

Abstract
The discovery of Toll-like receptors (TLRs), and their role in sensing infections represents one of the most seminal advances in immunology in recent years. It is now clear that TLRs play a fundamental role in innate recognition of microbes, and stimulate and tune the quality of the adaptive immune response. However, major knowledge gaps remain in our understanding of how TLRs regulate the development and persistence of T and B cell memory. Here, we review our current understanding of how TLR signaling shapes the adaptive immune response, and highlight unanswered questions, the solution of which will be imperative in the rational exploitation of TLRs in vaccine design and immune therapy.
Keywords: TLR, Dendritic Cells, plasmacytoid DCs, Th1/Th2, Th17
A central property of the mammalian immune system is its ability to generate the right type of immunity against a given pathogen, whilst maintaining tolerance to self tissues. The immune system senses microbes through multiple receptors collectively known as pattern recognition receptors (PRRs), which are encoded by germ-line genes and which recognize a diverse array of microbial structures, and activate innate immune cells including dendritic cells (DCs) [1,2]. PPRs include Toll-like receptors (TLRs), C-type lectin like receptors (CLRs), RIG-I like receptors (RLRs), and Nod-like receptors (NLRs). Immune cells as well as non-immune cells differentially express a wide range of TLRs, CLRs, RLRs and NLRs, which enable them to sense and respond to pathogens. DCs, the most efficient antigen-presenting cells in the immune system, have emerged as key players in initiating and regulating adaptive immune responses. Furthermore, emerging evidences also suggest that DCs are also critical in suppressing immune responses, and maintaining peripheral tolerance, through the generation of anergic or unresponsive T cells and/or regulatory T cells. Great progress has been made over the past decade in identifying and characterizing several PRRs, and in elucidating the mechanisms by which they regulate the type, quality and magnitude of the adaptive immune response. Although emerging evidence points to an important role for CLRs, NLRs, and RLRs in modulating adaptive immunity, much of our understanding comes from analyses of TLR function and their roles in modulating the magnitude and quality of T cell immunity against pathogens and vaccines. However, much less is known about the role of TLRs in controlling the magnitude, quality and duration of antibody responses. Furthermore, there is little understanding about how TLRs regulate the development and persistence of memory T and B cell responses. Here, we review our current knowledge of the role of TLRs in regulating adaptive immune responses against microbes and vaccines, and highlight some unanswered questions about the.
TLRs
TLRs represent a family of evolutionary conserved PRRs that recognizes a wide range of microbial components [3–8]. In mammals,11 different TLRs have been described and most of them are widely expressed by different cell types in the immune system including DCs, macrophages, NK cells, mast cells, neutrophils, B cells, T cells and by non-immune cells such as fibroblasts, epithelial cells and keratinocytes. Most TLRs (TLR 1, 2, 4, 5, 6, 10 and 11) are expressed on the cell surface, whereas other TLRs (TLR3, 7, 8, 9) are present within the endosomal compartments. Furthermore, TLRs are expressed as homodimers or as hetrodimers (TLR2 + TLR1 or TLR2+ TLR6), and each TLRs recognizes distinct microbial stimuli (Figure 1). For example TLR4 recognizes lipopolysaccharides(LPS) from most bacteria[9,10], TLR2 recognizes several bacterial cell wall components, including peptidogycans of Gram-positive bacteria, lipoproteins of Mycobacterium tuberculosis and lipoteichoic acids, and certain components of fungal cell wall [11,12], TLR5 recognizes flagellin [13] and TLR11, in mice, senses profilin from Toxoplasma gondii [14]. Intracellular receptors such as TLR3 senses viral dsRNA, TLR7/8 recognize viral ssRNA and small synthetic immune modulators such as imiquimod, R-848 and loxoribine; whereas TLR9 recognizes CpG motifs present in bacterial and viral DNA [15–17] (Figure 1).
Figure 1. Modulation of adaptive immune responses by Toll-like receptors (TLRs) and its adaptor proteins.
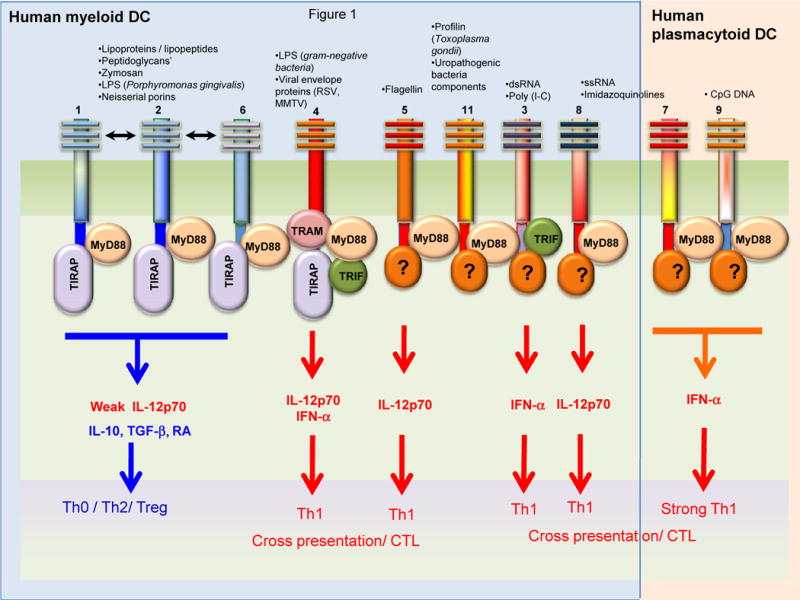
Activation of distinct TLRs in dendritic cells (DCs) results in the induction of distinct DC responses and adaptive immunities. In humans, TLRs 7 and 9 are expressed within the ER/phagolysosomes of pDCs, and signaling via these induces potent IFN-α, and induction of Th1 responses and cross-presentation of exogenous antigens to stimulate cytotoxic T cell (CTL) responses. In contrast, myeloid DCs in humans express TLR3 (in ER/phagolysosomal compartments), or TLR2 (heterodimerized with TLR1 or 6), or TLRs 3,4,5,8 and 11 on the surface. Stimulation via most TLRs induce potent IL-12p70 and Th1 responses. However, activation of TLR2 heterodimers (TLR2/1 or TLR2/6) produce relatively little IL-12 (p70) but robust IL-10, and skews the balance towards the Th0/Th2/T-regulatory phenotype. Interestingly, each TLR is associated with different adaptor proteins, which mediate distinct functions. For example, MyD88 signaling induces pro-inflammatory cytokines (IL-12, IL-6, and tumor necrosis factor) but not type I IFNs, and MyD88 does not upregulate costimulatory molecules. Signaling via TRIF or TRAM induces type I IFNs and upregulates costimulatory molecules.
Signals triggered by TLRs
TLR-mediated signaling is known to control antigen uptake, antigen presentation, DC maturation and cytokine production [18,19]. TLRs are type 1 transmembrane proteins and consist of three domains: a C-terminal leucine-rich repeat (LRR) that recognizes the microbial component, a central trans-membrane domain and an N-terminal cytoplasmic TIR (Toll/IL-1R homologous domain) signaling domain[7]. Interaction of TLRs with its specific ligands leads to the recruitment of intracellular TIR-domain containing adaptors such as MyD88, TIRAP, TRIF and TRAM through TIR-TIR interaction. This process results in recruitment of IRAK family proteins (IRAK1, IRAK2, IRAK4 and IRAK-M) and TNF receptor–associated factor 6 (TRAF6) to the receptor complex, leading to the activation of MAP kinases (ERK, JNK, p38) and transcription factors including NF-κB and AP-1 that are critical for the induction of various inflammatory cytokines and anti-inflammatory cytokine[20,21]. In addition, TLR-signaling induces the upregulation of various maturation markers such as CD80, CD83 and CD86, and the chemokine receptor CCR7.
Each TLR recruits a specific adaptor to activate different signaling pathways and transcription factors, which are critical for mounting specific response against pathogens. Most TLR, except TLR3, mediated signaling is dependent on MyD88. Thus, macrophages and DCs from MyD88 deficient mice have severe defects in the activation of NF-κB and JNK MAP kinase and inflammatory cytokines in response to various TLR ligands [3,4,20,22]. TLR3 signals through a MyD88 independent pathway involving TRIF. TLR4 signals through both the MyD88-dependent and the TRIF dependent pathways. In addition, TLR4 signaling selectively recruits TRAM adaptor protein [23,24]. Signaling through TLR3 is dependent on TRIF mediated activation of TBK1 and IRFs resulting strong induction of type I IFN. Studies with TRIF-deficient mice [25] or Lps2 mice (mutation in Trif gene)[10], have shown severe defects in both TLR3- and TLR4-mediated induction of INF-β and activation of IRF3 and type I IFNs. TLR4-MyD88 mediated signaling module leads to the activation of MAP kinases (ERK, p38, JNK) and NF-κB, whereas, TLR4-TRIF-mediated signaling module, Myd88-independent, activates IRF3 activation and induction of type I IFN-inducible genes. In addition, TRIF pathway also leads to delayed NF-κB activation and thus, mice deficient in both MyD88 and TRIF showed complete loss of NF-κB activation[25]. Similar to TRIF-deficient mice, TRAM-deficient mice showed defects in cytokine production specifically in response to the TLR4 ligand [24]. Furthermore, TLR2 and TLR4 are known to interact with TIRAP [26,27], although whether TIRAP mediated signaling results in some unique biological outcome is poorly understood.
An interesting feature of TLRs is that distinct TLRs are differentially expressed on distinct subsets of DCs. Thus, in humans TLRs 7 and 9 are expressed in plasmacytoid DCs, but not in myeloid DCs; in contrast, TLRs 2,3,4,5,8 are expressed in myeloid DCs [28]. Interestingly, activation of TLR7 and TLR9 in the plasmacytoid DCs, leads to the MyD88-dependent the phosphorylation and activation of the transcription factor IRF7, which is a master regulator of the production of type I IFNs [29]. Recent studies from several laboratories, including ours, have highlighted an important role for the PI3K-dependent activation of the mammalian target of rapamycin (mTOR, REF 27) signaling in promoting the production of type I IFNs by pDCs stimulated with TLR ligands or viruses (Figure 2)[31–33]. This is consistent with the observation that inhibition of PI3-kinase activation impairs the ability of pDCs to produce type I IFNs in response to TLR signaling [34]. Consistent with these observations, blocking mTOR signaling in pDCs with the immunosuppressive drug, severely impaired the IRF-7 activation and IFN-a/β production in response to CpG [32,33]. Furthermore, the kinases 4E-BPs [31]and S6K1/2 [33] act downstream of mTOR and play a critical role in regulating the TLR-signaling pathway. Thus, siRNA-mediated knock-down of S6K1/2 in pDCs or pDCs deficient in S6K1/2 display impaired activation of IRF-7 and IFN-α/β in response to CpG (Figure 2). Consistent with this findings, inhibition of mTOR signaling in APCs in vivo resulted in impaired immune response to yellow fever vaccine virus[32]. Further, these studies have shown that S6K1/2 and 4E-BPs regulate TLR-9 mediated signaling at two different levels. First, mTOR-S6K1/2 signaling pathway regulates the formation of TLR9-MyD88 complex and thereby influences subsequent activation of IRF-7[32]. Second, mTOR-4E-BP-pathway modulates the TLR-signaling pathway at the translational level by regulating IRF-7 mRNA translation. Consistent with this mechanism, pDCs-deficient in 4E-BP1 and 4E-BP2 produce higher levels of type I interferon[31]. Paradoxically, mTOR signaling appears to exert a rather different effect on TLR signaling in myeloid DCs; a recent study demonstrates that inhibition of mTOR signaling in bone marrow derived DCs results in upregulation of the anti-inflammatory cytokine interleukin 10 and inhibition of proinflammatory cytokines [35]. However, bone marrow derived DCs cultured with rapamycin have been shown to express enhanced levels of IL-1beta, which promotes upregulation of the receptor ST2L, (which is an IL-1R family member known to bind the Th2 inducing cytokine IL-33 and which is known to be a negative regulator of TLR signaling), and impair DC maturation[36]. Thus, the role of mTOR in regulating TLR signaling appears to be very context driven, and dependent on the subset of DC. This context dependency seems to be a recurring theme in TLR biology – for example, macrophages and myeloid DCs, but not pDCs, produce IL-10 in response to TLR stimulation [37]. Collectively this suggests that different TLRs uses distinct adaptor protein to mediate distinct signaling pathways. Importantly, TLR signaling appears to be very dependent on the subset of antigen presenting cells being studied.
Figure 2. Regulation of TLR-mediated type I IFN production in plasmacytoid DCs, by mTOR.
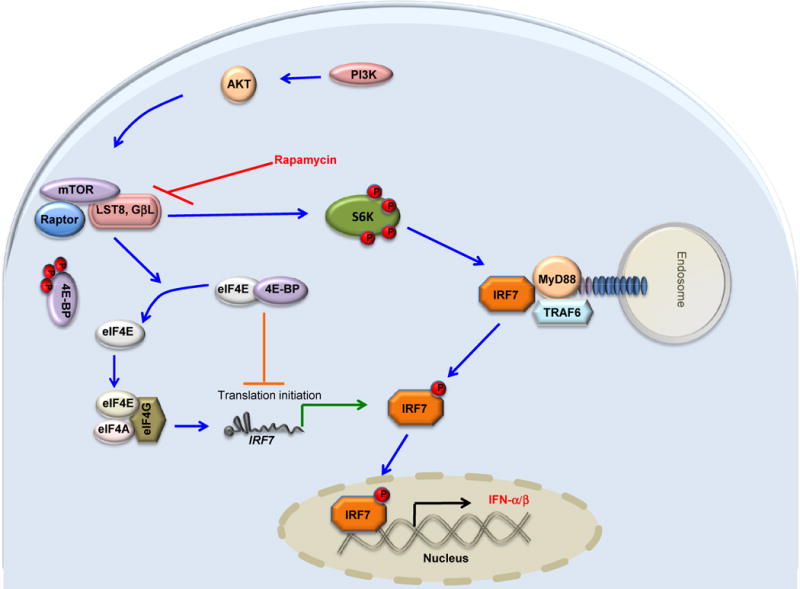
CpG or viral mediated activation of TLR9 leads to recruitment of MyD88 and TRAF6, and subsequent phosphorylation of IRF7, which promotes its translocation to the nucleus resulting in transcriptional activation of type I IFN genes. PI3K-AKT- signaling pathway activates mTOR which in-turn promotes TLR9-MyD88 complex formation through S6K phosphorylation and activation. Simultaneously, mTOR-mediated phosphorylation of 4E-BP results in dissociation of eukaryotic translation initiation factor (eIF)4E which binds to eIF4G/eIF4A complex and thus initiates more translation of IRF7 mRNA.
Suppressing TLR –signaling
Uncontrolled regulation of TLR-mediated signaling may lead to excessive or persistent inflammation and severe immune pathology to the host [8,38–43]. Several diseases including septic shock, autoimmunity, atherosclerosis, metabolic syndrome and gastric cancer have been linked to chronic or acute inflammatory response [8,42,44]. An increasing number of negative regulators exist that are known to dampen the degree and duration of the TLR-mediated inflammatory response. Most of these negative regulators are induced upon TLR activation and regulate the response at multiple levels [39,41,42]. PI3K is one such critical negative regulator of TLR signaling that acts at the early phase of TLR signaling and modulates the magnitude of the immune responses to pathogens [45,46]. Thus, macrophages and DCs from PI3K knockout mice (lacking the P85 regulatory subunit of class 1A PI3K) produce more IL-12 than wild type DCs in response to various TLR ligands. Furthermore, inhibition of PI3K activity using specific inhibitors enhanced the production of IL-12 in DCs in response to various TLR- ligands[46]. These observations suggest that PI3K is a critical regulator of IL-12 and modulates the Th1/Th2 balance. Thus mice deficient in PI3K−/− showed enhanced Th1 response and resistance to Leishmania major pathogen but displayed impaired Th2 response against intestinal nematode, Strongyloides venezuelensis. In DCs, PI3K is activated in response to various TLR ligands[45,46]. However, there are several studies using human monocyte derived DCs or mouse bone marrow derived DCs have shown that PI3K play positive role in the induction of inflammatory responses by regulating the NF-κB transactivation in response to various TLR-ligands [47,48].
Another key negative regulators of TLR signaling is the family of SOCS proteins, which are induced upon TLR activation [49]. For example, Pam3-cys mediated activation of TLR2/1 and LPS-mediated activation of TLR4 induces SOCS1 [50]. SOCS1-deficient macrophages produce greater amounts of IL-12 in response to LPS or Pam-3-Cys [51,52]. Further, forced expression of SOCS1 in macrophages suppressed the NF-ΚB activation and IL-12 production. Consistent with this observation, SOCS1-deficent mice are more susceptible to LPS-induced lethal effects[51]. Subsequent studies have shown that SOCS1 dampen TLR-mediated signaling by two different ways. First, SOCS1 directly interacts TIRAP and initiates it degradation through ubiquitination-proteosome pathway [53]. Second, SOCS1 directly interacts with p65 subunit of NF-κB and induces its proteosomal degradation and thereby suppressing NF-κB activation[53].
Our recent work has shown that zymsoan mediated TLR2 signaling induces both SOCS3 and SOCS1 in DCs via IL-10 and RA production which is critical for the suppression of induction of pro-inflammatory cytokines [54]. Thus, siRNA mediated knockdown of SOCS3 in DCs or blocking RAR- or IL-10R-mediated signaling in DCs, showed increased production of pro-inflammatory cytokines in response to zymosan skewing to Th-1 or Th-17 response[54]. Similarly, DCs from TLR2 knockout mice or IL-10 knock-mice showed reduced levels of SOCS3 and increased production of inflammatory cytokines response to zymosan [54].
In addition to SOCS family proteins, proteins such SIGIRR[55], IRAK-M[56], RP105[57], A20[58], ST2L[59], TRAF1[60] and TRAF4 [61]are implicated in negatively regulating TLR signaling. Mice deficient in SIGIRR[55], IRAK-M[56], ST2L, A20[58] and RP105 are hyper-responsive to LPS and secrete more inflammatory cytokines to various TLR ligands. Over expression of TRAF1 and TRAF4 can block TRIF-dependent and TRAF6-dependent activation of NF-κB.
ILT7 a member of the immunoglobulin-like family is selectively expressed in human plasmacytoid dendritic cells (pDCs), and associates with the signal adapter protein FcεR1γ to form a receptor complex. Cross-linking of ILT7 results in phosphorylation of Src family kinases and Syk kinase and induces a calcium influx, and inhibits the transcription and secretion of type I interferon and other cytokines. Therefore, the ILT7-Fc epsilonRI gamma receptor complex negatively regulates the innate immune functions of human pDCs [62]. In addition, human pDCs also express the blood dendritic cell antigen 2 (BDCA2) protein, a C type lectin which also complexes with FcεR1 . Signaling via this complex triggers a pathway similar to that observed downstream of the B cell receptor (BCR), involving Syk kinase and inhibits type I IFN production from pDCs [63].
Modulation of T helper responses by TLRs
TLRs exert a potent influence on the quality of the T helper responses. Engagement of TLRs on DCs and other cells of innate immune system, results in the activation of various signaling pathways and distinct programs of gene expression that dictate the type of adaptive immune response. There is also emerging evidence that T cells themselves may express specific TLRs, and that direct triggering on TLRs can occur. For example, murine CD8 T cells express TLR2, and costimulation of antigen-activated CD8 T cells with TLR2 ligands enhances their proliferation, survival, and effector functions, and significantly reduces their need for costimulatory signals delivered by DCs [64]. The types of cytokines and other factors secreted by DCs and other innate immune cells program the differentiation of newly primed CD4+ T cells into Th-1 or Th-2 or Th-17 effector cells or regulatory T cells (Treg) responses. There is a general consensus that signaling via most TLRs potently stimulate DCs to produce IL-12 (p70), which promotes IFN-γ producing Th-1 cells [3,7,8] (Figure 1). Thus, mice deficient in MyD88 exhibit a selective defect in Th1 responses upon immunization with protein ovalbumin plus Freunds Complete Adjuvant (FCA) [65]. One interpretation of this finding is that only Th1 responses are controlled by TLR signaling. However, since MyD88 knockout mice can still signal via the TRIF or TIRAP adaptor proteins, it is possible that these mediated Th2 responses.
In contrast certain TLR ligands are known to induce production of IL-10 which stimulates Th2 or T regulatory responses [5]. Furthermore, TLRs that induce strong production of TGF-β, IL-6 and IL-23 promote IL-17 producing Th-17 cells [12,66]. TLR4 ligand LPS, TLR9 ligand CpG DNA, TLR3 ligand poly (IC) and TLR7 ligands induce robust IL-12p70 production and mediate Th1 responses [3–5,22,41,67–71]. TLR ligands that mediate activation of p38 and JNK in DCs induce robust IL-12p70 and Th1 responses.
In the case of TLR2, there are reports of ligands activating either a Th1 or a Th2 or a T regulatory response. Although some TLR2 ligands induce IL-12p70 and stimulate Th1 response[11,72–74], accumulating evidence demonstrates that TLR2 mediated signaling can also stimulate Th2 or T regulatory response (Figures 1 & 3). Several studies using TLR2 specific ligands such as highly purified LPS from P. gingivalis [67,75], Pam-3cys [70,71,76,77], FSL-1[78], or certain helminth products [79,80], or other microbial stimuli[81], demonstrate that such TLR2 ligands induce robust IL-10 production and favor Th2 responses or T regulatory responses[70,75]. Work from our laboratory has shown that TLR2 mediated activation of DCs results in the sustained phosphorylation of ERK MAP kinase, which leads to phosphorylation of the AP-1 transcription factor c-Fos. This is critical in suppressing expression of the Th-1-inducing cytokine IL-12 and induction of IL-10, thereby favoring a Th-2 bias [70,71,82]. In fact, DCs from ERK1−/− mice produced enhanced levels of IL12p70 and reduced levels of IL-10 in response to TLR2 ligands[70]. Consistent with, ERK1−/− mice exhibited enhanced Th1 cell polarization and increased susceptibility to experimental autoimmune encephalomyelitis[82].
Figure 3.
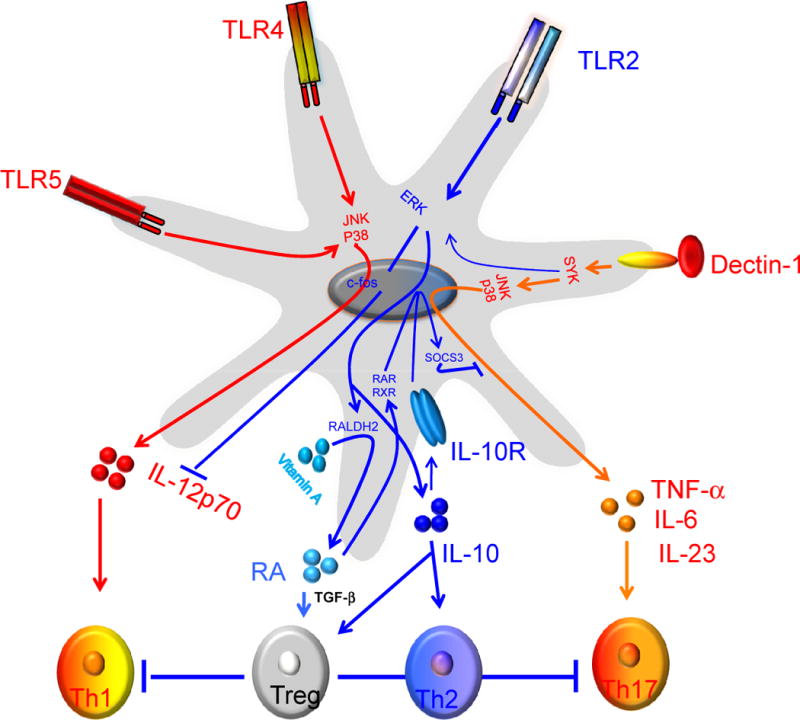
Programming DCs to induce Th1, Th2, Th17 or T regulatory responses. Signaling via TLR4 induces potent p38 and JNK1/2 MAPK activation which leads to the induction of interleukin-12 (IL-12) (p70). In contrast, Pam-3-cys, a TLR2/1 ligand, induce enhanced ERK 1/2 activation, which results in the stabilization of the transcription factor c-Fos that potently suppresses IL-12(p70) and enhances IL-10, thus favoring a Th2 bias. Interestingly, triggering DCs through TLR2/6 by zymosan efficiently induces ERK activation, which mediates induction of Raldh2. This results in the conversion of retinal to retinoic acid (RA), which then exerts an autocrine effect on DCs via RAR or RXR to induce SOSC3, which suppresses activation of p38 MAPK and proinflammatory cytokines. In contrast, zymosan mediated triggering of dectin-1in DCs promotes induction of proinflammatory cytokines IL-6 and IL-23 and thus mediates Th17 response.
Furthermore, zymosan, a particulate yeast cell wall derivative, that signals through several PRRs, including the heterodimeric TLR2 and 6 and the C-type lectin dectin-1 induces splenic DCs to produce the retinoic acid (RA) metabolizing enzyme RALDH2, as well as the anti-inflammatory cytokine IL-10, via a mechanism that is dependent on TLR2 signaling of ERK-MAPK[54,83,84] (Figure 3). Such zymosan stimulated splenic DCs induce robust activation of T regulatory cells. In the case of regulatory T-cell responses observed with zymosan in DCs, TLR2-mediated production of RA and IL-10, is critical for suppressing pro-inflammatory cytokine production through the induction of suppressor cytokine signaling 3 (SOCS3) [54]. Consistent with the above observation, DCs purified from ERK1-deficient mice showed defects in the induction of RALDH2, SOCS3 and production of IL-10 in response to zymosan [54]. Thus, activation of MAPK ERK1/2 plays an important role regulating the balance between Th1/Th2 polarization, or Th17/T-regulatory polarization by regulating the cytokine production by DCs. Consistent with this, a recent study by DePaolo et al, has shown that activation of TLR2/6 receptor in DCs using Yersinia pestis virulence factor LcrV and other ligands, induces tolerogenic DCs and T regulatory responses [85]. Finally, several studies have shown that systemic [86] or sublingual [77] injection of some TLR2 ligands induces anti-inflammatory cytokines and can suppress the inflammatory response. Injection of TLR2 ligand (BLP) systemically in mice suppressed the allergen-induced inflammation at peripheral sites such as lung and skin by regulating the inflammatory cytokine and expression of chemokine receptors on antigen presenting cells and other inflammatory cells[86].
Thus, TLRs exert a major influence on the cytokines produced by T-helper cells. Whether TLRs also regulate other parameters of T helper cell activation such as persistence, differentiation into central versus effector memory cells are not known. In this context, a recent study demonstrates that the ability of adjuvants to stimulate high-avidity T cell responses after protein vaccination correlates with TLR engagement [87]. Thus, the use of TLR-9 and TLR-4 agonists as adjuvants skewed TCR repertoire usage by increasing TCR selection thresholds and enhancing antigen-specific clonal expansion [87]. This suggests that, TLRs control the local accumulation of Th cells expressing TCR with the highest peptide MHC class II binding.
Modulating CD8+ T responses with TLRs
Stimulation of DCs via specific TLRs endows them with the capacity to cross-present exogenously acquired antigens and prime antigen-specific CD8+ T cells to differentiate into cytotoxic T cells (CTLs). For example, signaling via TLR3, 7, and 9 induces cross-presentation[88,89]. Furthermore, targeting antigens plus TLR ligands to specific DC subsets induces cross-presentation and stimulation of CD8+ T cells [90]. Interestingly, TLR3, 7, and 9 also induce type I IFNs, which enhance cross-presentation by DCs [91]. The involvement of TLRs in mediating CD8+ T cell responses to viruses is demonstrated by the fact that antigen-specific CD8+ T cell responses to the live attenuated yellow fever vaccine YF-17D, or to lymphocytic choriomeningitis virus [92,93]. Consistent with this`, TLR7 and 9 ligands have been used as vaccine adjuvants to enhance the antigen-specific CD8+ T cell responses to protein based vaccines[94], and DNA vaccines [95]. Importantly, the “quality” of the CD8+ T cell response, as measured by the generation of polyfunctional T cells that secrete IFN-γ, IL-2 and TNF-α, was enhanced with TLR ligands [94,95]. In contrast to these stimulatory effects of TLRs on cross presentation and the generation of antigen-specific CD8+ T cell responses, a recent study demonstrated that systemic activation of DCs by malaria infection, or systemic injection of TLR ligands, resulted in a downregulation of cross-presentation of exogenous antigens, without hindering presentation of endogenous antigens [96]. However, as with T-helper cells, the mechanisms by which TLRs regulate the generation and persistence of long lived memory CD8+ T cells, and effector versus central memory cells are not known.
Modulating B cell responses with TLRs
Vaccination can induce the production of antibodies for a lifetime, although mechanisms that remain poorly understood. Although the role of TLRs in modulating T cell responses is well understood, there is relatively little information about their role in modulating antibody responses. There is no question that immunization with protein antigen plus TLR ligands results in robust induction of antigen-specific antibody responses. A recent longitudinal study of malaria-naive individuals, which examined the development of the antigen-specific memory B cell responses to candidate malaria vaccines administered with or without CpG DNA, showed that CpG enhances the kinetics, magnitude, and longevity of the memory B cell response [97]. The mechanisms by which TLRs promote antibody responses and B cell memory are poorly understood. A recent study demonstrates that in mice, in addition to TLR signaling in DCs, TLR mediated stimulation, directly on B cells is also required for inducing robust antibody responses [98]. This is consistent with the fact that murine B cells can be stimulated in vitro by TLR4 and TLR9 ligands to proliferate and secrete antibody[99]. Our data suggest that mouse naïve B cells express and undergo polyclonal expansion and differentiation to almost all known TLRs except TLR5 and 8 [100]. Moreover, recent studies suggest that triggering of TLRs directly on B cells is also required for autoantibody production [101–104].
As is the case with T cells, little is understood about the role of TLRs in regulating memory B cell formation and maintenance. In contrast to murine B cells, naïve human B cells do not express TLR4 or TLR9 and hence do not respond directly to LPS or CpG. However, human memory B cells in the blood do express TLR9 and respond to CpG DNA [105], and consistent with this, cross-linking of BCR results in upregulation of TLR9 expression, and responsiveness to TLR9 ligands [105]. Furthermore, human memory B cells can proliferate and differentiate into plasma cells in response stimulation with CpG DNA, and consistent with this, plasma cells secreting antibodies to recall antigens are produced in vivo at levels proportional to the frequency of specific memory B cells, even several years after antigenic stimulation [106]. Thus, it was speculated that persistent polyclonal activation of memory B cells can maintain “serological memory” for a human lifetime [106].
Finally, although it is clear that TLR ligands can stimulate antibody responses, the question of whether TLR signaling is required for antibody responses was addressed by a recent study in which wild type and MyD88 and TRIF doubly deficient mice were immunized with haptenated protein antigens, or protein antigens plus conventional adjuvants such as alum, monphosophoryl lipid A (MPL), Freunds complete adjuvant (FCA), and incomplete Freunds adjuvant (IFA). These results demonstrates a lack of requirement for the TLR pathway for induction of antibody responses [107]. In contrast to this study, other studies show that MyD88-dependent signaling pathways in B cells are essential for effectively generating long-term Ab responses and implicate a role for TLR in the formation of long-term humoral immunity against polyoma virus [108], or in induction of IgG2a/c antibody responses to influenza virus [108], or induction of haemagglutination inhibition antibody titers to vaccination against H5N1 influenza vaccine [109].
TLR/TLR cross-talk
In general, innate sensing of microbes and viruses occurs not through a single TLR alone, but rather by several TLRs, each of which recognize a different component. Although, there is a considerable understanding of how specific ligands trigger individual TLRs, and the signaling pathways involved, the question of signals emanating from several TLRs are integrated to generate a cellular response against the pathogen is poorly understood [6]. Recent work has shown that simultaneous stimulation of human monocyte derived derived DCs with LPS (TLR4 agonist) and R484 (TLR7 agonist) results in a synergistic increase in the production of pro-inflammatory cytokines such as IL-12p70, IL-23, IL-6, TNF–α, and anti-inflammatory cytokine IL-10, compared to DCs stimulated with individual TLR4 ligand and TLR7 ligand [110]. TLR/TLR synergy is not specific to TLR4 + TLR7 as similar effects were seen with different TLR ligand combinations such as TLR3 or TLR4 ligand in combination with TLR7, TLR8 or TLR9[110–112]. Interestingly, synergy was apparent when simultaneously stimulating the MyD88-dependent and TRIF-dependent pathways. The impact of this synergy on adaptive immunity is beginning to be understood; a recent study demonstrates a synergistic enhancement of the T cell response in mice immunized with TLR ligands that activate the MyD99 and TRIF-dependent pathways [113].
Even though several studies have shown synergy with different TLR ligands, the molecular mechanism underlying this synergy remains undefined. One possibility is that activation of two TLRs simultaneously on DCs might lead to sustained activation of various MAPK and transcription factors that are critical for the production of pro-inflammatory cytokines. Studies have shown that stimulation of TLRs leads to activation IRF transcription factors that can directly regulate the IL-12 transcription [111]. Interestingly, activation of several TLRs was shown in the case live attenuated yellow fever vaccine-17D [93]. Interestingly, although absence of MyD88 resulted in greatly diminished Th1 and Tc1 responses, absence of TLR2 skewed the response more towards the Th1 response suggesting a regulatory role for these receptors in limiting the response. [93]. Thus, combinatorial activation of multiple TLRs can either synergize (e.g. TLR3 plus TLR7) [110], or cross regulate each other (e.g. TLRs 7, 8, 9 plus TLR2) [93]. Clearly, further work is required to understand the mechanistic basis of such combinatorial activation of TLRs, and the consequences for T and B cell responses.
Summary
As will be evident from the discussion above, several recent studies have provided important insights into how TLRs sense pathogens and vaccines program DC activation, to initiate and fine tune the adaptive immune response. Although it is clear that TLR signaling can program DCs to stimulate robust T and B cell responses, and tune the quality of such responses, several important questions remain (Table 1). For example, the extent to which TLRs are essential for, and regulate the induction and maintenance of long lived T and B cell memory responses is unknown. Furthermore, if and how TLRs regulate affinity maturation of the antibody response and the induction of long lived antibody forming plasma cells is not known. How innate sensing of pathogens or vaccines, via multiple PRRs is integrated to promote the “right” type of adaptive immunity is scarcely understood. Answers to these questions will be critical for the rational exploitation of TLRs in designing vaccine adjuvants that induce long lived T and B cell memory responses, and persistent antibody forming cells that secrete high affinity neutralizing antibody responses. Furthermore, a major unanswered question is the extent to which the “rules of TLR biology,” (determined by studying DCs or macrophages from spleens or lymph nodes) are applicable to other organs such as the intestine, liver or lung. In this context, a central problem in immunology is how, despite constant triggering of TLRs by the commensals in the intestine, the immune system does not become hyperactivated, resulting in overt inflammation and autoimmunity. The solutions to these problems represent a critical step on the path to designing effective mucosal adjuvants, and therapeutics measures against organ specific autoimmune diseases. Finally, the question of whether persistent chronic infections such as HIV, HCV or TB result in a persistent triggering of TLRs, and if so, how this would impact the innate and adaptive immune systems is unknown. Clearly, there is much to keep TLR biologists busy for years to come!
Table 1.
Some knowledge gaps in understanding how TLRs regulate adaptive immunity
| The role of TLRs in regulating the induction and maintenance of CD4+ and CD8+ T cell memory responses |
| The role of TLRs in regulating the induction and maintenance of the germinal center response, affinity maturation and generation of long lived antibody forming cells |
| How signals from multiple TLRs are integrated and the consequence for adaptive immunity |
| How signals from TLRs are integrated with signals from CLRs, NLRs, and RLRs, and the consequence for adaptive immunity |
| Organ-specific differences in TLR signaling (e.g. spleen versus intestines versus lung versus liver) |
| The mechanisms by which constitutive TLR signaling in the intestine are restrained from promoting overt inflammation and autoimmunity |
| The role of TLRs in regulating immunity to chronic infections. (i.e. the consequences of persistent TLR signaling by persistent chronic infections). |
Acknowledgments
We thank the generous support of the National Institutes of Health (grants R01 DK057665, R01 AI048638, U19 AI057266, U54 AI057157, N01 AI50019 and N01 AI50025) and the Bill & Melinda Gates Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM. Dendritic cells in vivo: a key target for a new vaccine science. Immunity. 2008;29:319–324. doi: 10.1016/j.immuni.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R, Janeway C., Jr Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- 5.Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 7.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Beutler B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol Rev. 2009;227:248–263. doi: 10.1111/j.1600-065X.2008.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 10.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 12.Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol Rev. 2008;226:112–131. doi: 10.1111/j.1600-065X.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 14.Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 15.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 16.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 17.McCluskie MJ, Krieg AM. Enhancement of infectious disease vaccines through TLR9-dependent recognition of CpG DNA. Curr Top Microbiol Immunol. 2006;311:155–178. doi: 10.1007/3-540-32636-7_6. [DOI] [PubMed] [Google Scholar]
- 18.Blander JM. Coupling Toll-like receptor signaling with phagocytosis: potentiation of antigen presentation. Trends Immunol. 2007;28:19–25. doi: 10.1016/j.it.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 20.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 24.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 26.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, McMurray D, Smith DE, Sims JE, Bird TA, O’Neill LA. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 27.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 28.Cao W, Liu YJ. Innate immune functions of plasmacytoid dendritic cells. Curr Opin Immunol. 2007;19:24–30. doi: 10.1016/j.coi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 30.Young DA, Nickerson-Nutter CL. mTOR–beyond transplantation. Curr Opin Pharmacol. 2005;5:418–423. doi: 10.1016/j.coph.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Colina R, Costa-Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, Makrigiannis AP, Bell JC, Sonenberg N. Translational control of the innate immune response through IRF-7. Nature. 2008;452:323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 32.Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, Pulendran B. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol. 2008;9:1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmitz F, Heit A, Dreher S, Eisenacher K, Mages J, Haas T, Krug A, Janssen KP, Kirschning CJ, Wagner H. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur J Immunol. 2008;38:2981–2992. doi: 10.1002/eji.200838761. [DOI] [PubMed] [Google Scholar]
- 34.Guiducci C, Ghirelli C, Marloie-Provost MA, Matray T, Coffman RL, Liu YJ, Barrat FJ, Soumelis V. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J Exp Med. 2008;205:315–322. doi: 10.1084/jem.20070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Horl WH, Hengstschlager M, Muller M, Saemann MD. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 36.Turnquist HR, Sumpter TL, Tsung A, Zahorchak AF, Nakao A, Nau GJ, Liew FY, Geller DA, Thomson AW. IL-1beta-driven ST2L expression promotes maturation resistance in rapamycin-conditioned dendritic cells. J Immunol. 2008;181:62–72. doi: 10.4049/jimmunol.181.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boonstra A, Rajsbaum R, Holman M, Marques R, Asselin-Paturel C, Pereira JP, Bates EE, Akira S, Vieira P, Liu YJ, Trinchieri G, O’Garra A. Macrophages and myeloid dendritic cells, but not plasmacytoid dendritic cells, produce IL-10 in response to MyD88- and TRIF-dependent TLR signals, and TLR-independent signals. J Immunol. 2006;177:7551–7558. doi: 10.4049/jimmunol.177.11.7551. [DOI] [PubMed] [Google Scholar]
- 38.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 39.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 41.Pulendran B. Variegation of the immune response with dendritic cells and pathogen recognition receptors. J Immunol. 2005;174:2457–2465. doi: 10.4049/jimmunol.174.5.2457. [DOI] [PubMed] [Google Scholar]
- 42.Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 2009;130:7–15. doi: 10.1016/j.clim.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–1407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 45.Fukao T, Yamada T, Tanabe M, Terauchi Y, Ota T, Takayama T, Asano T, Takeuchi T, Kadowaki T, Hata Ji J, Koyasu S. Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat Immunol. 2002;3:295–304. doi: 10.1038/ni768. [DOI] [PubMed] [Google Scholar]
- 46.Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, Koyasu S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 47.Re F, Strominger JL. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. 2001;276:37692–37699. doi: 10.1074/jbc.M105927200. [DOI] [PubMed] [Google Scholar]
- 48.Ishii KJ, Takeshita F, Gursel I, Gursel M, Conover J, Nussenzweig A, Klinman DM. Potential role of phosphatidylinositol 3 kinase, rather than DNA-dependent protein kinase, in CpG DNA-induced immune activation. J Exp Med. 2002;196:269–274. doi: 10.1084/jem.20020773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalpke A, Heeg K, Bartz H, Baetz A. Regulation of innate immunity by suppressor of cytokine signaling (SOCS) proteins. Immunobiology. 2008;213:225–235. doi: 10.1016/j.imbio.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 50.Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279:54708–54715. doi: 10.1074/jbc.M410992200. [DOI] [PubMed] [Google Scholar]
- 51.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, Seki E, Sato S, Takeuchi O, Takeda K, Akira S, Yamanishi K, Kawase I, Nakanishi K, Kishimoto T. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 52.Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 53.Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O’Neill LA, Hertzog PJ. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 54.Manicassamy S, Ravindran R, Deng J, Oluoch H, Denning TL, Kasturi SP, Rosenthal KM, Evavold BD, Pulendran B. Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med. 2009 doi: 10.1038/nm.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, Towne J, Sims JE, Stark GR, Li X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–927. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 57.Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt-Jones EA, Karp CL. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, McNally E, Pickart C, Ma A. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 59.Brint EK, Xu D, Liu H, Dunne A, McKenzie AN, O’;Neill LA, Liew FY. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5:373–379. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 60.Su X, Li S, Meng M, Qian W, Xie W, Chen D, Zhai Z, Shu HB. TNF receptor-associated factor-1 (TRAF1) negatively regulates Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling. Eur J Immunol. 2006;36:199–206. doi: 10.1002/eji.200535415. [DOI] [PubMed] [Google Scholar]
- 61.Takeshita F, Ishii KJ, Kobiyama K, Kojima Y, Coban C, Sasaki S, Ishii N, Klinman DM, Okuda K, Akira S, Suzuki K. TRAF4 acts as a silencer in TLR-mediated signaling through the association with TRAF6 and TRIF. Eur J Immunol. 2005;35:2477–2485. doi: 10.1002/eji.200526151. [DOI] [PubMed] [Google Scholar]
- 62.Cao W, Rosen DB, Ito T, Bover L, Bao M, Watanabe G, Yao Z, Zhang L, Lanier LL, Liu YJ. Plasmacytoid dendritic cell-specific receptor ILT7-Fc epsilonRI gamma inhibits Toll-like receptor-induced interferon production. J Exp Med. 2006;203:1399–1405. doi: 10.1084/jem.20052454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cao W, Zhang L, Rosen DB, Bover L, Watanabe G, Bao M, Lanier LL, Liu YJ. BDCA2/Fc epsilon RI gamma complex signals through a novel BCR-like pathway in human plasmacytoid dendritic cells. PLoS Biol. 2007;5:e248. doi: 10.1371/journal.pbio.0050248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mercier BC, Cottalorda A, Coupet CA, Marvel J, Bonnefoy-Berard N. TLR2 engagement on CD8 T cells enables generation of functional memory cells in response to a suboptimal TCR signal. J Immunol. 2009;182:1860–1867. doi: 10.4049/jimmunol.0801167. [DOI] [PubMed] [Google Scholar]
- 65.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 66.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 67.Pulendran B, Kumar P, Cutler CW, Mohamadzadeh M, Van Dyke T, Banchereau J. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol. 2001;167:5067–5076. doi: 10.4049/jimmunol.167.9.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pulendran B. Modulating vaccine responses with dendritic cells and Toll-like receptors. Immunol Rev. 2004;199:227–250. doi: 10.1111/j.0105-2896.2004.00144.x. [DOI] [PubMed] [Google Scholar]
- 69.Pulendran B. Modulating TH1/TH2 responses with microbes, dendritic cells, and pathogen recognition receptors. Immunol Res. 2004;29:187–196. doi: 10.1385/IR:29:1-3:187. [DOI] [PubMed] [Google Scholar]
- 70.Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, Maliszewski C, Akira S, Pulendran B. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. 2004;172:4733–4743. doi: 10.4049/jimmunol.172.8.4733. [DOI] [PubMed] [Google Scholar]
- 71.Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, Pulendran B. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol. 2003;171:4984–4989. doi: 10.4049/jimmunol.171.10.4984. [DOI] [PubMed] [Google Scholar]
- 72.Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, Maitland M, Norgard MV, Plevy SE, Smale ST, Brennan PJ, Bloom BR, Godowski PJ, Modlin RL. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–736. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 73.Kaisho T, Hoshino K, Iwabe T, Takeuchi O, Yasui T, Akira S. Endotoxin can induce MyD88-deficient dendritic cells to support T(h)2 cell differentiation. Int Immunol. 2002;14:695–700. doi: 10.1093/intimm/dxf039. [DOI] [PubMed] [Google Scholar]
- 74.Sugawara I, Yamada H, Mizuno S, Takeda K, Akira S. Mycobacterial infection in MyD88-deficient mice. Microbiol Immunol. 2003;47:841–847. doi: 10.1111/j.1348-0421.2003.tb03450.x. [DOI] [PubMed] [Google Scholar]
- 75.Jotwani R, Pulendran B, Agrawal S, Cutler CW. Human dendritic cells respond to Porphyromonas gingivalis LPS by promoting a Th2 effector response in vitro. Eur J Immunol. 2003;33:2980–2986. doi: 10.1002/eji.200324392. [DOI] [PubMed] [Google Scholar]
- 76.Redecke V, Hacker H, Datta SK, Fermin A, Pitha PM, Broide DH, Raz E. Cutting edge: activation of Toll-like receptor 2 induces a Th2 immune response and promotes experimental asthma. J Immunol. 2004;172:2739–2743. doi: 10.4049/jimmunol.172.5.2739. [DOI] [PubMed] [Google Scholar]
- 77.Lombardi V, Van Overtvelt L, Horiot S, Moussu H, Chabre H, Louise A, Balazuc AM, Mascarell L, Moingeon P. Toll-like receptor 2 agonist Pam3CSK4 enhances the induction of antigen-specific tolerance via the sublingual route. Clin Exp Allergy. 2008 doi: 10.1111/j.1365-2222.2008.03056.x. [DOI] [PubMed] [Google Scholar]
- 78.Kiura K, Kataoka H, Yasuda M, Inoue N, Shibata K. The diacylated lipopeptide FSL-1 induces TLR2-mediated Th2 responses. FEMS Immunol Med Microbiol. 2006;48:44–55. doi: 10.1111/j.1574-695X.2006.00119.x. [DOI] [PubMed] [Google Scholar]
- 79.van Riet E, Everts B, Retra K, Phylipsen M, van Hellemond JJ, Tielens AG, van der Kleij D, Hartgers FC, Yazdanbakhsh M. Combined TLR2 and TLR4 ligation in the context of bacterial or helminth extracts in human monocyte derived dendritic cells: molecular correlates for Th1/Th2 polarization. BMC Immunol. 2009;10:9. doi: 10.1186/1471-2172-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goh F, Irvine KM, Lovelace E, Donnelly S, Jones MK, Brion K, Hume DA, Kotze AC, Dalton JP, Ingham A, Sweet MJ. Selective induction of the Notch ligand Jagged-1 in macrophages by soluble egg antigen from Schistosoma mansoni involves ERK signalling. Immunology. 2008 doi: 10.1111/j.1365-2567.2008.02979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu T, He SH, Zheng PY, Zhang TY, Wang BQ, Yang PC. Staphylococcal enterotoxin B increases TIM4 expression in human dendritic cells that drives naive CD4 T cells to differentiate into Th2 cells. Mol Immunol. 2007;44:3580–3587. doi: 10.1016/j.molimm.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 82.Agrawal A, Dillon S, Denning TL, Pulendran B. ERK1−/− mice exhibit Th1 cell polarization and increased susceptibility to experimental autoimmune encephalomyelitis. J Immunol. 2006;176:5788–5796. doi: 10.4049/jimmunol.176.10.5788. [DOI] [PubMed] [Google Scholar]
- 83.Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, Kasprowicz DJ, Kellar K, Pare J, van Dyke T, Ziegler S, Unutmaz D, Pulendran B. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest. 2006;116:916–928. doi: 10.1172/JCI27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Karumuthil-Melethil S, Perez N, Li R, Vasu C. Induction of innate immune response through TLR2 and dectin 1 prevents type 1 diabetes. J Immunol. 2008;181:8323–8334. doi: 10.4049/jimmunol.181.12.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Depaolo RW, Tang F, Kim I, Han M, Levin N, Ciletti N, Lin A, Anderson D, Schneewind O, Jabri B. Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe. 2008;4:350–361. doi: 10.1016/j.chom.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McKimmie CS, Moore M, Fraser AR, Jamieson T, Xu D, Burt C, Pitman NI, Nibbs RJ, McInnes IB, Liew FY, Graham GJ. A TLR2 ligand suppresses inflammation by modulation of chemokine receptors and redirection of leukocyte migration. Blood. 2009 doi: 10.1182/blood-2008-08-174698. [DOI] [PubMed] [Google Scholar]
- 87.Malherbe L, Mark L, Fazilleau N, McHeyzer-Williams LJ, McHeyzer-Williams MG. Vaccine adjuvants alter TCR-based selection thresholds. Immunity. 2008;28:698–709. doi: 10.1016/j.immuni.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schwarz K, Storni T, Manolova V, Didierlaurent A, Sirard JC, Rothlisberger P, Bachmann MF. Role of Toll-like receptors in costimulating cytotoxic T cell responses. Eur J Immunol. 2003;33:1465–1470. doi: 10.1002/eji.200323919. [DOI] [PubMed] [Google Scholar]
- 89.Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, DiDonato J, Dziarski R, Akira S, Schoenberger SP, Raz E. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol. 2003;170:4102–4110. doi: 10.4049/jimmunol.170.8.4102. [DOI] [PubMed] [Google Scholar]
- 90.Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S, Soares H, Brimnes MK, Moltedo B, Moran TM, Steinman RM. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199:815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4:1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 92.Zhou S, Kurt-Jones EA, Mandell L, Cerny A, Chan M, Golenbock DT, Finberg RW. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol. 2005;35:822–830. doi: 10.1002/eji.200425730. [DOI] [PubMed] [Google Scholar]
- 93.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203:413–424. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wille-Reece U, Flynn BJ, Lore K, Koup RA, Kedl RM, Mattapallil JJ, Weiss WR, Roederer M, Seder RA. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc Natl Acad Sci U S A. 2005;102:15190–15194. doi: 10.1073/pnas.0507484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kwissa M, Amara RR, Robinson HL, Moss B, Alkan S, Jabbar A, Villinger F, Pulendran B. Adjuvanting a DNA vaccine with a TLR9 ligand plus Flt3 ligand results in enhanced cellular immunity against the simian immunodeficiency virus. J Exp Med. 2007;204:2733–2746. doi: 10.1084/jem.20071211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, Forehan SP, Mount A, Steptoe RJ, Shortman KD, de Koning-Ward TF, Belz GT, Carbone FR, Crabb BS, Heath WR, Villadangos JA. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- 97.Crompton PD, Mircetic M, Weiss G, Baughman A, Huang CY, Topham DJ, Treanor JJ, Sanz I, Lee FE, Durbin AP, Miura K, Narum DL, Ellis RD, Malkin E, Mullen GE, Miller LH, Martin LB, Pierce SK. The TLR9 ligand CpG promotes the acquisition of Plasmodium falciparum-specific memory B cells in malaria-naive individuals. J Immunol. 2009;182:3318–3326. doi: 10.4049/jimmunol.0803596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 99.Whitlock CA, Watson JD. B-cell differentiation in the CBA/N mouse. I. Slower maturation of mitogen and antigen-responsive B cells in mice expressing an X-linked defect. J Exp Med. 1979;150:1483–1497. doi: 10.1084/jem.150.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gururajan M, Jacob J, Pulendran B. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS ONE. 2007;2:e863. doi: 10.1371/journal.pone.0000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 102.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 103.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, Mackay F. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med. 2007;204:1959–1971. doi: 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008;29:249–260. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. 2003;101:4500–4504. doi: 10.1182/blood-2002-11-3569. [DOI] [PubMed] [Google Scholar]
- 106.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 107.Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science. 2006;314:1936–1938. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guay HM, Andreyeva TA, Garcea RL, Welsh RM, Szomolanyi-Tsuda E. MyD88 is required for the formation of long-term humoral immunity to virus infection. J Immunol. 2007;178:5124–5131. doi: 10.4049/jimmunol.178.8.5124. [DOI] [PubMed] [Google Scholar]
- 109.Geeraedts F, Goutagny N, Hornung V, Severa M, de Haan A, Pool J, Wilschut J, Fitzgerald KA, Huckriede A. Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by Toll-like receptor signalling. PLoS Pathog. 2008;4:e1000138. doi: 10.1371/journal.ppat.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Goriely S, Molle C, Nguyen M, Albarani V, Haddou NO, Lin R, De Wit D, Flamand V, Willems F, Goldman M. Interferon regulatory factor 3 is involved in Toll-like receptor 4 (TLR4)- and TLR3-induced IL-12p35 gene activation. Blood. 2006;107:1078–1084. doi: 10.1182/blood-2005-06-2416. [DOI] [PubMed] [Google Scholar]
- 112.Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, van Lieshout AW, Sprong T, van den Hoogen FH, van den Berg WB, Radstake TR. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005;52:2313–2322. doi: 10.1002/art.21278. [DOI] [PubMed] [Google Scholar]
- 113.Zhu Q, Egelston C, Vivekanandhan A, Uematsu S, Akira S, Klinman DM, Belyakov IM, Berzofsky JA. Toll-like receptor ligands synergize through distinct dendritic cell pathways to induce T cell responses: implications for vaccines. Proc Natl Acad Sci U S A. 2008;105:16260–16265. doi: 10.1073/pnas.0805325105. [DOI] [PMC free article] [PubMed] [Google Scholar]