels. Briefly, excised knee bone joints were cut into fine pieces with a sharp razor blade, and added to TBS buffer containing EDTA (20 mM) (0.5 mL buffer per 100 mg tissue), freeze-thawed, vortexed, and centrifuged to obtain clear supernatant for measuring FVIIa antigen or activity levels.
Ex vivo studies were conducted using blood drawn into factor Xa inhibitor, rivaroxaban (100 µg/ml blood) as an anticoagulant. Plasma was separated by centrifugation at 6, 000×g for 15 min.
Isolation of microparticles (MPs) from plasma, and TF activity assay
Microparticles from mouse plasma were isolated essentially as described earlier [33]. TF activity in MPs was measured by adding either mouse (in wild-type mice) or human FVIIa (in low TF and HTF mice) (10 nM), and human FX (175 nM), and measuring the amount of FXa generated at the end of 1 or 2 h activation period in a chromogenic assay. In some experiments, TF activity was measured both in the presence and absence of neutralizing TF antibodies to determine TF-specific coagulant activity.
Generation and isolation of cell-derived microparticles
Confluent monolayers of HUVEC cultured in 100 mm dish were infected with control and TF adenovirus [34] (10 moi/cell). Two days post-infection, cells were washed with buffer A (10 mM Hepes, 0.15 M NaCl, 4 mM KCl, 11 mM glucose, pH 7.5), and then treated with calcium ionomycin (10 µM) in buffer B (buffer A containing 5 mM CaCl2 and 1 mg/ml bovine serum albumin) for 20 min at 37°C. Supernatant was collected and centrifuged first at 200×g for 5 min to remove any cell debris, and then centrifuged for 1 h at 20,000×g at 4°C to sediment microparticles. Microparticle pellet was washed once with buffer A, resuspended in 100 µl of buffer A, and stored at 4°C until used. Characterization of microparticles derived from endothelial cells transfected with control adenovirus (TF− microparticles) and TF adenovirus (TF+ microparticles) in prothrombin activation assay revealed that their prothrombin activation potential was essentially equal, indicating that both TF− and TF+ MPs express equal amounts of phosphatidylserine. As expected, TF+ microparticles and not TF− microparticles effectively activated FX. TF concentration in TF+ microparticles was estimated in factor X activation assay using known concentrations of relipidated TF as a standard.
FVIIa antigen and activity assays
rFVIIa antigen levels were determined by ELISA using rabbit anti-human FVIIa as the capture antibody and biotinylated rabbit anti-human FVIIa as the detecting antibody. FVIIa clotting activity was measured in a FVIIa-specific clotting assay as described previously [25] using STart coagulation analyzer (Diagnostica Stago, Parsippany, NJ, USA). Briefly, 50 µl of sample was incubated with 50 µl of 100 nM soluble TF in 1 mM phospholipids (40% phosphatidyl choline/25% phosphatidyl serine/35% phosphatidyl ethanolamine) and 100 µl of FVII-deficient plasma (George King Biomedical Inc., Overland Park, KS, USA) for 3 min at 37°C, and clotting was initiated by the addition of 100 µl of 25 mM CaCl2. Varying known concentrations of rFVIIa (0.5 ng to 30 ng/ml for the ELISA; 0.25 to 50 ng/ml for the activity assay) were used to construct a standard curve.
Measurement of FVIIa-AT complexes
Majority of experiments described in this study required the measurement of human rFVIIa-mouse AT complex. The amount of rFVIIa-AT complex generated in our experimental system was determined by using an in-house developed ELISA, which is specific to measure the complex between human rFVIIa and mouse AT. Briefly, rabbit anti-human FVIIa antibodies (5 µg/ml) were coated onto 96-well ELISA plate for overnight at 4°C. After blocking the wells with 0.1% gelatin, diluted samples were added to the wells to capture rFVIIa. After 1 h incubation at room temperature, unbound material was removed, wells were washed 4 times, and then sheep anti–mouse AT antibody (5 µg/ml) was added to the wells for 1 h, followed by donkey anti-sheep biotinylated IgG (1∶500), and streptavidin ALP (1∶200 dilution) for 1 h each with ample washings of the wells prior to the addition of each reagent. Color was developed by using BluePhos microwell phosphatase substrate system, and measured at 650 nm. A background reading obtained in a zero min time point sample was subtracted from the readings of other time points. To generate rFVIIa-AT standard, a known concentration of rFVIIa (100 nM, 5 µg/ml) was incubated with soluble TF (200 nM), mouse AT (1 µM), and heparin (10 U/ml) overnight at 37°C, and the samples were subjected to non-reducing SDS-PAGE followed by immunoblot analysis to evaluate the extent of FVIIa-AT complex formation. The concentration of rFVIIa-AT in the standard was determined by the relative band intensities of rFVIIa-AT and free rFVIIa. Typically, more than 80% of FVIIa was in complex with AT. Varying concentrations of rFVIIa-AT complex (0.325 to 80 ng/ml, FVIIa-AT concentration depicts ng FVIIa in complex with AT) were used to generate a standard curve. The lower detection limit of the assay was between 0.25 to 0.5 ng ml−1 rFVIIa-AT complexes. The assay was specific to detect human FVIIa-mouse AT complex (substitution of free human FVIIa, mouse FVIIa, mouse AT or mouse control plasma in place of human FVIIa-mouse AT complex gave a reading similar to that of the blank (buffer), OD<0.1).
A similar procedure was used for measuring mouse FVIIa- mouse AT complexes or human FVIIa-human AT complexes. For this, ELISA plates were coated with either rabbit anti-mouse FVIIa antibodies or anti-human FVIIa antibodies to capture FVII/FVIIa from that particular species. The species-specific FVIIa-AT complexes were detected using either sheep anti-mouse AT or anti-human AT. It may be pertinent to note here that anti-mouse FVIIa antibodies do not cross react with human FVIIa and vice versa. Mouse FVIIa-AT standard was made by complexing recombinant mouse rFVIIa with mouse AT, and human FVIIa-AT standard was made by incubating human rFVIIa with human AT as described in the above paragraph.
Statistics
All in vitro and ex vivo experiments were repeated three or more times. For in vivo studies, 3 to 9 mice were used for each group. Analysis of a data set using statistical software (GraphPad, Prism vs. 4.03, La Jolla, CA, USA) where n was 8 or more passed the normality test (D’Agostino and Pearson omni bus normality test). The data were shown as the mean ± SEM. Statistical significance between the two experimental groups was determined by Students t-test. One-way analysis of variance was used to determine statistical significance among three groups.
Results
Formation of FVIIa-AT complex in buffer, plasma and blood
First, we compared the rate of rFVIIa-AT complex formation in buffer, plasma, and blood in the absence of exogenously added heparin or TF. We included the plasma concentration of AT in a buffer system, and rFVIIa (1 µg/ml) was added to all three systems. As shown in Fig. 1A, increasing amounts of rFVIIa-AT complex was formed with increase in times under all experimental conditions. However, only less than 1% of FVIIa was found to be in complex with AT in a buffer system at the end of 1 h incubation with AT (Fig. 1A). The rate of rFVIIa-AT complex formation was significantly higher in plasma compared in buffer (about 4-fold). The rate of rFVIIa-AT complex formation was 2-fold higher in blood, compared in plasma.
Figure 1. Comparison of FVIIa-AT complex formation in vitro and ex vivo conditions.
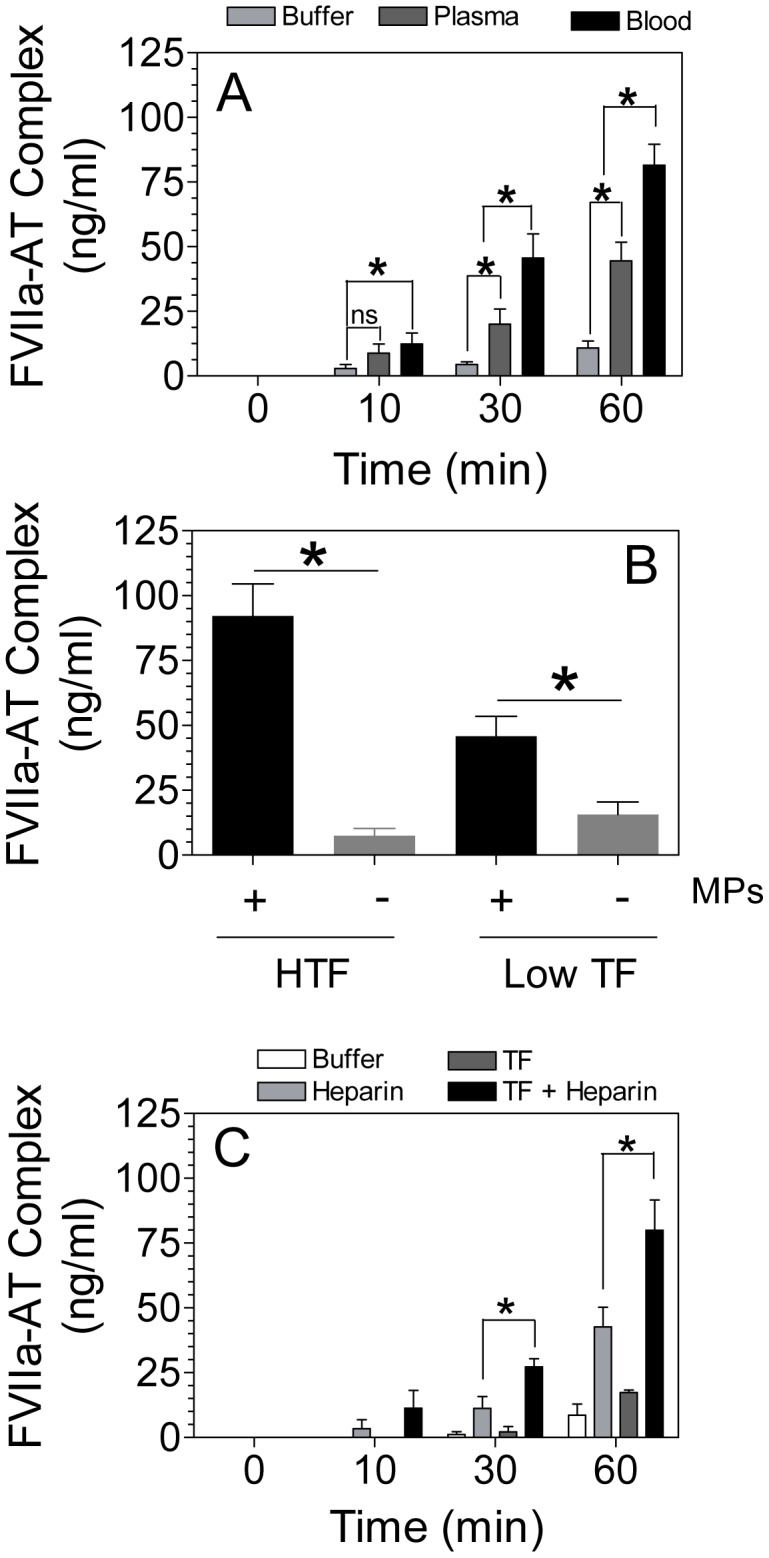
(A) Blood from wild-type mice was drawn into rivaroxaban as an anticoagulant. Plasma was separated by centrifugation at 6,000×g for 5 min. To a reaction system not containing plasma or blood, a plasma concentration of AT (125 µg/ml) was added to Tris-buffered saline (TBS) containing 1 mg/ml bovine serum albumin (BSA), 5 mM CaCl2 and 1 mM MgCl2. rFVIIa was added to all three reaction systems (buffer, plasma and blood) to a final concentration of 1 µg/ml. At various time points, an aliquot was removed from the reaction mixtures and diluted 1∶10 in TBS/BSA containing 10 mM EDTA, and frozen immediately until they were used for the assay. Levels of FVIIa-AT complex was measured in an ELISA as described in Methods (n = 6 to 9). (B) Plasma obtained from HTF and low TF mice was divided into two equal aliquots, and one of the aliquots was subjected to centrifugation 20,000×g for 1 h at 4°C to remove microparticles. rFVIIa (1 µg/ml) was added to both the plasmas, and FVIIa-AT complex generated at 60 min was determined. (C) rFVIIa (1 µg/ml) was incubated for varying times with AT (125 µg/ml) in TBS/BSA buffer containing 5 mM CaCl2+1 mM MgCl2 in the presence or absence of heparin (1 U/ml) and relipidated TF (100 pg/ml). FVIIa-AT levels were determined in an ELISA (n = 4). The concentration of FVIIa-AT (ng/ml) reflects ng of FVIIa complexed with AT. *Indicates that the compared values differ in statistically significant manner (P<0.05).
It is possible that increased levels of rFVIIa-AT generated in plasma and blood, compared to a buffer system, may be due to the presence of traces of TF in plasma and blood. To investigate this possibility, we first attempted to measure TF antigen levels in mouse plasma. However, the sensitivity of TF ELISA assay was not suitable for detecting picograms of TF that may be present in plasma. Next, we measured MP-associated TF activity in plasma in factor X activation assay, using known concentrations of relipidated TF as the standard. This assay revealed that plasma of wild-type mice contains ∼2 pg/ml TF (data not shown). Actual concentration of TF in plasma may be slightly higher since this assay does not take into account soluble TF and TF that may not associate with MPs. To determine the role of trace amounts of TF in plasma in rFVIIa inactivation by AT, first we attempted to inhibit plasma TF activity by using TF neutralizing antibodies. However, both TF antibodies and control IgG interfered in measuring FVIIa-AT complex levels in an ELISA. Therefore, next we compared the generation of rFVIIa-AT complexes in plasma of low TF and HTF mice. As shown in Fig. 1B, the formation of rFVIIa-AT complex in plasma of HTF mice was significantly higher than in plasma of low TF mice. Furthermore, removal of MPs from the plasma by centrifugation significantly reduced the levels of rFVIIa-AT formation in plasma. These data indicate that traces of TF in plasma significantly accelerate inactivation of FVIIa by AT.
The effect of low concentrations of TF on FVIIa-AT complex formation was examined further in a buffer system where rFVIIa was incubated with AT ± heparin (1 U/mL) in the presence or absence of relipidated TF (100 pg/ml). As shown in Fig. 1C, a low concentration of TF substantially enhanced the formation of FVIIa-AT complex both in the absence and presence of heparin. In the presence of both TF and heparin, we observed ∼10-fold increase in the rate of FVIIa-AT generation in comparison to a buffer system containing AT alone. However, even in the presence of heparin and TF, only less than 10% of rFVIIa was found to be in complex with AT at the end of 60 min incubation time (Fig. 1C).
In additional studies, to examine the role of MPs on FVIIa-AT complex formation in plasma, endothelial cell-derived MPs bearing TF or lacking TF were added to human plasma or whole blood supplemented with rFVIIa (1 µg/ml), and the levels of FVIIa-AT formed at the end of 1 h was measured. The concentration of MP TF chosen for this study was 30 pg/ml, an outer limit of MP-TF concentration that could possibly be present in patients with high thrombotic risk (see rev [35] for references). Although we found a modest, 20 and 50%, increase in FVIIa-AT levels in plasma supplemented with TF− and TF+ MPs, respectively, over the control plasma, this increase was not statistically significant (FVIIa-AT levels, ng/ml : no MPs, 6.14±1.45; TF− MPs, 8.23±2.84; TF+ MPs, 11.13±5.9; n = 6). Heparin (1 U/ml) increased FVIIa-AT levels in plasma by 4-fold, but the presence of TF− or TF+ MPs had no measurable influence on AT inactivation of FVIIa. Similarly, no differences were found in FVIIa-AT levels in whole blood in the presence or absence of MPs (data not shown).
Role of TF in generation of FVIIa-AT complex in vivo
First to determine the extent of FVIIa-AT complex formed in vivo in mice under basal conditions and the role of TF in generation of FVIIa-AT, we measured mouse FVIIa-AT complexes in plasma obtained from wild-type, HTF and low TF mice. Both wild-type and HTF mice contained similar levels of mouse FVIIa-AT complex (wild-type, 6.93±2.47 ng/ml, n = 6; HTF, 7.66±2.40 ng/ml, n = 8) whereas FVIIa-AT levels were substantially low in low TF mice (1.21±0.19 ng/ml, n = 12).
Next to compare the rate of AT inactivation of FVIIa in vivo vs. in vitro or ex vivo, and to determine the role of TF or other in vivo parameters in influencing AT inactivation of therapeutic concentrations of rFVIIa, low TF and HTF mice were administered with human rFVIIa (120 µg kg−1 body weight). Blood was collected at various time intervals following rFVIIa administration, and rFVIIa-AT complex and rFVIIa antigen levels in plasma were measured. As shown in Fig. 2A, rFVIIa complex formation with AT was evident as early as 1 min following rFVIIa administration in both the genotypes. rFVIIa-AT levels peaked at 60 min, and start to decline thereafter. At 24 h following rFVIIa administration, rFVIIa-AT complexes were barely detectable in plasma. The levels of rFVIIa-AT complex were significantly higher in HTF mice as compared with low TF mice at 30 and 60 min following rFVIIa administration (Fig. 2A). It may be pertinent to note here that measurement of TAT at these points showed no significant differences between HTF and low TF mice (TAT levels ng/ml at 30 min: low TF, 19.69±4.38; HTF 14.30±2.7; at 60 min: low TF, 24.27±9.78; HTF 18.93±4.7, n = 6).
Figure 2. Levels of FVIIa-AT generated in vivo in HTF and low TF mice following administration of rFVIIa.

HTF and low TF mice were injected with rFVIIa (120 µg/kg body weight) intravenously via the tail vein. FVIIa-AT complex (A) and FVIIa antigen (B) levels were determined in plasma obtained from these mice at various time points following rFVIIa administration (n = 4 or more). The small bar (−) in the figure represents expected FVIIa antigen levels, based on our previous study of rFVIIa clearance in wild-type mice using 125I-rFVIIa as a tracer [26], at 3, 5, 15, 30, 60, 120, 180, and 240 min following rFVIIa administration. The concentration of FVIIa-AT (ng/ml) reflects ng of FVIIa complexed with AT. *Indicates the value differs in statistically significant manner (P<0.05) between HTF and low TF mice at the given time point.
We also measured rFVIIa antigen levels in the plasma of HTF and low TF mice to investigate whether TF influences pharmacokinetics of rFVIIa, which in turn could influence the formation of rFVIIa-AT complex in vivo. As shown in Fig. 2B, no significant differences were found in rFVIIa antigen levels in the HTF and low TF mice, indicating that differences in rFVIIa-AT complex levels in these mice at 30 and 60 min is not due to the difference in the plasma rFVIIa antigen levels in these mice.
The comparison of rFVIIa antigen and rFVIIa-AT complex levels, particularly in samples obtained at 60 min or later following rFVIIa administration, suggests that rFVIIa antigen assay does not fully recognize rFVIIa in rFVIIa-AT complex. Thus, rFVIIa antigen levels were substantially lower than rFVIIa-AT complex levels at these later time points, and also lower than predicted rFVIIa antigens levels based on our earlier FVIIa clearance studies utilizing 125I-labeled rFVIIa. Here, it may be pertinent to note that we evaluated several other FVIIa antibodies, both prepared in-house and obtained commercially or from other investigators, and none of the antibodies recognized rFVIIa in complex with AT to the same extent as of free rFVIIa.
Role of EPCR in generation of FVIIa-AT complex in vivo
Recently, we have shown that FVIIa binds EPCR both in vitro and in vivo [19], [36]. Therefore, we investigated here whether FVIIa binding to EPCR influences FVIIa-AT complex formation in vivo. Human rFVIIa was injected into wild-type, EPCR over-expressing (EPCR-OE), and EPCR-deficient mice (EPCR-def) and the levels of rFVIIa-AT complex were measured. As shown in Fig. 3A, there were no significant differences in the levels of rFVIIa-AT complex generated among the wild-type, EPCR-def and EPCR-OE mice. We also measured endogenous FVIIa-AT levels in the above groups of mice (no rFVIIa was administered). These data also showed no significant differences in FVIIa-AT levels among the three groups (Fig. 3B). It may pertinent to note here that while human FVIIa binds murine EPCR, murine FVIIa does not bind murine EPCR in any significant manner [36].
Figure 3. Levels of FVIIa-AT generated in vivo in wild-type, EPCR-overexpressing (EPCR-OE), and EPCR-deficient (EPCR-def) mice.
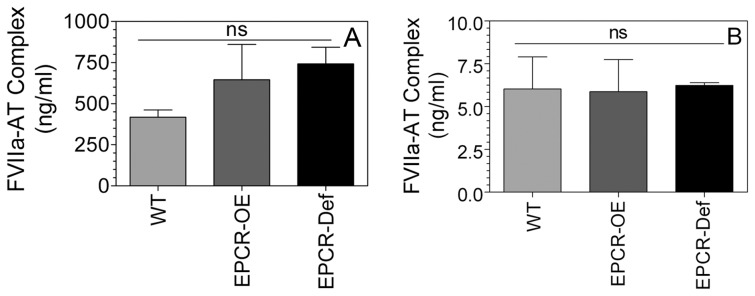
(A) Wild-type, EPCR-OE and EPCR-def mice were injected with rFVIIa (120 µg/kg body weight) intravenously via the tail vein. FVIIa-AT levels in plasma obtained from these mice at 60 min post-rFVIIa administration was measured in an ELISA assay (n = 3–4). (B) Endogenous FVIIa-AT levels. Plasma obtained from wild-type, EPCR-OE and EPCR-def mice that were not subjected to any treatment were used to measure endogenous FVIIa-AT levels. The concentration of FVIIa-AT (ng/ml) reflects ng of FVIIa complexed with AT. ns, not statistically significant as determined in one-way analysis of variance.
Effect of enhanced TF expression on FVIIa-AT complex formation in vivo
If traces of TF contribute to the generation of FVIIa-AT complex formation in vivo, then pathological conditions (e.g., sepsis, atherosclerosis etc.), where TF expression is upregulated in cells that come in contact with blood, may accelerate FVIIa-AT complex formation. Wang et al. [33] showed that TF MP levels were increased several fold in endotoxemic mice. To test the effect of increased expression of TF in vivo on FVIIa-AT generation, wild-type mice were injected with saline or LPS (5 mg/kg body weight), and 5 h after LPS administration blood was collected from these mice by cardiac puncture, and plasma was processed for measuring MPs procoagulant activity and mouse FVIIa-AT complexes. As shown in Fig. 4A, LPS administration markedly increased the procoagulant activity of plasma MPs. The increased procoagulant activity was due to the generation of TF+ MPs since the incubation of MPs with mouse TF mAb blocked the increased procoagulant activity. LPS administration increased the levels of FVIIa-AT complexes in plasma by about 50% compared to the saline control, but the difference is not statistically significant, probably due to a wider variation in FVIIa-AT levels in these mice. It may be important to note here that measuring endogenous murine FVIIa-AT complexes in LPS administered mice may not provide a complete picture of the role of TF on AT inactivation of FVIIa since FVIIa-AT complex formation is limited by the availability of FVIIa. LPS administration may not significantly increase FVIIa levels in vivo [37].
Figure 4. Measurement of microparticle TF procoagulant activity and FVIIa-AT levels in plasma following LPS administration in wild-type mice.
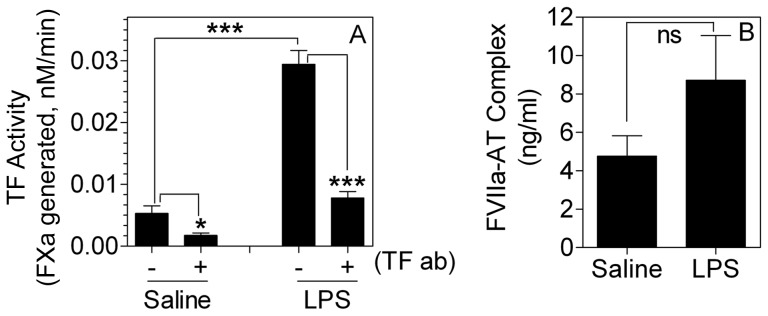
Wild-type mice (C57BL/6J) were injected with saline or LPS (5 mg/kg body weight) intraperitoneally. 5 h after saline or LPS administration, blood was obtained from these mice by cardiac puncture, and plasma samples were processed for isolation of microparticles to measure TF procoagulant activity (A) or for determination of FVIIa-AT levels (B). Procoagulant activity measured in factor Xa generation assay in the absence or presence of mouse TF mAb (10 µg/ml, preincubated for 30 min) (n = 5). *Indicates the values differ in statistically significant manner between the control and LPS-treated mice. The concentration of FVIIa-AT (ng/ml) reflects ng of FVIIa complexed with AT. *, P<0.05; ***, P<0.001; ns, not statistically significant.
Therefore, to investigate the effect of enhanced in vivo TF on AT inactivation of FVIIa more accurately, HTF mice were injected with LPS (5 mg/kg body weight), and after 5 h, human rFVIIa was given to these mice via tail vein injection, and the amount of rFVIIa-AT complex generated in plasma was measured. As shown in Fig. 5A, MP TF activity was increased by 6-fold in HTF mice administered with LPS compared to control HTF mice. Measurement of rFVIIa-AT complex in these mice showed a slight but statistically significant increase in rFVIIa-AT levels in LPS-challenged mice compared to control mice (Fig. 5B). We also measured the levels of endogenous mouse FVIIa-AT complexes generated in the above experimental system. LPS treatment increased the levels of endogenous mouse FVIIa-AT in these mice by 40% (data not shown).
Figure 5. Effect of increased TF expression on formation of FVIIa-AT complex in HTF mice administered with therapeutic concentrations of rFVIIa.
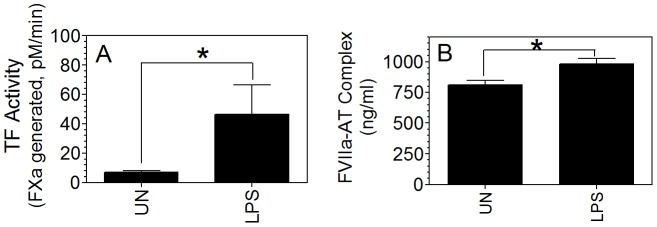
HTF mice were injected with LPS (5 mg/kg body weight) intraperitoneally. 5 h after LPS administration, rFVIIa (120 µg/kg body weight) was injected into these mice as well as unchallenged mice. 1 h following rFVIIa administration, blood was drawn from these mice by cardiac puncture, and plasma samples were processed for isolation of TF microparticles or determination of FVIIa-AT levels. (A) Microparticle TF activity; (B) FVIIa-AT levels (n = 4). The concentration of FVIIa-AT (ng/ml) reflects ng of FVIIa complexed with AT. *Indicates the values differ in statistically significant manner between the control and LPS-treated mice.
Role of TF in retention of rFVIIa activity and antigen in vivo
Recently, we have shown that rFVIIa entered into extravascular tissues via EPCR-dependent mechanism is retained in tissues for extended time periods [25]. It has been thought that TF present on extravascular cells may play a role in retention of FVIIa in tissues [25]. Our earlier analysis of TF expression by immunohistochemistry in various tissues of wild-type mice revealed the presence of TF in bone joints in the zone of calcified cartilage in the growth plate region and the mineralized bone [38]. To investigate the effect of TF in retaining FVIIa in bone joints, rFVIIa was administered to low TF and HTF mice, and seven days after rFVIIa administration, bone joints from these mice were collected, and rFVIIa antigen and activity levels in bone joints were measured. As shown in Fig. 6A, rFVIIa antigen levels were significantly higher in the bone joints of HTF mice as compared with low TF mice. Measurement of FVIIa activity in these samples showed that rFVIIa in the bone joint of HTF is functionally active. As with rFVIIa antigen level, FVIIa activity level was substantially higher in bone joints of HTF as compared to low TF mice (Fig. 6B).
Figure 6. Higher levels of FVIIa retained in bone joints of HTF compared to low TF mice.
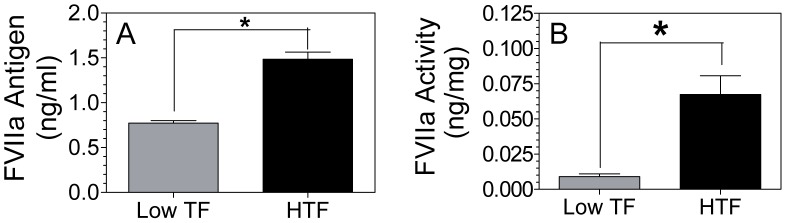
HTF and low TF mice were injected intravenously via the tail vein with rFVIIa (120 µg/kg body weight). Seven days later, bone joints were collected, bone joint fluids were eluted, and assayed for FVIIa antigen (A) and FVIIa activity (B) levels (n = 3). *Indicates the value differs in statistically significant manner (P<0.05) between HTF and low TF mice.
Discussion
The present study carried out with low TF mice expressing ∼1% of wild-type TF and HTF mice expressing ∼100% of wild-type TF indicates that TF contributes to the formation of FVIIa-AT complex in vivo. Increased levels of FVIIa-AT complex were found in plasma of HTF mice compared to low TF mice endogenously as well as after administration of a therapeutic concentration of rFVIIa. Further, the observation that increased intravascular exposure of TF by LPS administration to HTF mice moderately, but statistically significant fashion, increased FVIIa-AT levels in the plasma confirms that increased FVIIa-AT levels in plasma may reflect exposure of blood to increased levels of intravascular TF. Nonetheless, our data also indicate that the effect of TF on AT inactivation of FVIIa appears to be modest, and therefore, measurement of FVIIa-AT levels in plasma may not be a very sensitive indicator of intravascular TF exposure, and may lack robustness in accurately predicting hypercoagulable state. For example, despite a ∼100-fold difference in TF expression between HTF and low TF mice, differences in FVIIa-AT levels between the two genotypes following rFVIIa administration was less than 2-fold, that too only at 30 and 60 min following rFVIIa administration but not at later time points. Similarly, LPS administration increased MP-associated TF activity in plasma of HTF mice by more than 600% whereas it only led to a small increase (∼25%) in FVIIa-AT levels in plasma.
Recent studies of pharmacokinetics of rFVIIa in hemophilia patients or animals showed that the majority of rFVIIa clotting activity following rFVIIa intravenous administration was inhibited by AT by forming rFVIIa-AT complexes [11], [39], [40]. These results suggest that AT inhibition of rFVIIa controls drug duration in hemophilia treatment with rFVIIa. Furthermore, these data also showed that the rate of inhibition of rFVIIa by AT is significantly higher in vivo than in vitro [40]. Consistent with these data, we found that most of rFVIIa in circulation was in complex with AT at 60 min following rFVIIa administration as the levels of FVIIa-AT complex measured at this time were very similar to total FVIIa antigen levels in the circulation (compare Fig. 2A and 2B). Although TF contributes to the formation of rFVIIa-AT complex in vivo, it does not appear to influence the overall pharmacokinetics of rFVIIa as rFVIIa was cleared in a similar fashion in low TF and HTF mice. These data were also consistent with the earlier observation that showed clearance of FVIIa was unaffected by its inactivation with AT [41]. The observation that rFVIIa-AT complex formation was impeded, but not markedly impaired in low TF mice indicates that TF plays probably a minor role in FVIIa inactivation by AT in vivo. Overall, our data suggest that although TF contributes to FVIIa-AT generation in vivo, it may not be the principle player that regulate rFVIIa inactivation by AT in vivo.
Comparison of rFVIIa-AT complex formation in vitro, ex vivo and in vivo conditions show that only traces of rFVIIa was complexed with AT in vitro (in buffer) in the absence of heparin. Although rFVIIa-AT complex formation was higher in plasma and blood, relative to that in buffer, still only less than 10% of rFVIIa formed complex with AT even after 1 h of incubation time. A significantly higher level of rFVIIa-AT formation in blood, compared to plasma, indicates that blood cells may promote rFVIIa inactivation by AT. At present, blood cell types that are responsible for this are unknown. Addition of heparin and a low concentration of relipidated TF increased FVIIa-AT formation in a buffer system, reaching to comparable level obtained in the blood in ex vivo condition. At present, it is unclear whether heparin-like proteoglycans and/or TF on blood cells is responsible for increasing FVIIa-AT complex formation in blood. It may be pertinent to add here that exogenous addition of TF+ MPs to human slightly increased AT inactivation of FVIIa in ex vivo, but this increase was not statistically significant. It is possible that other clotting factors in blood or other components of blood may contribute for the increased FVIIa-AT generation in blood. Here, one should note that both clotting factors IX and X were shown to enhance the inhibition of FVIIa-TF by AT on a human bladder carcinoma cell line [42]. In contrast to in vitro and ex vivo, rFVIIa complex formation with AT was relatively rapid in vivo. Within one minute following rFVIIa administration, the formation of rFVIIa-AT complex was evident, and most of rFVIIa present in circulation at 60 min following rFVIIa administration was in complex with AT. It is unclear at present the exact mechanism(s) by which FVIIa was rapidly inactivated by AT in vivo.
Recently, we have shown that rFVIIa administered to mice rapidly associates with EPCR on vascular endothelium in vivo [25], [26], [36]. Therefore, it is entirely possible that FVIIa binding to EPCR could facilitate FVIIa inactivation by AT. However, studies conducted herein with EPCR-deficient mice and EPCR overexpressing mice (at least 8- or more fold higher EPCR expression over the wild-type [32]) clearly show that EPCR does not play a role in FVIIa-AT complex formation in vivo as we observed similar levels of rFVIIa-AT complex in these mice following rFVIIa administration.
Our recent studies showed that although most of the rFVIIa administered to mice was cleared rapidly from circulation with a half-life of less than 30 min, a small amount of rFVIIa enters into extravascular tissues, and this FVIIa was retained for extended time periods in tissues [25], [26], [38], [43]. Since most of the extravascular cells express TF, and FVIIa binds to TF with a high affinity, it had been thought that TF may be playing a role in retaining FVIIa entered into extravasculature [25]. Our present observation of increased levels of rFVIIa activity and antigen in bone joints of HTF mice compared to low TF mice even 7 days after rFVIIa administration clearly supports the above hypothesis that FVIIa entered into extravasculature associates with TF, and this association could retain FVIIa for extended time periods in extravascular tissues. The potential significance of this finding to hemostasis and hemophilia therapy with rFVIIa had been discussed earlier [25].
In summary, data presented in the manuscript show that TF contributes to FVIIa-AT complex formation in vivo. However, presence of traces of intravascular TF alone cannot explain the generation of high levels of FVIIa-AT complex rapidly in vivo following rFVIIa administration. EPCR does not influence FVIIa-AT complex formation in vivo. Finally, our data also show that TF plays a role in retaining FVIIa entered into extravasculature.
Acknowledgments
The authors are thankful to Charles T. Esmon, Oklahoma Medical Research Foundation, Oklahoma City, OK for providing EPCR-deficient and EPCR-overexpressing mice breeding pairs, and Jagan Sundaram, The University of Texas Health Science Center, Tyler, TX for breeding these mice. We thank Curtis Clark, The University of Texas Health Science Center, Tyler, TX, for his technical assistance in tail vein injections in initial experiments. We thank the late Walter Kisiel, University of New Mexico, Albuquerque, NM for providing affinity purified antibodies against human FVIIa; Mirella Ezban and Lars Peterson, Novo Nordisk, Denmark, for providing mouse FVIIa; and Daniel Kirchhofer, Genentech, CA, USA for providing rat anti-mouse TF mAb.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All available data is included in the manuscript.
Funding Statement
The work was supported by National Institutes of Health grant HL107483. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Rapaport SI, Rao LVM (1992) Initiation and regulation of tissue factor-dependent blood coagulation. Arterioscler Thromb 12: 1111–1121. [DOI] [PubMed] [Google Scholar]
- 2. Broze Jr GJ (1995) Tissue factor pathway inhibitor and the current concept of blood coagulation. Blood Coagul Fibrinolysis 6: S7–S13. [DOI] [PubMed] [Google Scholar]
- 3. Lawson JH, Butenas S, Ribarik N, Mann KG (1993) Complex-dependent inhibition of factor VIIa by antithrombin III and heparin. J Biol Chem 268: 767–770. [PubMed] [Google Scholar]
- 4. Rao LVM, Rapaport SI, Hoang AD (1993) Binding of factor VIIa to tissue factor permits rapid antithrombin III/heparin inhibition of factor VIIa. Blood 81: 2600–2607. [PubMed] [Google Scholar]
- 5. Hamamoto T, Yamamoto M, Nordfang O, Petersen JGL, Foster DC, et al. (1993) Inhibitory properties of full-length and truncated recombinant tissue factor pathway inhibitor (TFPI). Evidence that the third kunitz-type domain of TFPI is not essential for the inhibition of factor VIIa-tissue factor complexes on cell surfaces. J Biol Chem 268: 8704–8710. [PubMed] [Google Scholar]
- 6. Shigematsu Y, Miyata T, Higashi S, Miki T, Sadler JE, et al. (1992) Expression of human soluble tissue factor in yeast and enzymatic properties of its complex with factor VIIa. J Biol Chem 267: 21329–21337. [PubMed] [Google Scholar]
- 7. Rapaport SI, Rao LVM (1995) The tissue factor pathway: How it has become a “prima ballerina”. Thromb Haemost 74: 7–17. [PubMed] [Google Scholar]
- 8. Broze GJ Jr, Likert K, Higuchi D (1993) Inhibition of factor VIIa/tissue factor by antithrombin III and tissue factor pathway inhibitor. Blood 82: 1679–1680. [PubMed] [Google Scholar]
- 9. Rao LVM, Nordfang O, Hoang AD, Pendurthi UR (1995) Mechanism of antithrombin III inhibition of VIIa/tissue factor activity on cell surfaces. Comparison with tissue factor pathway inhibitor/factor Xa induced inhibition of factor VIIa/tissue factor activity. Blood 85: 121–129. [PubMed] [Google Scholar]
- 10. Smith SA, Antonaci FC, Woodhams BJ, Morrissey JH (2007) Factor VIIa-antithrombin complexes in human plasma. J Thromb Haemost 5: O–S-040 (abstract).. [Google Scholar]
- 11. Agerso H, Brophy DF, Pelzer H, Martin EJ, Carr M, et al. (2011) Recombinant human factor VIIa (rFVIIa) cleared principally by antithrombin following intravenous administration in hemophilia patients. J Thromb Haemost 9: 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Butenas S, Bouchard BA, Brummel-Ziedins KE, Parhami-Seren B, Mann KG (2005) Tissue factor activity in whole blood. Blood 105: 2764–2770. [DOI] [PubMed] [Google Scholar]
- 13. Giesen P, Rauch U, Bohrmann B, Kling D, Roque M, et al. (1999) Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci USA 96: 2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takahashi H, Satoh N, Wada K, Takakuwa E, Seki Y, et al. (1994) Tissue factor in plasma of patients with disseminated intravascular coagulation. Am J Hematol 46: 333–337. [DOI] [PubMed] [Google Scholar]
- 15. Spiezia L, Rossetto V, Campello E, Gavasso S, Woodhams B, et al. (2010) Factor VIIa-antithrombin complexes in patients with arterial and venous thrombosis. Thromb Haemost 103: 1188–1192. [DOI] [PubMed] [Google Scholar]
- 16. Spiezia L, Campello E, Gentilomo C, Gavasso S, Woodhams B, et al. (2011) Factor VIIa-antithrombin complexes in children with ischemic stroke. Thromb Res 128: 303–304. [DOI] [PubMed] [Google Scholar]
- 17. Simioni P, Spiezia L (2011) Factor VIIa-AT complex plasma levels and arterial thrombosis. Thromb Res 128: 507. [DOI] [PubMed] [Google Scholar]
- 18. Silveira A, Scanavini D, Boquist S, Ericsson CG, Hellenius ML, et al. (2012) Relationships of plasma factor VIIa-antithrombin complexes to manifest and future cardiovascular disease. Thromb Res 130: 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, Rao LV (2007) Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem 282: 11849–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Preston RJ, Ajzner E, Razzari C, Karageorgi S, Dua S, et al. (2006) Multifunctional specificity of the protein C/activated protein C Gla domain. J Biol Chem 281: 28850–28857. [DOI] [PubMed] [Google Scholar]
- 21. Lopez-Sagaseta J, Montes R, Puy C, Diez N, Fukudome K, et al. (2007) Binding of factor VIIa to the endothelial cell protein C receptor reduces its coagulant activity. J Thromb Haemost 5: 1817–1824. [DOI] [PubMed] [Google Scholar]
- 22. Sen P, Gopalakrishan R, Kothari H, Keshava S, Clark C, et al. (2011) Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood 117: 3199–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sundaram J, Keshava S, Gopalakrishan R, Esmon CT, Pendurthi UR, et al. (2014) Factor VIIa binding to endothelial cell protein C receptor protects vascular barrier integrity in vivo . J Thromb Haemost 12: 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nayak RC, Sen P, Ghosh S, Gopalakrishnan R, Esmon CT, et al. (2009) Endothelial cell protein C receptor cellular localization and trafficking. Blood 114: 1974–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clark CA, Vatsyayan R, Hedner U, Esmon CT, Pendurthi UR, et al. (2012) Endothelial cell protein C receptor-mediated redistribution and tissue-level accumulation of factor VIIa. J Thromb Haemost 10: 2383–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gopalakrishnan R, Pendurthi UR, Hedner U, Agerso H, Esmon CT, et al. (2012) Influence of endothelial cell protein C receptor on plasma clearance of factor VIIa. J Thromb Haemost 10: 971–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mackman N (2004) Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol 24: 1015–1022. [DOI] [PubMed] [Google Scholar]
- 28. Mackman N (2008) Tissue-specific hemostasis: role of tissue factor. J Thromb Haemost 6: 303–305. [DOI] [PubMed] [Google Scholar]
- 29. Parry GC, Erlich JH, Carmeliet P, Luther T, Mackman N (1998) Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest 101: 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pawlinski R, Tencati M, Holscher T, Pedersen B, Voet T, et al. (2007) Role of cardiac myocyte tissue factor in heart hemostasis. J Thromb Haemost 5: 1693–1700. [DOI] [PubMed] [Google Scholar]
- 31. Li W, Zheng X, Gu JM, Ferrell GL, Brady M, et al. (2005) Extraembryonic expression of EPCR is essential for embryonic viability. Blood 106: 2716–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li W, Zheng X, Gu J, Hunter J, Ferrell GL, et al. (2005) Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost 3: 1351–1359. [DOI] [PubMed] [Google Scholar]
- 33. Wang JG, Manly D, Kirchhofer D, Pawlinski R, Mackman N (2009) Levels of microparticle tissue factor activity correlate with coagulation activation in endotoxemic mice. J Thromb Haemost 7: 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kothari H, Nayak RC, Rao LV, Pendurthi UR (2010) Cystine186-cystine 209 disulfide bond is not essential for the procoagulant activity of tissue factor or for its de-encryption. Blood 115: 4273–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geddings JE, Mackman N (2013) Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood 122: 1873–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sen P, Clark CA, Gopalakrishnan R, Hedner U, Esmon CT, et al. (2012) Factor VIIa binding to endothelial cell protien C receptor: Differences between mouse and human systems. Thromb Haemost 107: 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Warr TA, Rao LVM, Rapaport SI (1990) Disseminated intravascular coagulation in rabbits induced by administration of endotoxin or tissue factor: effect of anti-tissue factor antibodies and measurement of plasma extrinsic pathway inhibitor activity. Blood 75: 1481–1489. [PubMed] [Google Scholar]
- 38. Gopalakrishnan R, Hedner U, Ghosh S, Nayak R, Allen TC, et al. (2010) Bio-distribution of pharmacologically administered rFVIIa. J Thromb Haemost 8: 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agerso H, Kristensen NR, Ostergaard H, Karpf DM, Hermit MB, et al. (2011) Clearance of rFVIIa and NN1731 after intravenous administration of Beagle dogs. Eur J Pharm Sci 42: 578–583. [DOI] [PubMed] [Google Scholar]
- 40. Petersen LC, Karpf DM, Agerso H, Hermit MB, Pelzer H, et al. (2011) Intravascular inhibition of factor VIIa and the analogue NN1731 by antithrombin. Br J Haematol 152: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petersen LC, Elm T, Ezban M, Krogh TN, Karpf DM, et al. (2009) Plasma elimination kinetics for factor VII are independent of its activation to factor VIIa and complex formation with plasma inhibitors. Thromb Haemost 101: 818–826. [PubMed] [Google Scholar]
- 42. Hamamoto T, Foster DC, Kisiel W (1996) The inhibition of human factor VIIa-tissue factor by antithrombin III-heparin is enhanced by factor X on a human bladder carcinoma cell line. Int J Hematol 63: 51–63. [DOI] [PubMed] [Google Scholar]
- 43. Gopalakrishnan R, Hedner U, Clark C, Pendurthi UR, Rao LV (2010) rFVIIa transported from the blood stream into tissues is functionally active. J Thromb Haemost 8: 2318–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All available data is included in the manuscript.