

Abstract
Sickle cell disease is a growing global health concern because infants born with the disorder in developing countries are now surviving longer with little access to diagnostic and management options. In Haiti, the current state of sickle cell disease/trait in the population is unclear. To inform future screening efforts in Haiti, we assayed sickle hemoglobin mutations using traditional hemoglobin solubility tests (HST) and add-on techniques, which incorporated spectrophotometry and insoluble hemoglobin separation. We also generated genotype data as a metric for HST performance. We found 19 of 202 individuals screened with HST were positive for sickle hemoglobin, five of whom did not carry the HbS allele. We show that spectrophotometry and insoluble hemoglobin separation add-on techniques could resolve false positives associated with the traditional HST approach, with some limitations. We also discuss the incorporation of insoluble hemoglobin separation observation with HST in suboptimal screening settings like Haiti.
Introduction
Sickle cell disease and sickle cell trait are common erythrocyte disorders that are most often caused by a point mutation (rs334, designated HbS) in the hemoglobin beta gene (HBB).1 This polymorphism causes hemoglobin to polymerize when deoxygenated, resulting in “sickle” shaped erythrocytes that adhere to the blood vessel walls. Individuals homozygous for the mutation (HbSS) have sickle cell disease and may experience chronic anemia, aplastic crisis, severe pain, cerebrovascular accidents, and splenic and renal dysfunction2; carrying only a single copy of the HBB rs334 polymorphism (HbAS) confers sickle cell trait, which is usually asymptomatic. Additional mutations in HBB can lead to sickle cell disease when coinherited with the HbS mutation, such as the hemoglobin C rs33930165 mutation (HbC).3,4 When properly diagnosed, sickle cell disease and its associated complications can be managed with proper hydration, pain killers, and penicillin prophylaxis to prevent infections.5
Sickle cell mutations are common in sub-Saharan Africans and descendant populations, where heterozygous individuals (HbAS) are protected against malaria.6,7 Previously, individuals born with sickle cell disease died within the first few years of life from infection.8 In recent decades, however, the life-expectancy for sickle cell disease individuals is increasing because of steady global advances in public health and access to nutrition.5,8,9 With more sickle cell infants surviving into childhood, sickle cell disease is now being considered an emerging global health concern, posing a challenge to low income countries that lack access to diagnostic tools, prophylaxis treatment (e.g., antibiotics), and additional treatments (i.e., hydroxyurea and blood transfusions).10 Therefore, it is crucial to identify best screening methods for detecting sickle cell hemoglobin in individuals to assess the status of sickle cell mutations in high risk populations.
Haiti, located in the Caribbean, is one of the poorest countries in the Western Hemisphere, with limited public health infrastructure. Given the high proportion of West African ancestry in Haiti, it is assumed that sickle cell disease/trait is prevalent in this country. However, there are limited data on the status of sickle cell disease/trait across the country. Three prior studies estimated the prevalence of sickle cell hemoglobin in Haitian populations to range from 8% to 15%.11–13 Specifically, the Pegelow and Mack study (1989) estimated the prevalence of HbS mutation at 8.0% in infants of Haitian immigrants in Miami using DNA genotyping methods.11 Randolph (2010) used a hemoglobin solubility test (HST) and estimated the prevalence of individuals with sickle hemoglobin (HBS) in northern Haiti at 15%.12 A recent study used high performance liquid chromatography to estimate the prevalence of HbS and HbC mutations to be around 11.3% and 2.6% in newborn infants, respectively13; this study is the first to use HST techniques and genotyping to detect sickle cell hemoglobin and sickle cell mutations to investigate best sickle cell screening practices in Haiti.
Screening for HBS can be accomplished with HSTs that are based on the principle of direct observation of turbidity, resulting from the insoluble HBS in the presence of phosphate or sulfate buffer solution. Several studies have explored modifications to the solubility test to suit a particular setting or purpose. For example, spectrophotometry has been explored as a way to quantify the resulting turbidity in the HST to predict the zygosity of the mutation, with notable success.14,15 Other studies were able to confirm the presence of sickle hemoglobin or predict the sickle cell mutation zygosity by characterizing the insoluble hemoglobin that aggregates on the surface of the HST solution after sitting several hours or after centrifugation.14,16,17 Here, we present the results of a sickle hemoglobin detection investigation conducted in Haiti between 2012 and 2013. Specifically, we identified HBS individuals using the traditional HST protocol (designated HSTtrad) and two add-on techniques that incorporate 1) spectrophotometry measurements (HSTspec) and 2) insoluble hemoglobin separation observation (HSTinsol), and we used genotype data as the gold standard to assess test performance. The results of our study are relevant to public health policy discussions in Haiti involving adequate and affordable sickle cell screening methods in Haiti.
Materials and Methods
Sample collection and processing.
This research was conducted as part of a larger investigation into malaria infection in Haiti. Samples were collected from patients, ranging from 2 to 80 years of age, visiting four health center sites in the West and Southeast Departments in Haiti: Blanchard Clinic in Terre Noire (BC), Hospital St. Croix in Leogane (HSC), a mobile clinic in Chabin (CN), and the Community Coalition for Haiti Clinic in Jacmel (JM). The BC and HSC consist of febrile patients, whereas CN and JM samples reflect the general clinic population. The CN sample likely includes more related individuals than the other sites, because mobile clinics usually serve a small local population. Additionally, parents often bring their entire families to visiting mobile clinics for general health check-ups when no other health facilities are available in the area or if it is uncertain when the mobile clinic will return.
Whole blood was collected by venipuncture for HST by heath center laboratory technicians or nurses. HSTs and HST add-ons were performed and evaluated by University of Florida researchers on site; researchers' evaluations were blinded to patient information. At BC and HSC, HSTs were conducted immediately after blood collection at the health center sites. At CN and JM, HSTs were conducted 2 weeks after blood was collected and stored in EDTA Vacutainer tubes at 4°C. Spectrophotometry readings of the HST samples were completed only for two collection sites, BC and HSC. The BC spectrophotometric analyses were conducted at the time of the HSTs and the HSC spectrophotometric analyses were conducted at the University of Florida Public Health Laboratory in Gressier, Haiti 3 to 6 hours after the initial HST. For all collection sites, whole blood was spotted on Whatman FTA cards (General Electric, Buckinghamshire, UK). For most cards, all four sample circles were filled for a total of 500 μL of blood. The FTA cards were transported to the University of Florida Genetics Institute (Gainesville, FL) for genotyping. The study was approved by the Haiti Ethical Review Board, University of Florida's IRB-01, and the Office of Research Protections, U.S. Army Medical Research and Materiel Command. Informed consent was obtained by physicians/health care workers on site from all adult participants and from the parents or legal guardians of minors.
Hemoglobin solubility test.
Two HST kits were used to screen individuals for HBS in this study. The SickleHEME kit (MichClone Associates, Inc, Madison Heights, WI) was used at HSC. The Pacific Hemostasis SickleScreen Kit (ThermoScientific, Middleton, WI) was used for the BC, CN, and JM samples. Assays were conducted according to the manufacturer's recommendations with the exception of the SickleHEME kit where 50 μL of blood was added to ∼4 mL HST solution instead of 20 μL blood and 2 mL solution. The different volumes of blood and buffer did not alter the assay (MichClone technical assistance, MichClone Associates), but were used to accommodate the measurement and setting limitations at HSC. The HST is based on the principle that HBS in its deoxygenated form is insoluble and will become turbid in concentrated phosphate buffer. The blood samples from the patients were compared with positive and negative controls from another HST kit (Sickle-STAT, Chembio Diagnostic Systems, Medford, WI). The level of turbidity was assessed against a white background with black lines such that the black lines were not visible in the HBS-positive samples. The HST only detects HBS and not any other abnormal hemoglobin because the test is based on the unique chemical reaction between HBS and concentrated phosphate.
Additions to hemoglobin solubility test protocol.
Spectrophotometry.
To determine whether spectrophotometry would be useful as a secondary confirmation of the HST, we collected spectrophotometric readings of the HST samples at two sites (BC and HSC) as a way of quantifying the HST turbidity results for further comparison with genotype data. A 1 mL aliquot of the blood-phosphate buffer mixtures from the HSTs was read at 540 nm in a Genesys 20 Model 4001/4 spectrophotometer (Thermo Scientific, West Palm Beach, FL). All readings were blanked to a HST-negative control. The same spectrophotometer was used at both sites.
Insoluble hemoglobin separation observation.
The HST samples were also left at room temperature for 5–8 hours. After that time, we noted which samples displayed separation of insoluble hemoglobin on the surface of the HST solution as previously described.14,16,17
Genotyping.
Genotyping was performed to use as a gold standard for comparisons of the HSTtrad protocol and the HSTspec and HSTinsol add-ons. Dried blood spot samples were collected for genotyping HbS, HbC, and hemoglobin E (HbE) mutations in the hemoglobin beta gene (HBB). Genomic DNA was prepared using two separate protocols. For BC, CN, and JM samples, 1.20 mm punches were collected and washed with FTA Purification Reagent (GE Healthcare, Buckinghamshire, UK) and water (3 washes each, 100 μL each). For HSC samples, genomic DNA was extracted using a QIAamp DNA Investigator Kit (Qiagen, Valencia, CA).
A portion of exons 1 and 2 of the HBB gene were amplified in a single reaction using the following primers: forward 5′ CCCCTTCCTATGACATGAACT′3 and reverse 5′ GGGTTTGAAGTCCAACTCCTA′3, as previously described.18 Promega Hot Start Kit (Promega, Madison, WI) was used with a final reaction volume of 25 μL and the following components: 1× Buffer, 1.5 mM MgCl2, 0.2nM each dNTP, 0.5 μM each primer, 2.5 U Taq, and 2 μL DNA extract or FTA punch. The temperature cycling protocol was as follows: 95°C for 1 min, and then 40 cycles of 95°C for 10 sec, 60.6°C for 30 sec, 72°C for 2 min followed by a final extension at 72°C for 7 min. The size of the amplicon was 744 base pairs (bp). Amplicons were sequenced at the University of Florida's Interdisciplinary Center for Biotechnology Research DNA Sequencing Core Laboratory with BigDye chemistry on an Applied Biosystems 3730 Genetic Analyzer, using the forward and reverse polymerase chain reaction (PCR) primers. Sequence data were aligned to the HBB reference sequence (GenBank; J00173) and scanned for mutations using Sequencher 4.10.1. (Gene Codes Corp., Ann Arbor, MI). The HbS mutation rs334 (glu6val), HbC mutations rs33930165 (glu6gln), rs33991472 (pro59his), rs33945705 (asp74asn) or (asp74tyr), and HbE mutation rs33950507 (glu26lys) were analyzed.
Statistical analysis.
We calculated the frequency and 95% confidence intervals (CIs) of HSTtrad and HSTinsol-positive and negative individuals, and the frequencies of each HBB mutation (i.e., HbS, HbC). For spectrophotometry, we calculated the overall mean and 95% CIs for all sites, within each site, and by HbS genotypes. We also compared spectrophotometry data across the sample site and HbS genotype using the Mann–Whitney Wilcoxon Test. To determine a cutoff for HbS group versus non-HbS group for HSTspec, we pooled the data from HSC and BC (the only two sites with HSTspec data) and performed bootstrap resampling of 80% of the pooled data (100,000 repetitions). For each bootstrap sample, a potential cutoff was determined as the midpoint/average of the highest HbS and lowest non-HbS spectrophotometry value (no overlap was observed for the HbS and non-HbS groups). The cutoffs for each resampling were averaged to create a final cutoff, which was then applied to the entire data set.
To assess the performance of each HBS detection method, we calculated the accuracy, sensitivity, and specificity of each method in comparison with the HbS genotype data. All statistical analyses were completed using SAS 9.2 (SAS Institute Inc., Cary, NC).
Results
Sample set.
A total of 212 individuals were included in the study. Table 1 lists the sample sizes for each collection site and data type. Out of the 212 individuals in the study, genotype and HST data were generated for 194 and 202 individuals, respectively. In addition, we obtained HSTspec data for 87 individuals at two collection sites, BC and HSC, and HSTinsol data for 156 individuals from HSC, CH, and JM.
Table 1.
Sample sizes for each collection site and data type*
| Collection site | Total | Genotype | HST | Spectrophotometry | Insoluble hemoglobin separation |
|---|---|---|---|---|---|
| Blanchard Clinic | 36 | 35 | 36 | 34 | − |
| Hospital St. Croix | 57 | 55 | 55 | 53 | 53 |
| Mobile Clinic - Chabin | 41 | 37 | 34 | − | 32 |
| CCH Clinic in Jacmel | 78 | 67 | 77 | − | 71 |
| Total | 212 | 194 | 202 | 87 | 156 |
HST = hemoglobin solubility test.
Data from hemoglobin solubility tests and add-on methods.
Of the 202 individuals screened for HBS using the HST kit, 19 (9.4%; 95% CI = 6.1–14.2) were positive and 183 (90.6%; 95% CI = 85.8–93.9%) were negative. By collection site, we observed 3 (8.3%; 95% CI = 2.9–21.8), 8 (14.6%; 95% CI = 7.6–26.2%), 2 (5.9%; 95% CI = 1.6–19.1%), and 6 (7.8%; 95% CI = 3.6–16.0%) HST-positive samples from BC, HSC, CN, and JM, respectively. Figure 1 shows a selection of the positive and negative HST results.
Figure 1.

A representative figure of the traditional hemoglobin solubility test (HST) with the blood samples in the phosphate buffer solution in transparent glass vials to allow the observation of turbidity that forms when sickle hemoglobin is present. Positive and negative samples are denoted by “+” and “−”, respectively.
The overall mean spectrophotometric value was 0.43 (95% CI = 0.37–0.48). The mean spectrophotometric values for BC and HSC samples were 0.58 (95% CI = 0.50– 0.67) and 0.32 (95% CI = 0.24–0.40), respectively. The distributions of spectrophotometric values were significantly different (P value < 0.001) between the two collection sites based on a Mann–Whitney Wilcoxon test.
We recorded the presence of insoluble hemoglobin formation at the top of the solution after 5–8 hours. Eleven out of 156 (7.1%, 95% CI = 3.8–12.6%) samples displayed this feature, three from HSC, two from CH, five from JM. Figure 2 gives an example of the separation of insoluble hemoglobin we observed in these samples.
Figure 2.
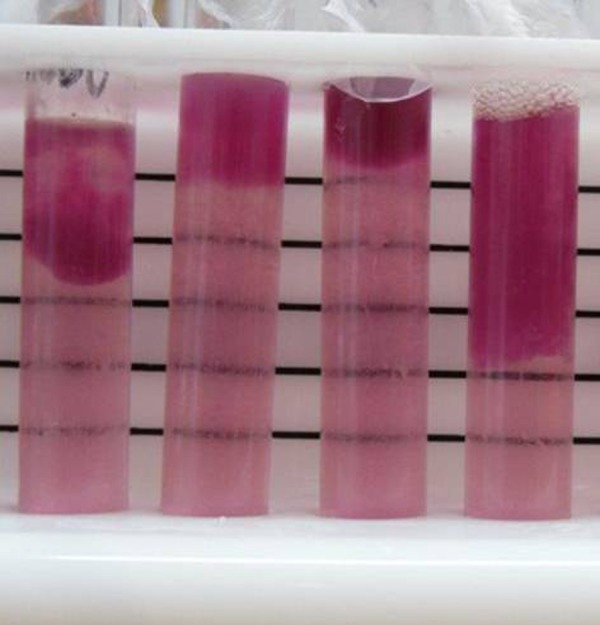
Examples of separation of insoluble hemoglobin in blood-phosphate buffer solution. Pictures were taken after spectrophotometry readings, which required a portion of the solution, and resulted in the variable volume of solution observed in this figure.
Genotype data.
Of the 194 samples that were genotyped for HBB mutations, one (0.5%, 95% CI = 0.0–2.9%) was homozygous for the HbS rs334 mutation and 14 (7.2%; 95% CI = 4.4–11.8%) were heterozygous for this mutation. One hundred seventy-nine samples (92.3%; 95% CI = 87.6–95.3%) did not carry the mutation. For the HbC rs33930165 mutation, one individual was homozygous for the mutation, 12 (6.2%; 3.4–10.8%) were heterozygous, and 181 (93.3%; 95% CI = 0.9–96.2%) did not carry the mutation. No individuals carried both the HbS and HbC mutations. No samples carried HbC mutations at rs33991472 or rs33945705, or the HbE mutation at rs33950507. Table 2 shows the frequency of the HBB genotypes for each collection site.
Table 2.
HBB allele frequencies for each collection site
| Collection site | n | HbAA | HbAS | HbSS | HbAC | HbCC |
|---|---|---|---|---|---|---|
| Blanchard Clinic | 35 | 30 (85.7%) | 3 (8.6%) | 0 (0.0%) | 1 (2.9%) | 1 (2.9%) |
| Hospital St. Croix | 55 | 45 (81.8%) | 4 (7.3%) | 0 (0.0%) | 6 (10.9%) | 0 (0.0%) |
| Mobile Clinic in Chabin | 37 | 35 (94.6%) | 2 (5.4%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| CCH Clinic in Jacmel | 67 | 56 (83.6%) | 5 (7.5%) | 1 (1.5%) | 5 (7.5%) | 0 (0.0%) |
| Total | 194 | 166 (85.6%) | 14 (7.2%) | 1 (0.5%) | 12 (6.2%) | 1 (0.5%) |
Comparing HST traditional and HST add-on data with genotype data.
The sensitivity and specificity for HSTtrad, HSTspec, and HSTinsol as compared with the HbS genotypic data are listed in Table 3.
Table 3.
Measures of performance of the traditional HST and HST add-on techniques*
| N | Accuracy | Sensitivity | Specificity | |
|---|---|---|---|---|
| HSTtrad | 184 | 96.7 | 92.9 | 97.1 |
| HSTspec | 87 | 100.0 | 100.0 | 100.0 |
| HSTinsol | 139 | 99.3 | 90.0 | 100.0 |
Genotype data were used as a comparison metric.
HST = hemoglobin solubility test.
HSTtrad.
Table 4 includes a comparison of the HbS genotype groups and HSTtrad results for the 184 individuals with both genotype and HST data. All of the samples carrying at least one HbS allele, i.e., HbAS and HbSS, were also positive according to the HSTtrad, with one exception (see Table 4, JM site). Of the 170 non-HbS individuals, five were HSTtrad-positive and 165 were HSTtrad-negative. In other words, the entire HbS group except one individual was positive for the HST, as expected, but five of the non-HbS group were also HSTtrad-positive.
Table 4.
Comparison of traditional hemoglobin solubility test results to HbS genotype group (based on HBB rs334 mutation)*
| HbS status | Traditional hemoglobin solubility test | |||
|---|---|---|---|---|
| Negative | Positive | Total | ||
| Blanchard Clinic | ||||
| Non-HbS | 32 | 0 | 32 | |
| HbS (heterozygote) | 0 | 3 | 3 | |
| Total | 32 | 3 | 35 | |
| Hospital St. Croix | ||||
| Non-HbS | 35 | 5 | 50 | |
| HbS (heterozygote) | 0 | 3 | 3 | |
| Total | 35 | 8 | 53 | |
| Mobile Clinic in Chabin | ||||
| Non-HbS | 28 | 0 | 28 | |
| HbS (heterozygote) | 0 | 2 | 2 | |
| Total | 28 | 2 | 30 | |
| CCH Clinic in Jacmel | ||||
| Non-HbS | 60 | 0 | 60 | |
| HbS (heterozygote) | 1 | 4 | 5 | |
| HbS (homozygote) | 0 | 1 | 1 | |
| Total | 61 | 5 | 66 | |
| All Collection Sites | ||||
| Non-HbS | 165 | 5 | 170 | |
| HbS (heterozygote) | 1 | 12 | 13 | |
| HbS (homozygote) | 0 | 1 | 1 | |
| Total | 166 | 18 | 184 | |
Table includes only samples with both genotype and hemoglobin solubility test (HST) data.
HbS = sickle cell hemoglobin.
HSTspec.
The mean spectrophotometry values and corresponding 95% CIs for HbS and non-HbS groups are listed in Table 5. To determine whether there was a measurable difference in the HST solution turbidity for the HbS group (HbAS, HbSS) and non-HbS group (i.e., HbAA, HbAC, HbCC), we compared the HbAS (no HbSS individuals were identified in BC or HSC samples) and non-HbS groups' spectrophotometric values using the Mann–Whitney Wilcoxon test. A significant difference was observed for both sites BC (P value < 0.001) and HSC (P value < 0.001). Figure 3 shows the distribution of spectrophotometric values across HST results and HbS genotypes.
Table 5.
Spectrophotometric values of hemoglobin solubility test solutions across HbS genotype group (based on HBB rs334 mutation) at two collection sites: Hospital St. Croix and Blanchard Clinic*
| HbS genotype group | Blanchard Clinic | Hospital St. Croix | ||||
|---|---|---|---|---|---|---|
| n | Mean | Range | n | Mean | Range | |
| Non-HbS | 32 | 0.52 | 0.210–0.780 | 50 | 0.28 | 0.00–0.626 |
| HbS | 3 | (0.34–0.70) | 0.827–1.637 | 3 | (0.24–0.33) | 0.790–1.180 |
| 1.26 | 0.940 | |||||
| (0.80–1.72) | (0.37–1.31) | |||||
These two sites were the only sites at which genotype and spectrophotometry data were collected. The 95% confidence intervals are shown in parentheses.
HbS = sickle cell hemoglobin.
Figure 3.
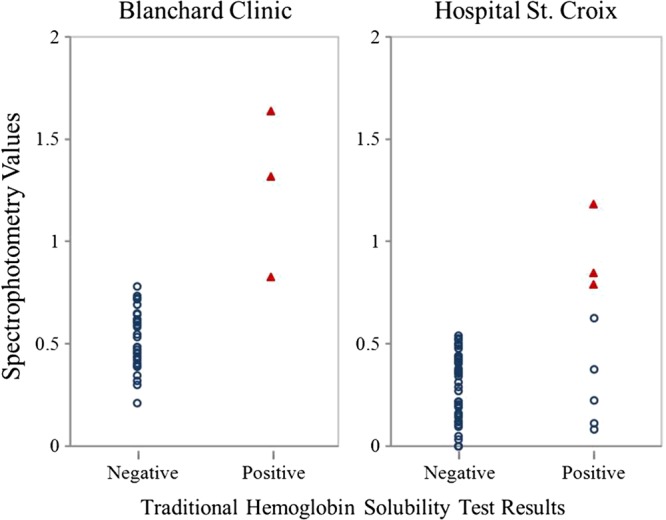
Distribution of spectrophotometric values across hemoglobin solubility test (HST) values by genotype results. Red triangles and open blue circles represent the sickle cell hemoglobin (HbS) and non-HbS group, respectively.
We compared the spectrophotometry and genotype results for BC and HSC and found no overlapping spectrophotometry values between HbS and non-HbS groups. The bootstrap resampling procedure produced a final cutoff of 0.785. After applying this cutoff to the entire data set, we observed that six individuals out of 87 individuals were HSTspec-positive. All HSTspec-positive individuals carried the HbS mutation; no HSTspec-negative individuals carried the HbS mutation. Specifically, the discordant individuals who were HSTtrad-positive but non-HbS, were HSTspec-negative.
HSTinsol.
Nine of the 11 samples with insoluble hemoglobin separation also had genotype data and those nine samples carried the HbS genotype as either HbAS or HbSS. The five HSTtrad-positive/non-HbS individuals were negative according to the HSTinsol protocol. There was one sample that was negative according to the HSTinsol protocol but did carry the HbS mutation (this sample was also negative with the HSTtrad protocol).
Discussion
In this study, we compared the performance of the traditional HST protocol and two add-ons to the traditional HST protocol for sickle hemoglobin detection in comparison to DNA genotyping for sickle cell alleles at the HBB gene. We found that the traditional HST protocol (HSTtrad) was, for the most part, able to correctly identify HbS versus non-HbS individuals, resulting in a high specificity (Table 3). There were six individuals who were wrongly classified for sickle hemoglobin according to the HSTtrad. Five of the six individuals, all from HSC, were positive based on HSTtrad but were negative by genotyping (i.e., they did not carry the HbS rs334 mutation). Several factors could have contributed to the disagreement between HbS genotype and HSTtrad results, including the warm and humid setting in which the HST samples were processed at HSC. From the patient perspective, it is possible that these individuals may have been dehydrated, which causes thickening of the hematocrit and can be mistaken for turbidity. In addition, several HST kit instructions warn that results could be inconclusive in patients with very low hemoglobin levels. Our results show no significant difference in hemoglobin levels between false HSTtrad-positives and negative samples (data not shown), suggesting that variable hemoglobin levels are not responsible for the five non-HbS individuals with HSTtrad-positive results. It is worth noting that a higher ratio of blood to phosphate buffer was used in the HST assays at HSC and this could potentially impact assay results, although manufacturer's technical representatives said the altered ratio should not matter. The other conflicting sample, collected from JM, was HSTtrad-negative/HSTinsol-negative but carried the HbS mutation. At the time of assay, it was noted that the HST sample for this individual was more turbid relative to other HSTtrad-negative samples. However, because some black lines were faintly visible at the bottom of the tube, this individuals was ultimately deemed HSTtrad-negative. Additional studies should be completed to determine what factors could have contributed to incomplete turbidity.
We also examined the performance of two HST add-on protocols, HSTspec and HSTinsol protocols. For HSTspec, we observed a significant difference between the spectrophotometry values of the HbS and non-HbS genotypes and non-overlapping spectrophotometric ranges for the genotypes, which supports previous investigations reporting spectrophotometry's usefulness in conjunction with the standard HST protocol. In addition, HSTspec values correctly identified the false HSTtrad-positives as sickle cell negative (in agreement with their non-HbS genotype). However, the spectrophotometric ranges for sickle cell positive and negative samples differed slightly by study site and the gap between positive and negative samples was very narrow when both sites were pooled, suggesting it may be difficult to establish a standard spectrophotometric cutoff for HBS detection if more samples and more sites are included. Furthermore, from a practical perspective, spectrophotometers are not readily available in many health centers in Haiti (we provided our own spectrophotometer for this study), suggesting that the HSTspec protocol may not be a feasible option in resource-limited settings.
The HSTinsol add-on to the HST may be a simple and inexpensive secondary detection technique. We observed a direct correlation between HbS genotype and HSTinsol results, i.e., insoluble hemoglobin only accumulated in the HbS group, suggesting that this observation could be a beneficial add-on to the HST protocol as a secondary detection of HBS in samples. Not only would this addition to the protocol improve the accuracy of the HST, but it is inexpensive, simple, and does not require any additional equipment or reagents, which are primary considerations in regions with limited resources.
Building on our comparisons of HST methods with genotype data, we can use our genotype data to provide the best current estimate of sickle cell allele frequencies in Haiti. Specifically, the frequencies of individuals carrying the HbS rs334 and HbC rs33930165 mutations are 7.2% and 6.9%, respectively. The HbS frequency estimate is similar to what was previously reported by Pegelow and Mack (1989)11 in their study on Haitian immigrants in Miami (8.0%), confirming that sickle cell mutations are prevalent in Haiti. Interestingly, in our study, only one individual (an infant) out of 194 genotyped had sickle cell disease (HbSS), which may suggest that many infants born with sickle cell disease are not surviving into adulthood in Haiti. Further studies in a larger sample are needed to determine the frequency of sickle cell disease in Haiti's general population to assess the survival rate of infants with sickle cell disease. Furthermore, in our study, we found the frequency of the HbC rs33930165 mutation was very close to the frequency of the HbS rs334 mutation. Given that the HbC rs33930165 mutation also leads to sickle cell disease when inherited with a single HbS mutation (for review, see Nagel RL, Fabry ME, and Steinberg NH)19 and the HST only identifies HbS mutations, future sickle cell disease/trait screening programs in Haiti should incorporate an additional method for detecting HbC mutations. Additionally, future studies in Haiti should investigate the frequency of other hemoglobin mutations, such as alpha and beta thalassemia mutations, that can lead to sickle cell disease when co-inherited with the HbS mutation.
In conclusion, we found that the HST protocol, though mostly accurate in comparison with DNA genotype results, could be improved by incorporating the insoluble hemoglobin separation observation. Incorporation of the insoluble hemoglobin add-on may be particularly useful in sickle cell screenings in resource-limited settings like Haiti as it does not require additional equipment or overly complicated analysis. Finally, we found that 14.4% of the individuals in our samples from Haiti carry sickle cell associated hemoglobin mutations (HbS rs334 or HbC rs33930165), emphasizing the need for wider screening for sickle cell associated mutations across Haiti. Given the frequency of HbC rs33930165 mutation in Haiti, we encourage exploration of possible HbC screening methods, in combination with the future sickle cell disease/trait screening efforts in Haiti. Our results add to the limited knowledge on sickle cell disease/trait in Haiti and contribute to the discussion on best screening practices for Haiti.
ACKNOWLEDGMENTS
We thank the following organizations and people for their support: Ministry of Sanitation and Public Practice (MSPP), Government of Haiti, Christianville Foundation in Gressier Haiti, Community Coalition for Haiti, Fish Ministries Haiti, Family Health Ministries, University of Florida, the U.S. Department of Defense, and Hospital St. Croix staff including Carol, Farrah, and Gillian for their help. We also thank Aida Miro, Thomas A. Weppelmann, Taifur Shovon, Halley Maloy, and Erin Leone for their laboratory assistance and/or productive discussions. Finally, we would like to thank all of the participants who made this research possible.
Disclaimer: The authors declare that they have no competing interests.
Footnotes
Financial Support: This material is based upon work supported by the Department of Defense Global Emerging Infections Surveillance & Response System (DoD-GEIS) grant no. C0607_12_UN awarded to BAO and a National Science Foundation Graduate Research Fellowship under grant no. DGE-0802270 awarded to TEC.
Authors' addresses: Tamar E. Carter and Connie J. Mulligan, University of Florida, Genetics Institute, Gainesville, FL, E-mails: tamarec@ufl.edu and cmulligan@ad.ufl.edu. Michael von Fricken and Bernard A. Okech, University of Florida, Emerging Pathogens Institute, Gainesville, FL, E-mails: MichaelVonfricken@epi.ufl.edu and bokech@ufl.edu. Jean R. Romain and Gladys Memnon, Hospital St. Croix, Leogane, Haiti, E-mails: roosevald2008@yahoo.fr and gmemnon@gmail.com. Yves St. Victor, Blanchard Clinic, Family Health Ministries, Terre Noire, Haiti, E-mail: jeanyvessaintvictor@yahoo.fr. Laura Schick, Community Coalition for Haiti, Jacmel Haiti, E-mail: laura@cchaiti.org.
References
- 1.Ingram VM. Gene mutations in human hemoglobin: the chemical difference between normal and sickle cell hemoglobin. Nature. 1957;180:326–328. doi: 10.1038/180326a0. [DOI] [PubMed] [Google Scholar]
- 2.Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151:839–845. doi: 10.1093/oxfordjournals.aje.a010288. [DOI] [PubMed] [Google Scholar]
- 3.Itano HA, Neel JV. A new inherited abnormality of human hemoglobin. Proc Natl Acad Sci USA. 1950;36:613–617. doi: 10.1073/pnas.36.11.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunt JA, Ingram VM. Abnormal human hemoglobins. IV. The chemical difference between normal human hemoglobin and hemoglobin C. Biochim Biophys Acta. 1960;42:409–421. doi: 10.1016/0006-3002(60)90818-0. [DOI] [PubMed] [Google Scholar]
- 5.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- 6.Allison AC. Protection afforded by sickle-cell trait against subtertian malareal infection. BMJ. 1954;1:290–294. doi: 10.1136/bmj.1.4857.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, Nahlen BL, Lal AA, Udhayakumar V. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet. 2002;359:1311–1312. doi: 10.1016/S0140-6736(02)08273-9. [DOI] [PubMed] [Google Scholar]
- 8.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–4336. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weatherall DJ, Clegg JB. Inherited hemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- 10.Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anemia in children under five, 2010–2050: modeling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10:e1001484. doi: 10.1371/journal.pmed.1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pegelow CH, Mack AK. Incidence of hemoglobins S and C in infants born in Miami to recent Haitian immigrants. Trop Geogr Med. 1989;41:316–319. [PubMed] [Google Scholar]
- 12.Randolph TR. Estimated prevalence of sickle cell in northern Haiti. Clin Lab Sci. 2010;23:79–83. [PubMed] [Google Scholar]
- 13.Rotz S, Arty G, Dall'amico R, De Zen L, Zanolli F, Bodas P. Prevalence of sickle cell disease, hemoglobin S, and hemoglobin C among Haitian newborns. Am J Hematol. 2013;88:827–828. doi: 10.1002/ajh.23510. [DOI] [PubMed] [Google Scholar]
- 14.Bowie LJ, Dohnal JC. Proportion of hemoglobin S in blood, as determined from solubility measurements. Clin Chem. 1983;29:325–328. [PubMed] [Google Scholar]
- 15.Randolph TR, Wheelhouse J. Novel test method (sickle confirm) to differentiate sickle cell anemia from sickle cell trait for potential use in developing countries. Clin Lab Sci. 2012;25:26–34. [PubMed] [Google Scholar]
- 16.Huntsman RG, Barclay GP, Canning DM, Yawson GI. A rapid whole blood solubility test to differentiate the sickle-cell trait from sickle-cell anemia. J Clin Pathol. 1970;23:781–783. doi: 10.1136/jcp.23.9.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serjeant BE, Serjeant GR. A whole blood solubility and centrifugation test for sickle cell hemoglobin: a clinical trial. Am J Clin Pathol. 1972;58:11–13. doi: 10.1093/ajcp/58.1.11. [DOI] [PubMed] [Google Scholar]
- 18.Waterfall CM, Cobb BD. Single tube genotyping of sickle cell anemia using PCR-based SNP analysis. Nucleic Acids Res. 2001;29:E119. doi: 10.1093/nar/29.23.e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev. 2003;17:167–178. doi: 10.1016/s0268-960x(03)00003-1. [DOI] [PubMed] [Google Scholar]