

Abstract
The preparation of typically thermodynamically unstable polymorphic structures is a challenge. However, solid surfaces are well established aids for the formation and stabilization of polymorphic structures within, for instance, organic electronics. In this study, we report the stabilization of a pharmaceutically relevant substance via a solid surface at ambient conditions. Form III of paracetamol, which is typically unstable in the bulk at standard conditions, can be stabilized with a model silica surface by a standard spin coating procedure followed by rapid heat treatment. Such a preparation technique allows the use of atomic force microscopy and grazing incidence X-ray diffraction measurements revealing detailed information on the morphology and structure of the polymorph. Furthermore, the results exhibit that this polymorph is stable over a long period of time revealing surface mediated stabilization. These findings demonstrate a novel approach to provide thermodynamic stability when applied to similar molecules with specific applications.
Short abstract
The stabilization of typically unstable polymorphs via a solid surface at ambient conditions is demonstrated using paracetamol and a model silica surface by utilizing a standard spin coating procedure followed by rapid heat treatment. The so-prepared films are analyzed in terms of their crystal structure, preferential alignment with respect to the surface, and crystal morphology.
The controlled preparation of polymorphic structures and different crystal morphologies has become a hot topic in several research areas such as pharmaceutical technology,1,2 crystal engineering,3,4 organic electronics5 as well as in material science.6 Various polymorphs of a single substance differ by means of their physical and chemical properties such as solubility, bioavailability, morphology, crystal structure, or even thermodynamic stability.7,8 Paracetamol, also known as acetaminophen (N-(4-hydroxyphenyl)acetamide), is widely used due to its antipyretic (fever depressant) and analgesic (painkiller) properties and is nowadays produced by many pharmaceutical companies at large scale.9 The commercially utilized polymorph form I has a monoclinic crystal structure, which is not suitable for direct compression into tablets due to the lack of slipping planes which are necessary for plastic deformation.10 In contrast, the orthorhombic paracetamol form II has well-defined slipping planes and undergoes plastic deformation upon compaction.10 The crystallization behavior was studied intensively over the last decades, but nevertheless the elusive form III was recently characterized experimentally by means of its crystal structure.11,12 A few procedures are known which describe the isolation and characterization of the thermodynamic less stable polymorph, e.g., nanoconfined in monoliths13,14 or confined between glass slides15 under the exclusion of air. It was shown that the confinement between glass slides is not necessary to isolate form III,16 but the handling of the elusive polymorph in an air environment at standard conditions is still nowadays a challenge. These confined preparation conditions however lack the ability to use techniques such as atomic force microscopy (AFM) or grazing incidence X-ray diffraction (GIXD) to get further information on the morphology and structures within layers as thin as a couple of nanometers. It is known that surfaces are able to induce specific polymorphs upon deposition.5,17,18 Even the occurrence of surface mediated phases can be observed with distinct properties from the bulk.5
In this work, we describe different approaches to prepare paracetamol thin films containing defined polymorphs at a solid model surface. For this purpose, paracetamol was dissolved in ethanol (EtOH) or tetrahydrofuran (THF) and spin coated on precleaned thermally oxidized silicon wafers. The usage of silicon wafers as model substrates allow exclusion of roughness induced effects on the crystallization behavior. Both as-prepared samples result in completely amorphous films, followed by different crystallization kinetics and crystal morphologies. While the EtOH spin coated samples crystallized in a time frame of 24 h, the THF prepared samples revealed spherulitic crystallization induction after approximately 5 min. Compared to the literature,16 the crystallization behavior in a confined environment is prolonged compared to crystallization at ambient conditions as it was done in this study (298 K, relative humidity 30%, 1 atm and under air). To prepare the elusive and thermodynamically unstable form III, a fresh and thus amorphous film was prepared from THF and was put in an oven at 383 K for 10 min without the exclusion of air. To provide optimal thermal contact, the oven was equilibrated whereby the fresh spun wafers were put on a heat-equilibrated alumina plate. The thermal conductivity of the used silicon wafer ensures fast thermal transfer, whereby the heat flow gradient is directed from the substrate toward the amorphous film. Variation in the preparation conditions showed that the rapid temperature increase was necessary to obtain form III. This allows the assumption that the crystallization in the oven occurs at the hot solid substrate further allowing the thermodynamic unstable polymorph to be entrapped and stabilized at the silica substrate even without the exclusion of air. It is well-known in the literature that form III is unstable at ambient conditions under air and interconverts into form II and I.16
GIXD measurements of the three samples are shown in Figure 1. The GIXD measurement in general allows netplanes which are close to the surface-normal to be detected within thick films19 (up to hundreds of nanometers for organic layers) as well as within thin films consisting of a monolayer.20 High intensity spots correspond to Bragg reflections which can be used to index the pattern and thus to identify their crystal structures. The measurements of the sample containing form I reveal ring-like Bragg reflections showing that the crystallites arrange like a random oriented powder; i.e., no preferred orientation is observed. The indexation shows that all rings are a result of paracetamol being in the thermodynamic stable polymorph form I with a monoclinic unit cell (Figure 2). The GIXD pattern of form II shows defined spots at qz = 0.0, 0.8, 1.7 nm–1 and various qxy. The indexation reveals that these spots are a result of paracetamol being in form II conformation with an orthorhombic unit cell. In addition, the crystallites of form II show a preferred orientation with respect to the surface, whereby the 001 plane is in contact with the surface. However, rings are also present within the pattern indicating random oriented form I domains. This shows that the sample contains two polymorphs simultaneously, even if the amount of form I is very low compared to form II. The form III sample reveals a GIXD pattern with spots being distinct from the previously observed ones. The indexation of the spots can be achieved by introducing a 021 contact plane with respect to the surface with an orthorhombic unit cell (compare Figure 2). The smearing of the Bragg spots shows that the mosaicity of the crystalline needles is relatively high. This means that the molecules have a certain degree of freedom to assemble at the silica surface which is different compared to form II with a nearly perfect alignment.
Figure 1.

Experimental grazing incidence X-ray diffraction (GIXD; wavelength λ = 0.9998 Å; incidence angle αi = 0.13°) patterns of all three polymorphs of paracetamol stabilized on SiOx (upper row) with the corresponding theoretical indexation (lower row) with form I having a powder character and form II and form III having a 001 or 021, respectively, contact plane.
Figure 2.
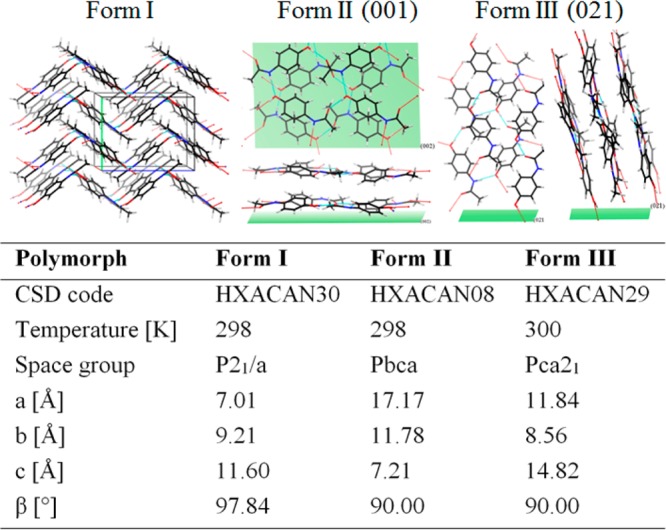
Visualization of the molecular arrangement within the crystal structures of paracetamol form I,21 II,21 and III11 together with the contact plane with respect of the SiOx surface. The table below summarizes the used crystal lattice parameters with their corresponding CSD code, space group, and corresponding temperature at which the experiments were performed.
In Figure 2, the visualization of the three different crystal structures of the surface stabilized polymorphs is shown with the corresponding crystal structure parameters, which were used for indexation. Form I is shown in its typical herringbone packing of the paracetamol sheets, whereby the delocalized π-orbitals of the phenyl units are oriented toward the methyl groups of the next molecule. The intersheet assembling of the molecules is a result of H-bonding, while the intrasheet connection (sheet–sheet stacking) completely lacks any of those. Form I is visualized with its monoclinic unit cell with no preferential alignment with respect to the surface due to its random orientation. The herringbone structure also is responsible for the mechanical stability and the disability for compaction required in tablet preparation.
In Figure 3 the morphologies of the three samples containing each polymorph of paracetamol at room temperature are shown with their corresponding height profiles. The AFM image of form I reveals the typical shape of monoclinic crystals with prismatic to plate-like morphology, whereby the crystallites are randomly rotated with respect to each other. Form II shows a distinct growth morphology, and plate-like structures are present. While the crystal morphologies are distinct, the height and consequently the coverage are very similar to form I and II, showing that diffusion in the upward direction from the surface is negligible. These two observed morphologies fit very well to previously observed experiments on paracetamol, where morphological differences are explained by different growth faces being dominant.22 The morphology of form III is distinct from the previous two forms, and needle-like structures are present. The coverage of the surface is strongly reduced, and consequently the heights of the needle-islands are higher compared to those of form I and II even though the nominal film thickness of the spin coated sample was very similar. The height of the needles is around 450 nm, which is about three times larger compared to the previous observed heights of form I and II.
Figure 3.
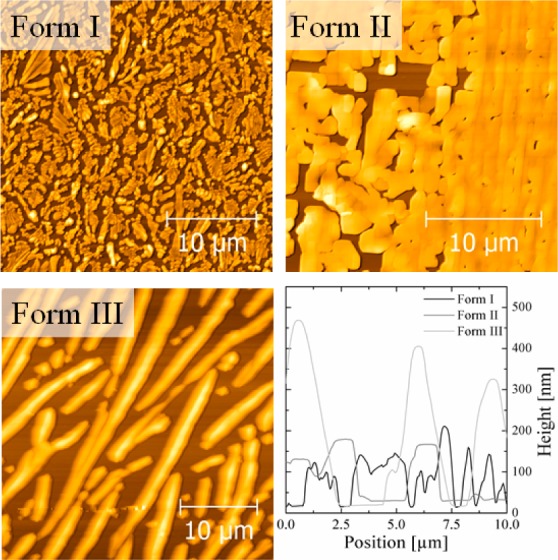
Atomic force microscopy height images of paracetamol form I, II, and III and the corresponding horizontal texture cuts taken in the middle of each image (lower right corner).
Differences in the crystal structure and alignment are also reflected within their morphologies. The structure of form II is shown in a 001 orientation (Figure 2, green plane) which is parallel to the silica surface. This visualization reveals that the paracetamol molecules arrange in a way that maximizes their contact areas with the silicon surface. Further, the delocalized π-orbitals are slightly tilted toward the surface, but due to the large contact area, also the polar groups are in the highest possible contact to interact with the semipolar silica. Again intersheet H-bonds are visible, while intrasheet bonds of the hereby flatly arranged paracetamol sheets are not present (slipping plane of form II). According to the literature, form II exhibits a plate-like growth whereby the slowest growing crystal face is the 001, which is in excellent agreement with our observations.22 These findings indicate that growing into the a- and b-axis direction of the orthorhombic unit cell is equally likely and more favorable compared to the stacking into the c-axis direction. The arrangement of paracetamol molecules within the orthorhombic form III unit cell with respect to the 021 direction indicates that the molecules form hydrogen bonds toward the silicon surface and that the molecules stand nearly perpendicular to the substrate (Figure 2). Again the intersheet connection exhibits H-bonds, while the intrasheet stacking lacks any of those. The upright alignment of the form III sheets is in very good agreement with the observed needle-like morphology which was measured with AFM (Figure 3), whereby the paracetamol sheets seem to grow into the c-axis of the orthorhombic unit cell, or the molecules are perpendicular to the long needle axis. Further, the stability and reproducibility of the surface mediated stabilization of all three polymorphs of paracetamol was investigated. During the course of the synchrotron experiment, fresh prepared samples as well as 4-week-old samples were characterized revealing the same diffraction patterns. The samples were stored at ambient conditions (25 °C, relative humidity ∼30%, 1 atm and under air environment), which reflects the potential of surface mediated stabilization. In addition, AFM investigations of the older samples did not reveal any significant change in the morphology, indicating that there is no rearrangement upon storage.
The surface mediated polymorph stabilization which was studied within this work using paracetamol is in excellent agreement with Ostwald’s step rule which states that the least thermodynamic stable polymorph crystallizes first.16 In particular, this means that a system moves to a thermodynamic equilibrium from an initial high energy state, whereby the least stable polymorph crystallizes first and rearranges stepwise into the different polymorphs (form III > form II > form I) due to changes within the free energy. Further, the different preparation conditions and the preferential alignment of the paracetamol molecules with respect to the surface indicate that the intensive (e.g., temperature, viscosity, chemical potential, density, etc.) and extensive (e.g., Gibbs free energy, entropy, mass, number of molecules, etc.) parameters of the solvent, analyte, and substrate in use are of crucial importance during surface mediated stabilization. EtOH exhibits a relatively low vapor pressure compared to THF. This suggests that EtOH solvent residues remain within the amorphous film, whereby molecule diffusion promotes the rearrangement in a thermodynamic stable configuration even if the viscosity of EtOH is about 2 times higher. Contrarily, the vapor pressure of THF is about 3 times larger compared to EtOH at standard conditions, meaning that THF molecules evaporate fast allowing the thermodynamic metastable polymorph II to be stabilized at the silica surface at 298 K. A shorter time frame typically means that formation of a thermodynamically less stable form is favored in accordance with other literature reports.23,24 The unstable forms II and III most likely develop due to the fast processing condition within the THF solutions. A rapid temperature increase to 383 K of an amorphous paracetamol film spin coated from THF results in polymorph III being stabilized at the silica surface. An increase in temperature typically reduces the H-bonding interaction strength which reduces the affinity for the molecules to interact with the substrate. Furthermore, higher temperatures mean that the molecules require more space on account of molecular vibration. Both effects favor the formation of upright standing molecules on top of the substrate surface which in the case of paracetamol means additionally assembling into form III.
By comparing the observed alignment of paracetamol form III with different literature statements, it can be seen that the orientation of form III crystallites is highly influenced by the surface in use even if different preparation methods indicate similar growth kinetics of form III. In a recent publication,25 the authors state that form III, when confined in self-ordered anodic aluminum oxide nano tubes (AAO), preferentially crystallizes with its 001 plane being parallel to the AAO interface. This alignment indicates that no H-bonds are formed between the AAO host and the paracetamol molecules. Contrarily, our investigations strongly indicate the formation of H-bonds between the semipolar silica substrate and the paracetamol molecules with the 021 reflection plane being parallel to the surface. The differences in the preparation method aside, this direct comparison indicates the strong influence of the surface energetic properties of the surfaces in use; i.e., variation of the surface induces a distinct growth behavior. A previous work26 suggested that along the a-axis of paracetamol only short-range order is present, which also corresponds to the slowest growth direction in the calculated morphology of form III.22 This is in excellent agreement with the observed morphology of paracetamol form III revealing preferential growth in the needle axis of the c-direction of the orthorhombic unit cell.
Acknowledgments
We thank the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation at the beamline BM25b. This research was funded by the Austrian Science Fund (FWF) [P25541-N19].
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Werzer O.; Kunert B.; Roblegg E.; Zimmer A.; Oehzelt M.; Resel R. Surface Induced Order of Solution Processed Caffeine Needles on Silica and Muscovite Mica. Cryst. Growth Des. 2013, 1331322–1328. [Google Scholar]
- Neumann M. A.; Perrin M.-A. Can crystal structure prediction guide experimentalists to a new polymorph of paracetamol?. CrystEngComm 2009, 11112475–2479. [Google Scholar]
- Blagden N.; de Matas M.; Gavan P. T.; York P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Delivery Rev. 2007, 597617–630. [DOI] [PubMed] [Google Scholar]
- Lee T.; Hung S. T.; Kuo C. S. Polymorph farming of acetaminophen and sulfathiazole on a chip. Pharm. Res. 2006, 23112542–2555. [DOI] [PubMed] [Google Scholar]
- Werzer O.; Boucher N.; de Silva J. P.; Gbabode G.; Geerts Y. H.; Konovalov O.; Moser A.; Novak J.; Resel R.; Sferrazza M. Interface induced crystal structures of Dioctyl-Terthiophene thin films. Langmuir 2012, 28228530–8536. [DOI] [PubMed] [Google Scholar]
- Thankaraj Salammal S.; Balandier J.-Y.; Arlin J.-B.; Olivier Y.; Lemaur V.; Wang L.; Beljonne D.; Cornil J.; Kennedy A. R.; Geerts Y. H. Polymorphism in Bulk and Thin Films: The Curious Case of Dithiophene-DPP (Boc)-Dithiophene. J. Phys. Chem. C 2014, 118, 657–669. [Google Scholar]
- Rodríguez-Spong B.; Price C. P.; Jayasankar A.; Matzger A. J.; Rodríguez-Hornedo N. r. General principles of pharmaceutical solid polymorphism: A supramolecular perspective. Adv. Drug Delivery Rev. 2004, 563241–274. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [DOI] [PubMed] [Google Scholar]
- Beames J. M.; Hudson A. J. Jet-cooled spectroscopy of paracetamol. Phys. Chem. Chem. Phys. 2010, 12164157–4164. [DOI] [PubMed] [Google Scholar]
- André V.; da Piedade M. F. M.; Duarte M. T. Revisiting paracetamol in a quest for new co-crystals. CrystEngComm 2012, 14155005–5014. [Google Scholar]
- Perrin M. A.; Neumann M. A.; Elmaleh H.; Zaske L. Crystal structure determination of the elusive paracetamol Form III. Chem. Commun. (Cambridge) 2009, 22, 3181–3. [DOI] [PubMed] [Google Scholar]
- Gaisford S.; Buanz A. B.; Jethwa N. Characterisation of paracetamol form III with rapid-heating DSC. J. Pharm. Biomed. Anal. 2010, 533366–70. [DOI] [PubMed] [Google Scholar]
- Beiner M.; Rengarajan G.; Pankaj S.; Enke D.; Steinhart M. Manipulating the crystalline state of pharmaceuticals by nanoconfinement. Nano Lett. 2007, 751381–1385. [DOI] [PubMed] [Google Scholar]
- Rengarajan G.; Enke D.; Steinhart M.; Beiner M. Size-dependent growth of polymorphs in nanopores and Ostwald’s step rule of stages. Phys. Chem. Chem. Phys. 2011, 134821367–21374. [DOI] [PubMed] [Google Scholar]
- Di Martino P.; Conflant P.; Drache M.; Huvenne J.-P.; Guyot-Hermann A.-M. Preparation and physical characterization of forms II and III of paracetamol. J. Therm. Anal. Calorim. 1997, 483447–458. [Google Scholar]
- Burley J. C.; Duer M. J.; Stein R. S.; Vrcelj R. M. Enforcing Ostwald’s rule of stages: Isolation of paracetamol forms III and II. Eur. J. Pharm. Sci. 2007, 315271–276. [DOI] [PubMed] [Google Scholar]
- Werzer O.; Stadlober B.; Haase A.; Flesch H. G.; Resel R. Evaluation of organic sub-monolayers by X-ray based measurements under gracing incident conditions. Eur. Phys. J. Appl. Phys. 2009, 462 10.1051/epjap/2009038. [DOI] [Google Scholar]
- Werzer O.; Stadlober B.; Haase A.; Oehzelt M.; Resel R. Full X-ray pattern analysis of vacuum deposited pentacene thin films. Eur. Phys. J. B 2008, 664455–459. [Google Scholar]
- Moser A.; Novak J.; Flesch H.-G.; Djuric T.; Werzer O.; Haase A.; Resel R. Temperature stability of the pentacene thin-film phase. Appl. Phys. Lett. 2011, 9922221911–221911–3. [Google Scholar]
- Smits E. C.; Mathijssen S. G.; Van Hal P. A.; Setayesh S.; Geuns T. C.; Mutsaers K. A.; Cantatore E.; Wondergem H. J.; Werzer O.; Resel R. Bottom-up organic integrated circuits. Nature 2008, 4557215956–959. [Google Scholar]
- Nichols G.; Frampton C. S. Physicochemical characterization of the orthorhombic polymorph of paracetamol crystallized from solution. J. Pharm. Sci. 1998, 876684–693. [DOI] [PubMed] [Google Scholar]
- Beyer T.; Day G. M.; Price S. L. The prediction, morphology, and mechanical properties of the polymorphs of paracetamol. J. Am. Chem. Soc. 2001, 123215086–5094. [DOI] [PubMed] [Google Scholar]
- Wedl B.; Resel R.; Leising G.; Kunert B.; Salzmann I.; Oehzelt M.; Koch N.; Vollmer A.; Duhm S.; Werzer O. Crystallisation kinetics in thin films of dihexyl-terthiophene: the appearance of polymorphic phases. RSC Adv. 2012, 2104404–4414. [Google Scholar]
- Lercher C.; Resel R.; Balandier J.-Y.; Niebel C.; Geerts Y. H.; Sferrazza M.; Gbabode G. Effects of temperature on the polymorphism of α, ω-dioctylterthiophene in thin films. J. Cryst. Growth 2014, 386, 128–134. [Google Scholar]
- Graubner G.; Rengarajan G. T.; Anders N.; Sonnenberger N.; Enke D.; Beiner M.; Steinhart M. Morphology of porous hosts directs preferred polymorph formation and influences kinetics of solid/solid transitions of confined pharmaceuticals. Cryst. Growth Des 2013, 14178–86. [Google Scholar]
- Peterson M. L.; Morissette S. L.; McNulty C.; Goldsweig A.; Shaw P.; LeQuesne M.; Monagle J.; Encina N.; Marchionna J.; Johnson A. Iterative high-throughput polymorphism studies on acetaminophen and an experimentally derived structure for form III. J. Am. Chem. Soc. 2002, 1243710958–10959. [DOI] [PubMed] [Google Scholar]
- Hammersley A. P.FIT2D V9.129 Reference Manual V3.1; ESRF: Grenoble, 1998.
- Hammersley A. P.FIT2D: An Introduction and Overview; ESRF: Grenoble, 1997. [Google Scholar]
- Hammersley A.; Svensson S.; Hanfland M.; Fitch A.; Hausermann D. Two-dimensional detector software: from real detector to idealised image or two-theta scan. Int. J. High Pressure Res. 1996, 144–6235–248. [Google Scholar]
- Hammersley A. P.; Svennson S. O.; Thompson A. Calibration and correction of spatial distortions in 2D detector systems1994.
- Moser A.; Werzer O.; Flesch H.-G.; Koini M.; Smilgies D.-M.; Nabok D.; Puschnig P.; Ambrosch-Draxl C.; Schiek M.; Rubahn H.-G. Crystal structure determination from two-dimensional powders: A combined experimental and theoretical approach. Eur. Phys. J. Spec. Top. 2009, 167159–65. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.