

Abstract
Aldo-keto reductase 1C3 (AKR1C3) is an enzyme involved in metabolizing prostaglandins (PGs) and sex hormones. It metabolizes PGD2 to 9α11β-PGF2, diverting the spontaneous conversion of PGD2 to the PPARγ agonist, 15-Deoxy-Delta-12,14-prostaglandin J2 (15d-PGJ2). AKR1C3 is overexpressed in various malignancies, suggesting a tumor promoting function. This work investigates AKR1C3 expression in human non-melanoma skin cancers, revealing overexpression in squamous cell carcinoma (SCC). Effects of AKR1C3 overexpression were then evaluated using 3 SCC cell lines. AKR1C3 was detected in all SCC cell lines and its expression was upregulated in response to its substrate, PGD2. Although attenuating AKR1C3 expression in SCC cells by siRNA did not affect growth, treatment with PGD2 and its dehydration metabolite, 15d-PGJ2, decreased SCC proliferation in a PPARγ-dependent manner. In addition, treatment with the PPARγ agonist pioglitazone profoundly inhibited SCC proliferation. Finally, we generated an SCC cell line that stably overexpressed AKR1C3 (SCC-AKR1C3). SCC-AKR1C3 metabolized PGD2 to 9α11β-PGF2 12 fold faster than the parent cell line and was protected from the anti-proliferative effect mediated by PGD2. This work suggests that PGD2 and its metabolite 15d-PGJ2 attenuate SCC proliferation in a PPARγ-dependent manner, therefore activation of PPARγ by agonists such as Pioglitazone may benefit those at high risk of SCC.
Keywords: Aldo-keto reductase, PPARγ, Prostaglandin, Squamous cell carcinoma, Skin cancer
Introduction
Aldo-Keto Reductase 1C3 (AKR1C3) belongs to a large superfamily of NAD(P)(H)-dependent AKR enzymes which metabolize various substrates including prostaglandins and sex hormones [1]. In humans, the AKR1C subfamily consists of 4 enzymes termed AKR1C1, AKR1C2, AKR1C3 and AKR1C4 which despite their extensive structural overlap, exhibit different substrate binding preferences [2].
The role of AKR1C3 in steroid hormone metabolism includes the conversion of the relatively weak androgen Δ4-androstene-3,17-dione to its potent derivative, testosterone, as well as the reduction of the weak estrogen, estrone, to the more potent steroid, 17β-estradiol [2]. AKR1C3 can therefore indirectly regulate steroid hormone receptor activation under physiological and pathological conditions. With respect to this, previous work has shown that AKR1C3 is over-expressed in hormone-dependent cancers such as prostate, breast and endometrium suggesting that the capacity of AKR1C3 to convert weak sex hormones to their more potent derivatives may generate proliferative signaling to support tumor progression [3–8].
Unlike other AKR1C members, AKR1C3 can synthesize PGF2α from PGH2, an arachidonic acid derivative synthesized by cyclooxygenase [9]. Its role in prostaglandin metabolism also includes the conversion of PGD2, a pro-inflammatory lipid mediator, to 9α11β-PGF2 [9–11]. Thus, via its involvement in PG metabolism, AKR1C3 may regulate biological and pathological responses in hormone-independent tissues. In fact, AKR1C3 is overexpressed in various cancers originated from non-steroidogenic tissues such as kidney [12], central nervous system [13], pancreas, gastrointestinal tract and lung [14, 15]. AKR1C3 can activate carcinogens [16], modulate oxidative stress [17], and its polymorphisms is associated with risk for childhood leukemia [18] and bladder cancer [19]. Collectively, these observations suggest a tumor promoting function for AKR1C3 which is beyond its function in sex hormone metabolism. Interestingly, other AKR1C enzymes including AKR1C1 and AKR1C2 have been shown to be inducible in skin following UV light exposure which is the main cause of non-melanoma skin cancers [20].
This work investigates the expression and function of AKR1C3 in non-melanoma skin cancers. Results suggest the expression of AKR1C3 may facilitate squamous cell carcinoma growth in the setting of inflammation, and that the PPARγ agonist pioglitazone may be a useful treatment to decrease SCC growth.
Materials & Methods
SCC cell lines
Freshly isolated SCC tumors from sun-exposed areas were excised from patients by Moh’s surgery. In order to eliminate any possible surface contaminants, tumors were submerged in 70% ethanol for 10 minutes followed by 3 washes in sterile PBS. Using a scalpel, the outer surface of the tumor, including all necrotic areas was discarded and the viable tumor core was isolated and transferred to a sterile 30 mm tissue culture dish. The tumor core was very finely minced and suspended in a solution of 2000 units of Collagenase type 1 (MP Biomedicals, Cat # 195109) reconstituted in 1 ml KGM (Invitrogen Cat#17005042) and 1ml sterile DPBS and allowed to dissociate for 2hrs at 37°C. Processed tissue transferred to a 15ml conical tube and spun at 1000 rpm for 4 minutes. The supernatant was aspirated, and the tumor was resuspended in 2 ml of 0.25% trypsin-EDTA solution and trypsinized at 37°C for 30 more minutes. The trypsin reaction was quenched with 2ml FBS, the suspension filtered through a 100μm filter and then spun at 1000 rpm for 8 minutes to form a pellet. The supernatant was aspirated and the pellet was resuspended in KGM and transferred to collagen (Pure Col, Advanced BioMatrix) pre-coated T-25 flasks and allowed to adhere. Fibroblasts were removed from the initial culture by brief trypsinization that allowed fibroblasts, but not SCC cells to detach. Cells were routinely fed every 3–5 days with serum-free KGM containing 5ng/ ml EGF, 20μg/ml BPE, 100 U/ml penicillin and 100 μg/ml streptomycin. SCC-13 cell line was purchased from Harvard Skin Disease Research Center (http://rheinwaldlab.bwh.harvard.edu/cell.html) and maintained under the same cultivating conditions described above.
Immunohistochemistry
Samples were prepared and stained for AKR1C3 using the ImmunoCruz Staining System (Santa Cruz Biotechnology, Santa Cruz, CA) and ABC kit (Vector Laboratories, Burlingame CA) according to the manufacturer’s protocol using 1:50 dilution of the monoclonal anti-human AKR1C3 antibody. Sections were washed in TBST 3 times followed by 10 minutes incubation in AEC reagent (Vector Laboratories, Burlingame CA). Positive immunoreactive cells, identified by red stain, were visualized using a light microscope.
Cellular AKR1C3 activity
Cellular enzymatic activity of AKR1C3 was assessed by measuring the conversion of PGD2 to 9α11β-PGF2 using ELISA (catalog: 516521, Cayman Chemical, Ann Arbor, MI) as previously described [21]. Cells were seeded in 6 well plates and allowed to reach 90% confluence. Cells were treated with 1μM PGD2 (0.5ml/well) in growth medium without supplements for the indicated times. 9α11βPGF2 in each condition was determined in supernatants by ELISA following manufacturer directions and results were normalized to total protein. Each condition was evaluated for 9α11βPGF2 by averaging a minimum of 2 OD measurements.
Cell proliferation assessment by Click-iT assay
Cell proliferation was determined using Click-iT assay according to the protocol provided with Click-iT EdU Imaging Kit (Invitrogen #C10337). Cells were plated in chamber slides in growth media (KGM + additives) until 60% confluent. Cells were then treated as indicated. Proliferating cells were visualized using a fluorescence microscope (Nikon Eclipse E800 equipped with a Spot RT3 camera, Nikon, Melville, NY). Pictures were obtained using Spot advanced software (Diagnostic Instruments, Sterling Heights, MI). Five random fields from each condition were captured at X10 magnification and the number of proliferating cells normalized to total cell count in each field was determined. Results were averaged and presented as percent proliferation compared with DMSO treated controls.
Generation of AKR1C3-overexpressing cells (SCC-AKR1C3)
The AKR1C3-pLNCX2 retroviral construct was kindly provided by Dr. Trevor Penning (University of Pennsylvania, Philadelphia, PA). HEK293T packaging cells and additional reagents were kindly provided by Dr. Glynis Scott and Dr. Lei Xu (University of Rochester, Rochester, NY). AKR1C3-pLNCX2 or empty vector was added to HEK293T packaging cells and media from these cells was collected and added to SCCAM1 cells. Stable transfectants were selected by the inclusion of 0.6mg/ml neomycin to the growth medium.
Western blot analysis
Protein extraction and western blot procedures were carried out as previously described [21]. For all experiments 50μg protein was used and the primary antibodies were diluted as follows: Anti-AKR1C3 1:1000; anti-human β-actin 1:2000.
Reagents
Monoclonal mouse anti-human AKR1C3 (ab49680) and polyclonal rabbit anti-human β-actin (ab8227) were obtained from Abcam (Abcam, Cambridge, MA). Common Antibody Diluent purchased from BioGenex (BioGenex, San Ramon, CA). PGD2, 9α11β-PGF2, 15d-PGJ2 and GW9662 purchased from Caymen Chemical (Caymen Chemical, Ann Arbor, MI) and dissolved in dimethyl sulfoxide (DMSO) to give a stock concentration of 10mM.
Statistical analysis
The results are presented as means ± SEM. The appropriate t-test was performed using Microsoft Excel or GraphPad Prism. A P-value < 0.05 was considered statistically significant.
Results
AKR1C3 is overexpressed in SCC while not detected in the tumor mass of BCC
To investigate the expression of AKR1C3 in non-melanoma skin cancers, 6 human SCCs and 7 BCCs were analyzed by immunohistochemistry. Strong immunoreactivity with AKR1C3 antibody (red) was observed in the tumor mass of all SCCs while its expression in tumor nodules of all the tested BCCs was undetectable (figure 1a). In normal skin samples, modest immunoreactivity was most evident in the upper epidermal layers. Staining of the tumor stroma and vasculature of both types of cancer was also moderately positive.
Figure 1. AKR1C3 overexpression in tumor mass of SCC and its induction by PGD2.

(a) AKR1C3 expression was evaluated in six cases of SCC, seven cases of BCC and six cases of normal buttock skin by immunohistochemical staining. Representative figures demonstrating AKR1C3 expression in normal skin and within the tumor mass (T) and tumor stroma (S) of SCC and BCC. Strong immunoreactivity was observed in SCC while undetected in BCC. Normal skin showed moderate expression mainly in suprabasal layers (arrows). Bar=30μM. (b) expression of AKR1C3 in cell lines SCCAM1, SCCJV and SCC13. To evaluate the expression of AKR1C3 in the different SCC cell lines, cells cultured at confluence for 24 hours following by protein extraction and western blot analysis (n=3; representative blot is shown). (c) PGD2-induced AKR1C3 expression in SCC cells. SCC cells were treated for 24 hours with various doses of PGD2 or its AKR1C3-mediated metabolite 9α11βPGF2 as indicated and AKR1C3 expression was assessed by western blot. A representative experiment in which SCCAM1 was used is shown.
AKR1C3 is expressed in SCC cell lines and is up-regulated by its substrate, PGD2
To further investigate the possible role of AKR1C3 in SCC, AKR1C3 expression was investigated in 3 SCC cell lines derived from sun-exposed skin compared with normal keratinocytes. Expression of AKR1C3 protein was detected in all SCC cell lines (figure 1b). It was also observed that AKR1C3 expression was dependent on SCC cell confluence with stronger expression noted in highly confluent cells (not shown). To evaluate the effects of AKR1C3 on SCC proliferation, its expression was knocked down using siRNA and SCC cell proliferation was assessed by ClickiT assay. Despite profound knockdown (varied between 39.5–84%) of AKR1C3 expression in the SCC cultures, proliferation was not altered compared with mock-transfected cells (Figure 2S). In addition, we tested the effects of steroid hormones and PGs which are metabolized by AKR1C3 on its expression. We found that PGD2 but not 9α11β-PGF2 induced AKR1C3 expression in each cell line (representative data shown in figure 1c), while testosterone and 17β-estradiol had no effect (data not shown).
PGD2 and 15d-PGJ2 reduce SCC cell proliferation in a PPARγ-dependent manner
Following the observation that AKR1C3 expression is upregulated in SCC cells specifically in response to PGD2, we evaluated the effects of PGD2 and its metabolites on SCC proliferation. Each SCC cell line was treated for 24 hours with PGD2, its AKR1C3-mediated metabolite 9α11β-PGF2, or with the PGD2 spontaneous dehydration product, 15d-PGJ2. Proliferation was then assessed using a Click-iT assay. Representative detailed dose response data obtained in our SCCAM1 cell line are shown (figure 2a). 15d-PGJ2 and PGD2 but not 9α11β-PGF2 inhibited cell proliferation dose-dependently. Treatment with 15d-PGJ2 reduced proliferation more effectively than PGD2 at all tested concentrations where 5μM of 15d-PGJ2 completely eliminated SCC proliferation. In contrast, 9α11β-PGF2 had no significant effect on SCC proliferation at any tested dose.
Figure 2. PGD2 and 15d-PGJ2 attenuated SCC cell proliferation in a PPARγ-dependent manner.
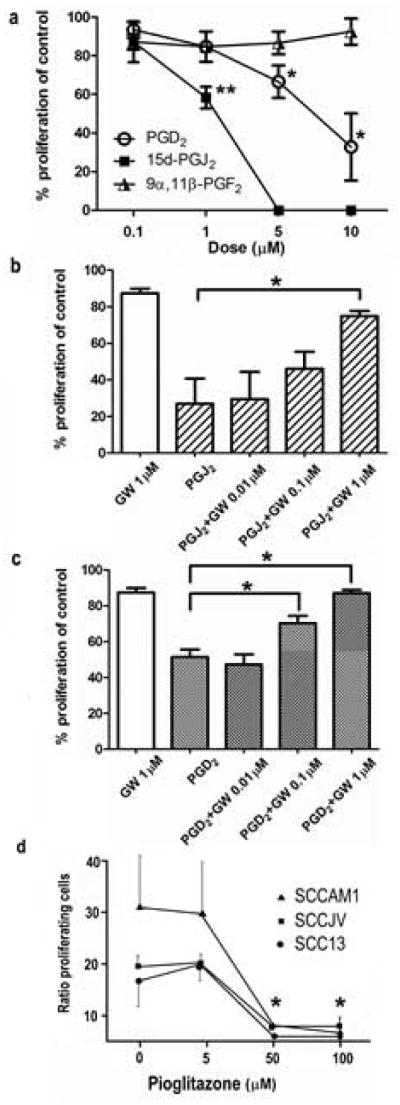
(a) SCCAM1 cells were treated with the indicated concentration of PGD2, 15d-PGJ2, 9α11β-PGF2 or DMSO for 24 hours followed by cell proliferation assessment by Click-iT assay. Data presented is the average of 3 experiments (±SEM), *P<0.005, **P=0.012. Similar trends were observed in SCCJV and SCC13 cell lines (not shown). To study the involvement of PPARγ activation in the anti-proliferative effects mediated by 15d-PGJ2 and by its precursor PGD2, SCCAM1 cells were treated with 1.5μM 15d-PGJ2 (b) or 7μM PGD2 (c) in the presence of the indicated concentration of the PPARγ specific inhibitor GW9662 (GW). Cell proliferation was assessed by Click-iT assay. n=3, *P<0.05. Similar trends were obtained with SCCJV and SCC13 cells (not shown). (d) The effects of Piogliazone on SCC proliferation. Each SCC cell line was treated for 24h as indicated and cell proliferation was assessed by Click-iT assay. (n=3)
15d-PGJ2 is a spontaneous dehydration product of PGD2 and a potent activator of PPARγ. To evaluate whether the anti-proliferative effects induced by PGD2 and 15d-PGJ2 were mediated by PPARγ activation, cells were treated with 1.5μM 15d-PGJ2 or 7μM of PGD2 in the presence of the PPARγ inhibitor, GW9662, and cell proliferation was assessed (Figure 2b and 2c). The presence of PGD2 and 15d-PGJ2 reduced SCC proliferation to 51% and 27% of DMSO-treated control cells, respectively. Although treatment with 1μM GW9662 itself modestly reduced SCC proliferation compared to DMSO treated controls, its combination with PGD2 and 15dPGJ2 dose-dependently reversed the anti-proliferative effects mediated by both prostaglandins. The presence of 1μM GW9662 partially restored the proliferation rate of SCC cells treated with 15dPGJ2 and completely reversed the anti-proliferative effects mediated by PGD2.
Since the proliferation of all SCC lines was profoundly inhibited by treatment with 15d-PGJ2, we next tested the effects of the PPARγ agonist, Pioglitazone, on SCC proliferation. Each SCC line was treated with Pioglitazone as indicated for 24 hours and cell proliferation was evaluated as before (fig 2d). Little effect was evident at the lowest dose tested, but exposure to 50 or 100μM Pioglitazone effectively inhibited cell growth. Taken together, this data implies that the anti-proliferative effect mediated by PGD2 is due to its spontaneous conversion to 15d-PGJ2 followed by activation of PPARγ.
AKR1C3 over-expressing SCC cells are protected from the anti-proliferative effects of PGD2
To evaluate whether AKR1C3 overexpression protects SCC cells from PGD2 in vitro, we used SCCAM1 cells to generate an SCC cell line that stably over-expresses AKR1C3 by means of retroviral infection. The SCC cell line was infected with either AKR1C3 over-expressing retroviral construct (SCC-AKR1C3) or with an empty vector (SCC-control). AKR1C3 over-expression in SCC-AKR1C3 cells was confirmed by western blot analysis (Figure 3a). To verify that the over-expressed AKR1C3 is enzymatically active, we compared the capacity of SCC-AKR1C3 and SCC-control cells to mediate the AKR1C3 specific conversion of exogenous PGD2 to 9α11β-PGF2 by ELISA (Figure 3b). Concentrations of 9α11β-PGF2 were markedly higher in supernatants of SCC-AKR1C3 cells compared with SCC-controls at all tested time points.
Figure 3. Over-expression of AKR1C3 in SCCAM1 cells increases their capacity to metabolize PGD2 to 9α11β-PGF2 and protects against PGD2 anti-proliferative effects.

(a) SCCAM1 cells were stably infected with either an AKR1C3 over-expressing retroviral construct (SCC-AKR1C3) or with empty vector (SCC-control). Over-expression of AKR1C3 was confirmed by western blot, n=2. (b) Over-expression of AKR1C3 in SCC cells increases their capacity to metabolize PGD2 to 9α11βPGF2. Cells were treated with PGD2-containing media for the indicated times and 9α11β-PGF2, was measured by ELISA (n=2). (c) SCC-AKR1C3 or SCC-control cells were incubated for 24 hours with various doses of PGD2 as indicated. Cell proliferation was then assessed by Click-iT assay. n=3, *P=0.0026.
Finally, the effect of AKR1C3 overexpression on proliferation of SCC cells treated with PGD2 was evaluated. SCC-AKR1C3 and SCC-control cells were treated with the indicated doses of PGD2 for 24 hours, and proliferation was assessed by Click-IT assay. While treatment with 10μM PGD2 reduced SCC-control proliferation to 44% of control, SCC-AKR1C3 cells retained approximately 70% of their proliferative capacity (Figure 3c). This resistance to PGD2 suggests that AKR1C3 overexpression in SCCs may protect tumors from the anti-proliferative effects mediated by this PG.
Discussion
AKR1C3 has been mostly studied in malignancies of classical steroidogenic tissues such as breast and prostate [3, 7, 8, 22]. This is due to its involvement in generating potent sex hormones such as testosterone and 17β-estradiol. However, recent work has shown altered AKR1C3 expression in tumors of various tissues such as lung, brain and kidney suggesting a tumerogenic function beyond its role in sex hormone metabolism [12, 13].
In this work we characterized the expression levels of AKR1C3 in non-melanoma skin cancers, demonstrating that AKR1C3 was markedly expressed by malignant SCC cells while its expression was undetected in tumor nodules of BCC. This finding aligns with previous observations showing that AKR1C3 overexpression was detected in malignancies with a more differentiated phenotype. For example, high AKR1C3 expression was detected in renal cell carcinoma, papillary urothelial carcinoma, glial neoplasms, meningiomas, lung SCC and adenocarcinoma compared with reduced or undetectable AKR1C3 expression in tumors with more embryonic phenotypes of the same tissue [12, 13, 15].
The current work provides further support for AKR1C3 being a marker of tumors with more differentiated characteristics, and suggests it may be worthwhile to assess AKR1C3 as a prognostic marker for cutaneous SCC.
The tumor mass of SCC is comprised of a heterogeneous population of malignant keratinocytes at various stages of differentiation. Different levels of AKR1C3 expression were detected within the epithelial SCC mass, with stronger immunoreactivity found in areas of moderately differentiated keratinocytes. Interestingly, this distribution pattern aligns with its relatively higher expression in the spinous and granular layers of normal epidermis and in differentiated cultured keratinocytes [21].
To study the role of AKR1C3 in cutaneous SCC, we established two new skin SCC cell lines from tumors arising on sun-exposed skin (SCCAM1 & SCCJV) and used one existing SCC line (SCC13). We further confirmed the epithelial origin as well as malignant potential of the newly established lines (Figure S1). Our SCC cells expressed keratin 5 (figure S1a), maintained a proliferative phenotype for as many as 70 passages and were able retain their proliferative capacity following an exposure for several days to concentrations of calcium sufficient to induce terminal differentiation of normal keratinocytes (figure S1b). Furthermore, subcutaneous injections of the SCC cell lines into immunocompromised mice resulted in the formation of SCC-like tumors in vivo (figure S1c). Taken together, these cell lines meet typical criteria for malignancy validating them as a model for studying molecular mechanisms in skin SCC [23–25].
Our work demonstrates that AKR1C3 is expressed in each cell line studied; however, attenuation of its expression did not modulate SCC cell growth (figure S2). Our examination therefore looked at the way in which SCC cells might respond to substrates and products formed by the action of AKR1C3. Under our experimental conditions, estrogen and testosterone had no effect on SCC proliferation or on the enzyme expression (data not shown). Since AKR1C3 is also involved in PG metabolism, we examined the effects of relevant prostaglandins on SCC proliferation and this enzyme expression. Interestingly, among the tested PGs, only PGD2, a substrate of AKR1C3, induced its upregulation in SCC cells suggesting the existence of a positive feedback loop wherein in the presence of high PGD2 concentrations, SCC cells up-regulate its metabolizing enzyme. It would be interesting to evaluate whether PGD2 induces AKR1C3 in other cancer cell lines especially of hormone-dependent malignancies such as breast and prostate. Upregulation of AKR1C3 due to high PGD2 levels in the microenvironment of hormone-dependent cancers may indirectly result in increased potent sex hormone synthesis leading to excess activation of proliferative signaling pathways.
Large numbers of inflammatory cells with the capacity to synthesize and secrete the pro-inflammatory mediator PGD2 are often observed in SCC tumors. Increased numbers of mast cells, the major PGD2 secreting cell type, have been detected in SCC of the lip [26], oral cavity [27], cervix [28] and esophagus [29]. Parizi et al assessed the number of mast cells in SCC of the mouth compared with SCC of the skin. They found the total number of mast cells were elevated in the tumor site compared with a marginal area, and that SCC of the skin demonstrated higher mast cell concentrations, in some cases more than 200 cells/mm2 [30]. Therefore, it is reasonable to assume the presence of high levels of PGD2 within the tumor microenvironment.
PGD2 is a relatively unstable mediator, with an apparent half-life of 30 minutes in plasma [31], after which it is spontaneously dehydrates to form 15d-PGJ2, a potent physiological PPARγ agonist [32]. The current work shows that PGD2 and its spontaneous dehydration product 15d-PGJ2, but not 9α11β-PGF2, an AKR1C3-mediated metabolite of PGD2, significantly attenuate SCC cell proliferation. Treatment with the selective PPARγ inhibitor GW9662 reversed the anti-proliferative effects of both mediators, supporting the role of PPARγ activation in this effect.
To model the observed overexpression of AKR1C3 in SCC and to test its possible function in modulating prostaglandin-induced changes in SCC proliferation, we generated AKR1C3 overexpressing SCC cells (SCC-AKR1C3) which metabolized PGD2 to 9α11β-PGF2 faster than parental cells (12.3 fold on average). Since 9α11β-PGF2 and 15d-PGJ2 share the same precursor, the observed elevation of 9α11β-PGF2 in SCC-AKR1C3 cells treated with PGD2 must also be accompanied by a reduction of 15d-PGJ2 levels. This relationship has been documented in similar experiments done in AKR1C3 overexpressing MCF-7 breast cancer cells. Using LC/MS analysis, the authors showed a decrease in the amount of spontaneous dehydration of PGD2 to 15d-PGJ2 by instead producing an accumulation of 9α11β-PGF2 [11]. Because of their increased capacity to convert exogenous PGD2 to 9α11β-PGF2, SCC-AKR1C3 cells can better maintain their proliferative capacity following PGD2 challenge, compared with parental SCC-control cells. Statistically significant differences between the proliferation of SCC-AKR1C3 and SCC-control cells were only observed at relatively high concentrations of PGD2. This could be explained by the robust capacity of PGD2 to induce endogenous expression of AKR1C3 in SCC-control cells which likely partially protected them from PGD2 effects during treatment.
Since 15d-PGJ2 was able to inhibit SCC growth, we wanted further evidence that this action was mediated by PPARγ activation. Recent work has demonstrated the potential contribution of PPARγ to skin cancer in a mouse photocarcinogenesis model [33]. Pioglitazone is an oral medication utilized in clinical practice for treatment of diabetes. We therefore tested the PPARγ activator Pioglitazone on SCC proliferation in our cell lines. These experiments showed Pioglitazone profoundly inhibited SCC cell line proliferation (Figure 2d). Daily dosing with Pioglitazone produces serum levels over time which are similar to those used in the experiments reported here (50 μM). In addition, pioglitazone has two active metabolites [34, 35]. When tested for selectivity of activation for all PPARs, Pioglitazone was found to be a modest activator of PPARα, a robust activator of PPARγ with little effect on other PPARs [36]. The efficacy of Pioglitazone in suppressing proliferation in the 3 cell lines tested here suggests that it may be effective clinically for individuals with a high risk of developing skin cancer, such as individuals with transplants on immunosuppressive therapy. More in vitro work testing SCC cell lines is needed.
We propose a model (figure 4) in which over-expression of AKR1C3 in SCC can benefit malignant cells due to their enhanced capacity to metabolize PGD2 to 9α11β-PGF2, resulting in decreased levels of the anti-proliferative and pro-apoptotic metabolite 15d-PGJ2 within the tumor microenvironment. The profound effects observed here on SCC proliferation by agents that influence PPARγ activation support the idea that manipulation of PPARγ activity in individuals prone to skin cancer may have therapeutic benefit.
Figure 4. AKR1C3 overexpression in SCC protects against the anti-proliferative effects mediated by PGD2- a proposed model.
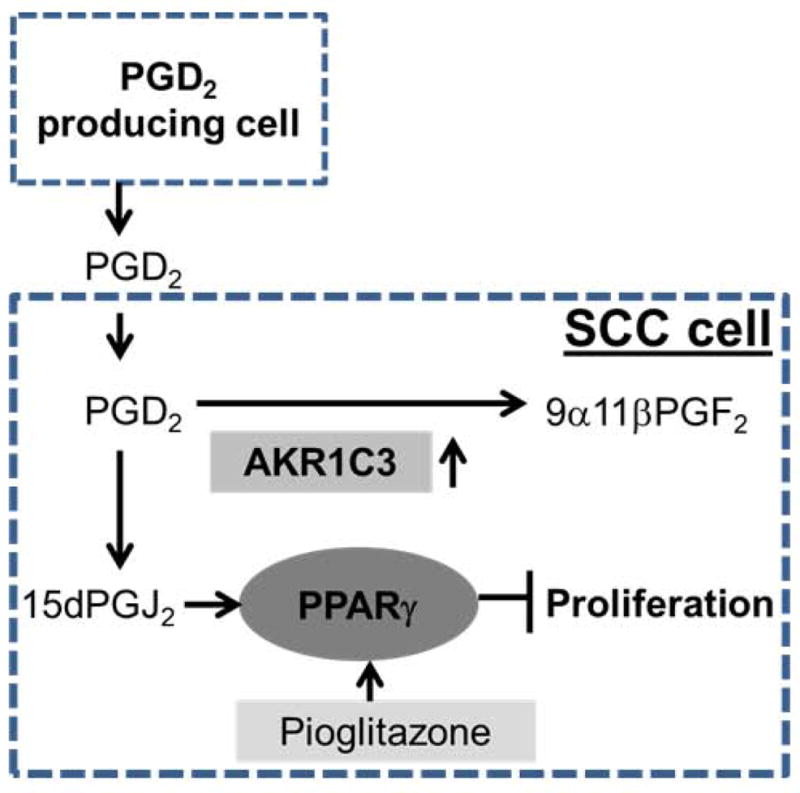
PGD2 is produced by either activated mast cells or other cell types within the tumor microenvironment of SCCs. PGD2 is non-enzymatically converted to 15d-PGJ2. This spontaneous metabolite of PGD2 inhibits SCC proliferation in a PPARγ-dependent manner. The model proposes that when SCC cells overexpress the PGD2 metabolizing enzyme AKR1C3, they are protected from 15dPGJ2–mediated inhibition of proliferation, instead producing 9α11βPGF2. In addition, our work suggests that direct activation of PPARγ via Pioglitazone inhibits SCC proliferation and therefore this drug may be further evaluated for treating cutaneous SCC patients.
Supplementary Material
Figure S1: Establishment and Characterization of SCC cell lines
In order to investigate the role of AKR1C3 in SCC, we used two newly established SCC cell lines derived from human SCC arising on sun-exposed skin (SCCAM1 and SCCJV) and one existing SCC cell line (SCC13). The new cell lines express keratin 5 (Figure S1a) and have been successfully maintained in the laboratory for up to 70 passages. In addition, our main SCC cell line SCCAM1 was able to retain its proliferative capacity following calcium-induced terminal differentiation challenge as compared with primary human keratinocytes (PHK; figure S1b; the other SCC cell lines were not tested). The tumorigenicity of all cell lines was also evaluated in vivo by subcutaneously injecting 5 × 106 cells per site into both flanks of two NOD.Cg-Prkdcscid ll2rgtm1Wjl/SzJ immunodeficient mice (at least 4 tumor injections per cell line) and tumor growth was monitored weekly for up to 18 weeks. All cell lines were found to form tumors consistently. Histological examination of excised SCCAM1-derived tumors showed moderately differentiated SCC-like tumor morphology, resembling the morphology of the original tumor (figure S1c; histology of other tumors derived from SCCJV and SCC13 was not assessed).
Figure S2: Attenuation of AKR1C3 by siRNA does not affect SCC proliferation.
SCC cells were seeded in 6 well plates and treated with 10nM of either AKR1C3 stealth siRNA (oligo ID: HSS112670; Invitrogen) or non-specific siRNA for 24 hours. Protein was extracted and AKR1C3 knock down was evaluated by western blot analysis (figure S2a). Protein KO was assessed in ImageJ by measuring the AKR1C3 band intensity normalized to actin. To assess whether AKR1C3 plays a direct role in SCC proliferation the different SCC cells were treated with either 10nM of AKR1C3 siRNA or non-specific siRNA for 24 hours followed by proliferation assessment using Click-iT assay as described in methods (shown is an average +/− SEM of 3 experiments). No direct effect of AKR1C3 knock down on SCC cell proliferation was noted as a P-value lower than 0.05 (t-test) was not met (Figure S2b).
Acknowledgments
Alon Mantel and Amanda Carpenter-Mendini performed and analyzed all experiments besides the ones showed in figures 1b, 2d and S2 that were done by JoAnne VanBuskirk. SCCAM1, SCC-AKR1C3 and SCC-control established by Alon Mantel. SCCJV cell line was generated by JoAnne VanBuskirk. Manuscript was written and edited by Alon Mantel and Alice Pentland.
This work was supported by National Institutes of Health Grant NIH RO1CA117821. We gratefully acknowledge the discussions, materials and help from Drs. Glynis Scott and Lei Xu of the University of Rochester. AKR1C3 plasmid was kindly provided by Dr. Trevor Penning, University of Pennsylvania.
Footnotes
Conflict of interests: none.
References
- 1.Jez JM, Penning TM. The aldo-keto reductase (AKR) superfamily: an update. Chemico-biological interactions. 2001;130–132(1–3):499–525. doi: 10.1016/s0009-2797(00)00295-7. [DOI] [PubMed] [Google Scholar]
- 2.Penning TM, et al. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351(Pt 1):67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrns MC, Penning TM. Type 5 17beta-hydroxysteroid dehydrogenase/prostaglandin F synthase (AKR1C3): role in breast cancer and inhibition by non-steroidal anti-inflammatory drug analogs. Chem Biol Interact. 2009;178(1–3):221–7. doi: 10.1016/j.cbi.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanbrough M, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer research. 2006;66(5):2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 5.Rizner TL, et al. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Molecular and cellular endocrinology. 2006;248(1–2):126–35. doi: 10.1016/j.mce.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Yepuru M, et al. Steroidogenic Enzyme AKR1C3 Is a Novel Androgen Receptor-Selective Coactivator that Promotes Prostate Cancer Growth. Clin Cancer Res. 2013;19(20):5613–25. doi: 10.1158/1078-0432.CCR-13-1151. [DOI] [PubMed] [Google Scholar]
- 7.Dozmorov MG, et al. Elevated AKR1C3 expression promotes prostate cancer cell survival and prostate cell-mediated endothelial cell tube formation: implications for prostate cancer progression. BMC Cancer. 2010:10, 672. doi: 10.1186/1471-2407-10-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamid AR, et al. Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol Med. 2012:18, 1449–55. doi: 10.2119/molmed.2012.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komoto J, et al. Prostaglandin F2alpha formation from prostaglandin H2 by prostaglandin F synthase (PGFS): crystal structure of PGFS containing bimatoprost. Biochemistry. 2006;45(7):1987–96. doi: 10.1021/bi051861t. [DOI] [PubMed] [Google Scholar]
- 10.Matsuura K, et al. Identification of a principal mRNA species for human 3alpha-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. Journal of biochemistry. 1998;124(5):940–6. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 11.Byrns MC, et al. Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. J Steroid Biochem Mol Biol. 2010;118(3):177–87. doi: 10.1016/j.jsbmb.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azzarello JT, et al. Expression of AKR1C3 in renal cell carcinoma, papillary urothelial carcinoma, and Wilms’ tumor. Int J Clin Exp Pathol. 2009;3(2):147–55. [PMC free article] [PubMed] [Google Scholar]
- 13.Park AL, et al. Differential expression of type 2 3alpha/type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) in tumors of the central nervous system. Int J Clin Exp Pathol. 2010;3(8):743–54. [PMC free article] [PubMed] [Google Scholar]
- 14.Chang TS, et al. Expression of aldo-keto reductase family 1 member C3 (AKR1C3) in neuroendocrine tumors & adenocarcinomas of pancreas, gastrointestinal tract, and lung. Int J Clin Exp Pathol. 2013;6(11):2419–29. [PMC free article] [PubMed] [Google Scholar]
- 15.Miller VL, et al. Aldo-keto reductase family 1 member C3 (AKR1C3) is expressed in adenocarcinoma and squamous cell carcinoma but not small cell carcinoma. Int J Clin Exp Pathol. 2012;5(4):278–89. [PMC free article] [PubMed] [Google Scholar]
- 16.Lan Q, et al. Oxidative damage-related genes AKR1C3 and OGG1 modulate risks for lung cancer due to exposure to PAH-rich coal combustion emissions. Carcinogenesis. 2004;25(11):2177–81. doi: 10.1093/carcin/bgh240. [DOI] [PubMed] [Google Scholar]
- 17.Birtwistle J, et al. The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: implications for leukemogenesis. Mutat Res. 2009;662(1–2):67–74. doi: 10.1016/j.mrfmmm.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 18.Liu CY, et al. Maternal and offspring genetic variants of AKR1C3 and the risk of childhood leukemia. Carcinogenesis. 2008;29(5):984–90. doi: 10.1093/carcin/bgn071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Figueroa JD, et al. Bladder cancer risk and genetic variation in AKR1C3 and other metabolizing genes. Carcinogenesis. 2008;29(10):1955–62. doi: 10.1093/carcin/bgn163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marin YE, Seiberg M, Lin CB. Aldo-keto reductase 1C subfamily genes in skin are UV-inducible: possible role in keratinocytes survival. Exp Dermatol. 2009;18(7):611–8. doi: 10.1111/j.1600-0625.2008.00839.x. [DOI] [PubMed] [Google Scholar]
- 21.Mantel A, et al. Aldo-keto reductase 1C3 is expressed in differentiated human epidermis, affects keratinocyte differentiation, and is upregulated in atopic dermatitis. J Invest Dermatol. 2012;132(4):1103–10. doi: 10.1038/jid.2011.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penning TM, Byrns MC. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci. 2009:1155, 33–42. doi: 10.1111/j.1749-6632.2009.03700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rheinwald JG, Beckett MA. Defective terminal differentiation in culture as a consistent and selectable character of malignant human keratinocytes. Cell. 1980;22(2 Pt 2):629–32. doi: 10.1016/0092-8674(80)90373-6. [DOI] [PubMed] [Google Scholar]
- 24.Parkinson EK, et al. Differential effects of complete and second-stage tumour promoters in normal but not transformed human and mouse keratinocytes. Carcinogenesis. 1984;5(8):1071–7. doi: 10.1093/carcin/5.8.1071. [DOI] [PubMed] [Google Scholar]
- 25.Yang LC, Ng DC, Bikle DD. Role of protein kinase C alpha in calcium induced keratinocyte differentiation: defective regulation in squamous cell carcinoma. J Cell Physiol. 2003;195(2):249–59. doi: 10.1002/jcp.10248. [DOI] [PubMed] [Google Scholar]
- 26.Costa NL, et al. Density and migration of mast cells in lip squamous cell carcinoma and actinic cheilitis. Histology and histopathology. 2009;24(4):457–65. doi: 10.14670/HH-24.457. [DOI] [PubMed] [Google Scholar]
- 27.Sharma B, et al. Immunohistochemical evaluation of mast cells and angiogenesis in oral squamous cell carcinoma. Indian J Dent Res. 2010;21(2):260–5. doi: 10.4103/0970-9290.66655. [DOI] [PubMed] [Google Scholar]
- 28.Jing Y, et al. Distribution and histochemical characteristics of mast cells in stroma of the cervix squamous cell carcinoma. Chin Med J (Engl) 1993;106(9):698–702. [PubMed] [Google Scholar]
- 29.Tomita M, et al. Association of mast cells with tumor angiogenesis in esophageal squamous cell carcinoma. Dis Esophagus. 2001;14(2):135–8. doi: 10.1046/j.1442-2050.2001.00171.x. [DOI] [PubMed] [Google Scholar]
- 30.Parizi AC, et al. A comparison between the concentration of mast cells in squamous cell carcinomas of the skin and oral cavity. An Bras Dermatol. 2010;85(6):811–8. doi: 10.1590/s0365-05962010000600006. [DOI] [PubMed] [Google Scholar]
- 31.Schuligoi R, et al. PGD2 metabolism in plasma: kinetics and relationship with bioactivity on DP1 and CRTH2 receptors. Biochem Pharmacol. 2007;74(1):107–17. doi: 10.1016/j.bcp.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Shibata T, et al. 15-deoxy-delta 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J Biol Chem. 2002;277(12):10459–66. doi: 10.1074/jbc.M110314200. [DOI] [PubMed] [Google Scholar]
- 33.Sahu RP, et al. Mice lacking epidermal PPARgamma exhibit a marked augmentation in photocarcinogenesis associated with increased UVB-induced apoptosis, inflammation and barrier dysfunction. International journal of cancer. Journal international du cancer. 2012;131(7):E1055–66. doi: 10.1002/ijc.27562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christensen ML, et al. Single- and multiple-dose pharmacokinetics of pioglitazone in adolescents with type 2 diabetes. Journal of clinical pharmacology. 2005;45(10):1137–44. doi: 10.1177/0091270005279578. [DOI] [PubMed] [Google Scholar]
- 35.Deng LJ, Wang F, Li HD. Effect of gemfibrozil on the pharmacokinetics of pioglitazone. European journal of clinical pharmacology. 2005;61(11):831–6. doi: 10.1007/s00228-005-0042-6. [DOI] [PubMed] [Google Scholar]
- 36.Seimandi M, et al. Differential responses of PPARalpha, PPARdelta, and PPARgamma reporter cell lines to selective PPAR synthetic ligands. Analytical biochemistry. 2005;344(1):8–15. doi: 10.1016/j.ab.2005.06.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Establishment and Characterization of SCC cell lines
In order to investigate the role of AKR1C3 in SCC, we used two newly established SCC cell lines derived from human SCC arising on sun-exposed skin (SCCAM1 and SCCJV) and one existing SCC cell line (SCC13). The new cell lines express keratin 5 (Figure S1a) and have been successfully maintained in the laboratory for up to 70 passages. In addition, our main SCC cell line SCCAM1 was able to retain its proliferative capacity following calcium-induced terminal differentiation challenge as compared with primary human keratinocytes (PHK; figure S1b; the other SCC cell lines were not tested). The tumorigenicity of all cell lines was also evaluated in vivo by subcutaneously injecting 5 × 106 cells per site into both flanks of two NOD.Cg-Prkdcscid ll2rgtm1Wjl/SzJ immunodeficient mice (at least 4 tumor injections per cell line) and tumor growth was monitored weekly for up to 18 weeks. All cell lines were found to form tumors consistently. Histological examination of excised SCCAM1-derived tumors showed moderately differentiated SCC-like tumor morphology, resembling the morphology of the original tumor (figure S1c; histology of other tumors derived from SCCJV and SCC13 was not assessed).
Figure S2: Attenuation of AKR1C3 by siRNA does not affect SCC proliferation.
SCC cells were seeded in 6 well plates and treated with 10nM of either AKR1C3 stealth siRNA (oligo ID: HSS112670; Invitrogen) or non-specific siRNA for 24 hours. Protein was extracted and AKR1C3 knock down was evaluated by western blot analysis (figure S2a). Protein KO was assessed in ImageJ by measuring the AKR1C3 band intensity normalized to actin. To assess whether AKR1C3 plays a direct role in SCC proliferation the different SCC cells were treated with either 10nM of AKR1C3 siRNA or non-specific siRNA for 24 hours followed by proliferation assessment using Click-iT assay as described in methods (shown is an average +/− SEM of 3 experiments). No direct effect of AKR1C3 knock down on SCC cell proliferation was noted as a P-value lower than 0.05 (t-test) was not met (Figure S2b).