

Abstract
NUT midline carcinoma (NMC) is an aggressive subtype of squamous cell carcinoma that typically harbors BRD4/3-NUT fusion oncoproteins that block differentiation and maintain tumor growth. In 20% of cases NUT is fused to uncharacterized non-BRD gene(s). We established a new patient-derived NMC cell line (1221) and demonstrated that it harbors a novel NSD3-NUT fusion oncogene. We find that NSD3-NUT is both necessary and sufficient for the blockade of differentiation and maintenance of proliferation in NMC cells. NSD3-NUT binds to BRD4, and BRD bromodomain inhibitors induce differentiation and arrest proliferation of 1221 cells. We find further that NSD3 is required for the blockade of differentiation in BRD4-NUT-expressing NMCs. These findings identify NSD3 as a novel critical oncogenic component and potential therapeutic target in NMC.
Introduction
Hematopoietic and mesenchymal malignancies are often characterized by translocation-associated fusion oncoproteins that block differentiation, whereas many epithelial cancers are typified by multiple sequential mutations that progress in a multistep pathway to carcinogenesis. One exception of an epithelial carcinoma that is driven by a fusion-oncogene is NUT midline carcinoma (NMC). NMC is defined by chromosomal rearrangement of the NUT gene (aka NUTM1), which is most commonly fused to the BET family genes BRD4 and BRD3 (1, 2), defined by the presence of dual bromodomains and an extraterminal (ET) domain. BRD-NUT oncoproteins’ primary mechanism is to block differentiation and maintain cells in a highly proliferative, poorly differentiated state. This poorly differentiated cancer is far more aggressive than even small cell carcinoma of the lung, with a median survival of 6.7 months (3), and there exist no effective treatment options.
Recent excitement in small molecule BET inhibitors arose from the demonstration of the therapeutic targeting of BRD-NUT oncoproteins in NMC in vitro and in pre-clinical models (4). This has led to a clinical trial using the GSK BET inhibitor drug, GSK-525762A, now enrolling NMC and other solid tumors (http://www.clinicaltrials.gov/ct2/show/NCT01587703?term=NMC&rank=1). BET inhibitors are acetyl-histone mimetics that target the acetyl-histone binding pocket of BET protein chromatin-reading bromodomains, such as those of BRD2, 3, 4 and T (4, 5). BET inhibitors induced differentiation and proliferation arrest of NMC, and are a potential form of differentiation therapy in this disease. However, it is not known how interference with chromatin binding leads to inhibition of the blockade of differentiation by BRD-NUT oncoproteins, because the mechanism by which BRD-NUT blocks differentiation is unclear. Evidence suggests that deregulation of MYC expression by BRD-NUT may be key to the blockade of differentiation (6), but it remains to be determined whether BRD-NUT acts directly or indirectly.
Known functional domains of BRD4 that are present in BRD-NUT fusions may give clues to its function. Wild type BRD4 binds to acetylated histones and the positive transcriptional elongation factor, P-TEFb with its bromodomains (7), and is associated with transcriptional activation of target genes (7, 8). Although the function of NUT, an entirely unstructured protein, is unknown, it binds to and activates the histone acetyltransferase (HAT), p300 (9). Both of the bromodomains, and the p300-binding domain are present in BRD-NUT oncoproteins. This has led to the hypothesis that BRD-NUT fusion oncoproteins tether HATs and transcriptional co-factors to chromatin via their bromodomains, leading to a feed-forward process of acetylation and recruitment that results in sequestration of these factors away from pro-differentiation genes to pro-growth genes, such as MYC (2, 9).
The role of the ET domain and its binding proteins has not been investigated in the context of BRD-NUT oncoproteins. Here we describe a novel fusion in a NUT-variant NMC between the methyltransferase protein, NSD3, that has been previously shown to associate with the ET domains of BET proteins (8), and NUT. The finding suggested that NSD3 may be a key component of the BRD-NUT oncogenic complex. Thus, we investigated the oncogenic role of NSD3 in this NUT-variant NMC as well as more typical BRD4-NUT NMCs.
Results
A Novel NSD3-NUT Fusion in NUT Midline Carcinoma
A poorly differentiated squamous cell carcinoma of the mediastinum (Figure 1A) metastatic to the femur of a 12 year old girl was referred to us for molecular diagnostic testing for NUT midline carcinoma. Immunohistochemical analysis revealed diffuse nuclear expression of the NUT protein, a feature that is diagnostic of NMC (Figure 1B (10)). Fluorescent in situ hybridization (FISH) demonstrated rearrangement of the NUT gene locus on chromosome 15q14, however neither BRD4 nor BRD3 rearrangement were detected. Discarded live tumor tissue from a metastatic focus in the patient’s lung was collected under institutional review board approval through the NUT midline carcinoma registry (www.NMCRegistry.org). From this tissue the first known NUT-variant cell line, 1221, was established. To determine the putative partner gene to NUT, we performed comprehensive RNA-sequencing on RNA purified from 1221. We identified an in-frame transcript fusing the 5’ coding sequence of NSD3 (exons 1–7) to exons 2–7 of NUT (Figure 1C). Expression of the NSD3-NUT fusion oncoprotein was verified by immunobloting with an antibody to NUT, revealing an approximately 200kDa band that is similar in size to BRD3-NUT, but smaller than BRD4-NUT (Figure 1D). Knockdown using siRNAs targeting NSD3 led to a disappearance of the putative NSD3-NUT band, as did siRNAs targeting NUT, confirming the identity of the NSD3 and NUT portions of the NSD3-NUT fusion protein (Figure 1E). Genomic fusion of the NSD3 and NUT genes was confirmed by FISH, demonstrating bring-together of NUT and NSD3 probes (Figure 1F). Likewise, the expression of an NSD3-NUT mRNA was demonstrated by reverse transcriptase PCR (RT-PCR, Figure 1G). Cytogenetic analysis of the 1221 cell line was consistent with a NSD3-NUT fusion, revealing a t(8;15)(p12;q15) translocation, and metaphase FISH demonstrated localization of the NUT probe near the NSD3 chromosomal region (8p11.23) (Supplementary Figures S1–2). Several additional aberrations of unknown significance were also present.
Figure 1. A novel NSD3-NUT fusion is identified in NUT midline carcinoma.

(A) Histology of the NMC from which the 1221 cell line was derived reveals a very poorly differentiated tumor (400× magnification).
(B) Immunohistochemistry of the tumor using the anti-NUT monoclonal antibody, C52 (400× magnification).
(C) RNA-sequencing reads spanning the junction of NSD3 and NUT.
(D) Immunoblot of three NMC cell lines and 293T control cells stained with AX.1 polyclonal antibody to NUT.
(E) Immunoblot of the 1221 cell line 48h following transfection with control (CTRL), NSD3, and NUT siRNAs stained with the AX.1 antibody to NUT.
(F) NSD3-NUT dual color bring-together fluorescent in situ hybridization assay (1000× magnification) using BAC probes telomeric (3’) to NUT (green), and BAC probes centromeric (5’) to NSD3 (red) as depicted in the chromosomes 8 and 15 ideograms. Yellow arrows indicate NSD3-NUT fusions.
(G) Gel electrophoresis of PCR of TC-797 and 1221 cell lines with (+) and without (−) reverse transcriptase reaction.
(H) Schematic of the NSD3-NUT predicted encoded protein in comparison with NSD3, NUT, and BRD4-NUT. Abbreviations: PWWP, Pro-Trp-Trp-Pro motif; PHD, PHD finger (Plant Homeo Domain); SET, Su(var)3–9, Enhancer-of-zeste and Trithorax domain; C/H rich, Cys–His-rich region; NLS, nuclear localization sequence; NES, nuclear export signal sequence; Bromo, bromodomain; ET, extra-terminal domain. Arrows indicated breakpoints.
(I) NSD3 dual color split-apart fluorescent in situ hybridization assay using BAC probes flanking NSD3, as depicted in the chromosome 8 ideogram, depicted in three NMCs designated cases 1–3. All photomicrographs are identical magnification (1000×).
The fusion sequence is predicted to encode a 1694 amino acid protein containing amino acids 1–569 of NSD3, and 8–1132 of NUT. Interestingly, the NSD3 portion of the fusion protein lacks the SET domain and contains only its PWWP domain, whereas nearly all of NUT is included in the fusion, as is typical in NMC (1, 11, 12) (Figure 1H). The NSD3-NUT fusion bears no resemblance to NUP98-NSD3/NSD1 fusion oncogenes that have been previously described in leukemia (13, 14), all which fuse NUP98 to the 3’ end of NSD3/NSD1 containing their SET, PHD, and C/H rich domains.
NSD3-NUT is a Recurrent Form of NMC
We next sought to determine whether NSD3 is a recurrent NUT-fusion partner in NMCs, thus we performed a dual color NSD3 split-apart FISH assay on several NUT-variant cases. Four of eight non-BRD3/BRD4-NUT NMC cases (including the index case) demonstrated rearrangement of NSD3 and NUT, suggesting a frequent incidence of NSD3-NUT amongst NUT-variant cases (Figure 1I).
NSD3-NUT is Required for the Blockade of Differentiation and Maintenance of Proliferation in 1221 NMC Cells
The recurrent existence of NSD3-NUT in NMCs suggested that it may function similarly to BRD-NUT by blocking differentiation and maintaining proliferation of NMC cells (2). We thus knocked down endogenous expression of NSD3-NUT in 1221 cells to determine its effect on growth and differentiation. Seventy-two hours following knockdown, 1221 cells exhibited differentiation as evidenced by increased keratin expression, an epithelial differentiation marker, by immunofluorescence (Figure 2A–B), and decreased proliferation as measured by Ki-67 fraction (Figure 2C) and cell number (Figure 2D). Notably, knockdown of wild type NSD3 using siRNAs directed toward the 3’ aspect of NSD3 that is not included in the NSD3-NUT fusion gene had no effect on differentiation (Figure 2B). These findings indicate that NSD3-NUT serves to block differentiation and maintain proliferation of 1221 cells.
Figure 2. NSD3-NUT is required for the blockade of differentiation and maintenance of proliferation in 1221 NMC cells.

(A) High throughput 384 well plate immunofluorescent assay of keratin using the DAPI nuclear counterstain in 1221 cells 72 h following transfection with control, NUT, or NSD3 siRNAs. Representative photos are identical magnification (400X).
(B) Using the high-throughput assay in (A) quantitative analysis of keratin intensity was compared in 1221 cells 72 h following transfection with control, NUT, NSD3-5’ (targets both NSD3-NUT and NSD3-full length), and NSD3-3’ (targets the NSD3 portion not included in NSD3-NUT). Two different siRNAs were used for each gene or region targeted. Representative results from one of three biological replicates, each performed in triplicate, are shown. Error bars indicate the mean ± SD of the triplicate wells.
(C) Proliferation assay (Ki-67 fraction) using the high-throughput assay comparing 1221 cells transfected with control, NUT, and NSD3 siRNAs. Shown are averages of three biological replicates, each performed in triplicate. Error bars indicate the mean ± SD of the three biological replicates.
(D) Cell number using the high-throughput assay comparing 1221 cells transfected with control, NUT, and NSD3 siRNAs. Shown are averages of three biological replicates, each performed in triplicate. Error bars indicate the mean ± SD of the three biological replicates. * p<0.01; **p < 0.05.
Wild type NSD3 is Required for the Blockade of Differentiation in BRD4-NUT-expressing NMC Cells
NSD3 is one of several proteins that have been shown to bind the ET domain of BET proteins, (8). Thus we hypothesized that NSD3 may have an oncogenic role in NMC through its interaction with BRD4-NUT’s retained ET domain. It is noted that the BRD3 and BRD4 fusions with NUT in the BRD-NUT NMCs occur 3’ to the ET domain, thus the ET domain is always included as part of the fusion protein(1, 2). We therefore tested whether NSD3 is required for the blockade of differentiation in BRD4-NUT-expressing NMC cells. In three different patient-derived BRD4-NUT+ NMC cell lines, TC-797 (15), PER-403 (16), and 8645 (17), siRNA-knockdown of NSD3 resulted in differentiation, as measured by increased expression of the terminal squamous differentiation marker, Involucrin (Figure 3A–C). This was accompanied by morphologic differentiation, as evidenced by flattening and enlargement of cells (Figure 3B, and Supplementary Figure S3), as well as mild-to-moderate decreased proliferation quantified by Ki-67 staining (Figure 3D) in all cell lines. Moreover, induced expression of the ET domain fused to a nuclear localization sequence (NLS) in a tet-inducible NMC derivative cell line, 797TRex, exhibited a dominant negative effect on BRD4-NUT function, inducing differentiation morphologically and immunophenotypically (Figure 3E). In addition, induction of ET domain expression also negatively affected the proliferation rate of TC-797 cells, whereas the growth of heterologous, non-NMC cells, U2OS or 293T, was unaffected (Figure 3F) The findings indicate that expression of wild type NSD3 protein and the ET domain of BRD4-NUT are required for the blockade of differentiation in BRD4-NUT+ NMC. The specific requirement of the ET domain for the oncogenic function of BRD4-NUT is evidenced by its conservation in all characterized BRD-NUT fusions (1, 2, 12, 18, 19), including uncommon splice variants(16, 18), and by the lack of growth inhibition induced by ET domain expression in non-BRD-NUT-expressing cell lines (Figure 3F).
Figure 3. Wild type NSD3 is required for the blockade of differentiation in BRD4-NUT-expressing NMC cells.

(A) Immunoblots of BRD4-NUT-positive NMC cell lines TC-797, PER-403, and 8645 120h following transfection with control and NSD3 siRNAs stained with the terminal squamous differentiation marker, involucrin, using GAPDH as loading control.
(B) Representative photomicrographs of TC-797s 120 h following transfection with either control, or NSD3 siRNAs stained either with hematoxylin and eosin (H&E) for morphology, or involucrin immunohistochemistry. All photos are identical magnification (400×).
(C) Quantitative RT-PCR of NSD3 levels 24 h following transfection of control or NSD3 siRNAs. Primers were either 5’ of the breakpoint (NSD3-5’ primers), or 3’ of the breakpoint (NSD3-3’ primers) with NUT. Results are of a single biological replicate performed in triplicate. Error bars indicate the mean ± SD of the triplicate wells.
(D) Proliferation assay (Ki-67 fraction) comparing BRD4-NUT-positive TC-797, 8645, and PER-403 NMC cells transfected with control and NSD3-6 siRNAs. Three hundred cells were counted per cell block.
(E) 797TRex cells induced to express FLAG-tagged NLS-ET domain of BRD4 for 120 h. Immunoblot was stained with anti-involucrin (Inv), anti-FLAG, or anti-GAPDH (left). Cell block preparations were H&E stained, or subjected to involucrin immunohistochemistry (right). All photos are identical magnification (400×).
(F) Cell viability assay (CellTiter-Glo) of 797TRex, 293TRex, and U2OSTRex cells induced to express FLAG-tagged NLS-ET domain for 120h. Results are the average of three biological replicates, each performed in quadruplet and normalized to the negative control (ethanol vehicle control) for each cell line. Error bars indicate the mean ± SD of the three biological replicates. Immunoblot demonstrating NLS-FLAG-ET expression was stained with anti-FLAG, or anti-GAPDH (right).
The N-terminus of NSD3 associates with BRD4 and BRD4-NUT
Because the ET domain is retained in BRD-NUT oncoproteins, we predicted that the interaction of NSD3 with BRD4 would be preserved when co-expressed with BRD4-NUT. BRD4-NUT normally localizes to discrete nuclear foci by immunofluorescence and immunohistochemistry. We found that the HA-tagged portion of NSD3 present in NSD3-NUT (NSD3Tr, corresponding to amino acids 1–569 of NSD3) co-localized with BRD4-NUT foci (Figure 4A). Moreover, HA-tagged NSD3, NSD3-NUT, and NSD3Tr (Figure 4B) co-immunoprecipitated BRD4 in C33A cervical carcinoma cells. In reciprocal experiments, HA-tagged constructs of BRD4 and BRD4-NUT, but not NUT, were able to co-immunoprecipitate NSD3 (Figure 4C). Of note, the multiple NSD3 isoforms seen in this blot all contain the N-terminal domain of NSD3 (NSD3Tr) that is present in the NSD3-NUT fusion protein that interacts with BRD4 (Figure 4B). The findings indicate that NSD3 does associate with BRD4-NUT. To determine the role of the association of NSD3 with BRD4-NUT in the blockade of differentiation, NSD3Tr was expressed in 797TRex cells, and was found to induce differentiation (Figure 4D). The findings, coupled with the dominant negative effects of ET domain expression (Figure 3E–F), suggest that the interaction of NSD3 with BRD4-NUT may be required for the blockade of differentiation. In support of this, other known interactors of the ET domain, including CHD4, ATAD5, GLTSCR1, and JMJD6 (MCB), were knocked down in TC-797 cells, but failed to induce differentiation (Supplementary Figures S4A–B). While the findings suggest that NSD3-ET domain interaction is required for the blockade of differentiation in NMC, they are not conclusive, because other unknown interactors with either of these domains may be critical.
Figure 4. The N-terminus of NSD3 associates with BRD4 and BRD4-NUT.

(A) Immunofluorescence microscopy of 797TRex cells induced to express the HA-tagged portion of NSD3 included in NSD3-NUT (NSD3Tr) for 24 h stained with anti-NUT monoclonal antibody (red), and anti-HA monoclonal antibody (green).
(B) Immunoblot of anti-HA immunoprecipitations of tet-repressor-positive C33A cell (C33A-6TR) lysates following induction of expression of HA-tagged NSD3 variants, HA-NSD3 (full length), HA-NSD3-NUT and HA-NSD3-tr (NSD3 portion of the NSD3-NUT fusion protein). Indicated proteins were detected using anti-HA, and anti-Brd4 antibodies. The smaller bands are degraded protein.
(C) Immunoblot of anti-HA immunoprecipitations of C33A-6TR lysates following induction of expression of HA-tagged NUT, BRD4, and BRD4-NUT constructs stained with anti-HA, -NSD3, -p300, and -actin antibodies. To identify the NSD3-specific bands, lysates from TC-797s subjected to siRNA knockdown of NSD3 are shown.
(D) Immunoblot of 797TRex lystes 120h following induction of expression of BioTAP-tagged NLS-fusion construct of NSD3Tr stained with anti-involucrin, -PAP (recognizes the protein A moiety of the BioTAP tag), and –GAPDH antibodies.
NSD3 is required for BRD4-NUT foci formation
The function of BRD4-NUT foci is unknown; however, it has been demonstrated that they are intensely enriched with BRD4-NUT and factors associated with transcription, including RNA polymerase II, the histone acetyl-transferase (HAT), p300, the transcriptional elongation complex P-TEFb, and active histone marks (9, 20). Based on these observations, it is believed that BRD4-NUT foci are important to the function of BRD4-NUT on transcriptional regulation (9, 20), though this has not been tested directly. To determine what role, if any, NSD3 has in the formation of BRD4-NUT foci, we knocked it down in TC-797 cells and quantified foci number as compared with control siRNA transfected cells. We found that the number of BRD4-NUT foci was significantly reduced within 24 hours following siRNA knockdown of NSD3 (Figure 5A–B). Reduction of BRD4-NUT foci post-NSD3 knockdown was not accompanied by a reduction of total BRD4-NUT protein levels (Figure 5C), indicating that the effect is not due to loss of BRD4-NUT. Thus, the findings indicate that NSD3 is required for BRD4-NUT focus formation, suggesting a key role in BRD4-NUT complex or aggregate formation.
Figure 5. BRD4-NUT foci are dependent on NSD3.

(A) Immunofluorescence microscopy of TC-797 cells 24 h following transfection with control or NSD3-6 siRNAs stained with monoclonal antibody to NUT. All photos are identical magnification (1000×).
(B) Quantitation of BRD4-NUT foci was performed in triplicate and the averages of the three experiments. Error bars indicate the mean ± SD of triplicate experiments. *p < 0.005.
(C) Immunoblot of TC-797 lysates 24 h following transfection with control, NSD3-6, or NUT siRNAs stained with anti-NUT polyclonal antibody, AX.1.
NSD3-NUT can Replace BRD4-NUT in the Blockade of Differentiation
Because NSD3 is critical for BRD4-NUT function, we investigated whether NSD3-NUT was functionally equivalent to BRD4-NUT and as such replaces BRD4-NUT’s function in blocking differentiation. To test this, we induced expression of a Bio-TAP tandem tagged NSD3-NUT construct with tetracycline in 797TRex cells subjected to BRD4-NUT knockdown using an siRNA targeting the 3’ untranslated region of NUT not present in the NSD3-NUT introduced gene. Indeed, induction of expression of NSD3-NUT in 797TRex cells almost completely abrogated differentiation and proliferation arrest in cells subjected to knockdown of endogenous BRD4-NUT. The ability of NSD3-NUT to block differentiation was demonstrated morphologically, showing a lack of flattening (Figure 6A), and immunophenotypically, showing markedly reduced involucrin expression (Figure 6A–B), as compared with cells not induced to express NSD3-NUT (ethanol vehicle control-treated cells). The ability of NSD3-NUT to maintain proliferation was demonstrated by a Ki-67 fraction similar to scrambled siRNA-transfected cells, and markedly greater than ethanol vehicle control-treated cells subjected to BRD4-NUT knockdown (Figure 6C).
Figure 6. NSD3-NUT can replace the function of BRD4-NUT to block differentiation.
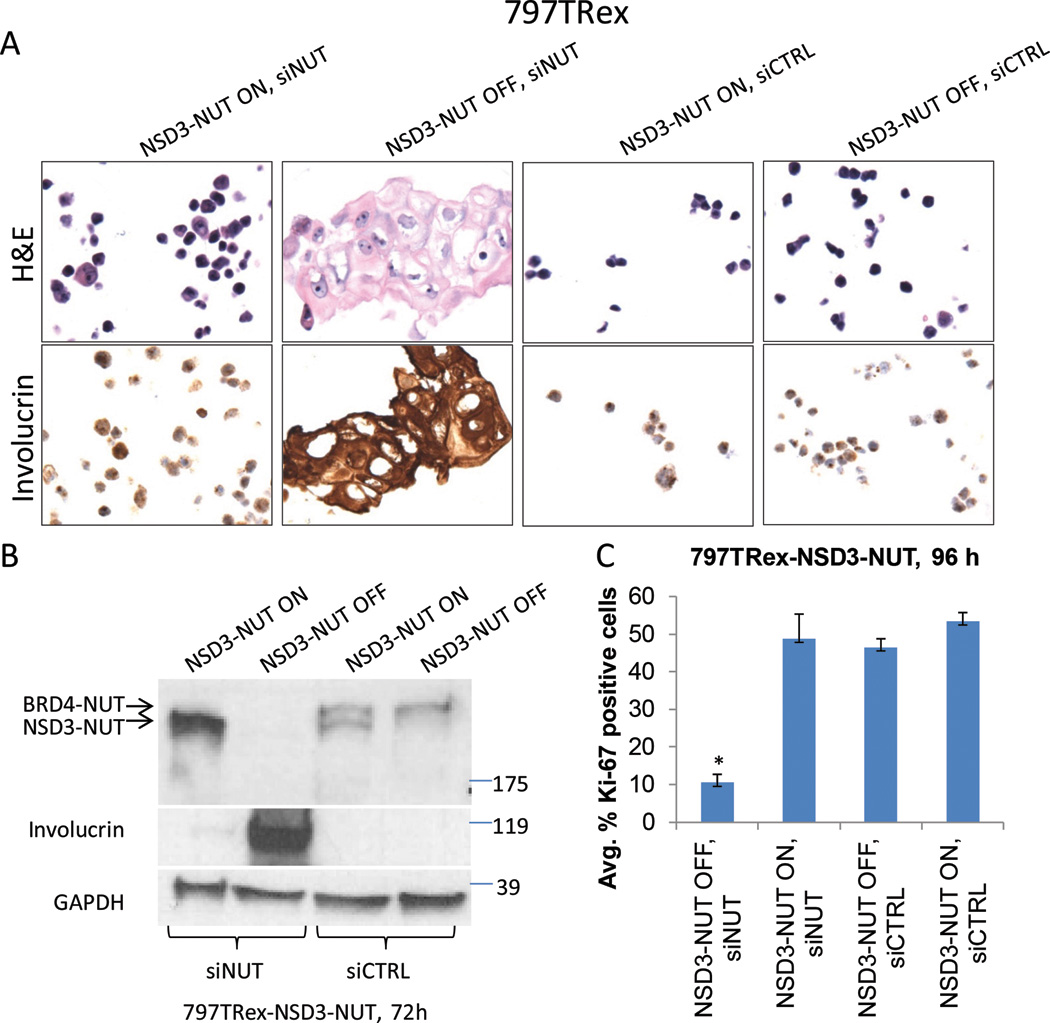
(A) H&E and anti-involucrin immunohistochemistry micrographs of 797TRex cells with tetracycline (ON), or treated with vehicle (OFF) to express NSD3-NUT 120 h following transfection with either control or NUT 3’UTR siRNA. All photos are identical magnification (400×).
(B) Immunoblots using lysates corresponding to the experiment in (A) were performed for the differentiation marker, involucrin, NSD3-NUT, and BRD4-NUT using antibodies to NUT.
(C) Quantification of immunohistochemical Ki-67 proliferation fraction of 797TRex cells induced to express NSD3-NUT 120 h following transfection with either control or NUT 3’UTR siRNA as in (A). Results are the average of three biological replicates performed using the 384-well high throughput assay as in Figure 2A, each performed in triplicate. Error bars indicate the mean ± SD of the three biological replicates. *p <0.0001.
BET Inhibitors Arrest Proliferation and Induce Differentiation of NSD3-NUT-expressing NMC Cells
The existence of NSD3 as a NUT-fusion oncogene partner, whose encoded protein is also an important functional member of BRD4 and BRD4-NUT complexes, is reminiscent of the oncogenic mechanism of MLL-fusion associated leukemia (21). Thus, we surmised that the oncogenic function of NSD3-NUT may depend on its interaction with BRD4 as a component of a chromatin-modifying complex with similar function to BRD4-NUT. Indeed, siRNA knockdown of BRD4, both long and short isoforms, induces differentiation of 1221 cells (Figure 7A), and treatment of 1221 cells with the BET inhibitor, JQ1, results in differentiation and arrested proliferation, in a dose-dependent manner (Figure 7B–D). These findings, together with the functional interchangeability of NSD3-NUT and BRD4-NUT, provide evidence that NSD3-NUT utilizes the chromatin-reading function of BRD4. Moreover, these data provide a sound rationale for treatment of patients with NSD3-NUT-positive NMCs using BET inhibitors.
Figure 7. BRD4 inhibition arrests proliferation and induces differentiation of NSD3-NUT-expressing NMC cells.

(A) Using the 384-well plate high-throughput assay exhibited in Figure 2A, quantitative analysis of keratin intensity was compared in 1221 cells 72 h following transfection with control versus BRD4 siRNAs. Representative results from one of three biological replicates, each performed in triplicate, are shown. Error bars indicate the mean ± SD of triplicate wells.
(B) Using the high-throughput assay (above), quantitative analysis of keratin intensity was compared in 1221 cells 72 h following treatment with a dose range of JQ1 versus DMSO vehicle control. Results are the average of three biological replicates performed using the 384-well high throughput assay, each performed in triplicate. Error bars indicate the mean ± SD of the three biological replicates.* p <0.01
(C) Representative immunofluorescence microscopy of 1221 cells treated as in (B), with vehicle control or 500nM JQ1 for 72h. All photos are identical magnification (400×).
(D) Cell number using the high-throughput assay comparing 1221 cells 72 h following treatment with increasing concentrations of JQ1 versus DMSO vehicle control. Results are the average of three biological replicates, each performed in triplicate. Error bars indicate the mean ± SD of the three biological replicates.* p <0.01
Discussion
The Role of NSD3 in NMC
A number of recent variant translocations have been described in NMC, illustrating the heterogenous nature of this disease (18, 22–24). In all of these previously described variants, both NUT and BET genes are fused. In this study, we describe a novel fusion gene in NMC that does not include a BET protein, but rather a BET-binding protein, NSD3. We find that the NSD3-NUT fusion oncogene encodes a protein that is both necessary and sufficient for the blockade of differentiation in NMC. We also find that wild type NSD3, which binds to BRD4 in non-neoplastic cells, also binds to BRD4-NUT and is required for the blockade of differentiation by more common BRD4-NUT-expressing NMCs. The presence of a fusion oncoprotein involving constituents of a single oncogenic complex is well documented in cancer, and the existence of NSD3-NUT speaks to the importance of NSD3 and its association with BRD4-NUT. NSD3, also known as WHSC1L1, is a histone methyl-transferase that belongs to the mammalian Nuclear SET Domain-containing (NSD) protein family of SET domain-containing methyltransferases, which also includes NSD1 and NSD2 (WHSC1/MMSET). Both NSD3 and NSD2 are known to bind the ET domain of BRD4, thus the dominant negative phenotype of ET and NSD3Tr expression in NMC cells is evidence that this interaction of BRD4 with NSD3 may be critical to BRD4-NUT function. However, it is not clear that the methyltransferase activity of NSD3 is needed for BRD4-NUT function as it is for NUP98-NSD3 fusions (14), because the NSD3 portion of NSD3-NUT lacks the SET domain. Moreover, knockdown of full-length NSD3 in the 1221 cells (Figure 2B) does not induce differentiation, again suggesting that NSD3’s SET domain is not required for the differentiation blockade. Thus, it appears that the critical portion of NSD3 is its N-terminal, ET-binding domain in NSD3-NUT-expressing NMC. Combining this data with the fact that BRD4 expression and interaction with chromatin is required for NSD3-NUT function (Figure 7), we have devised a model whereby the NSD3 portion links NSD3-NUT to BRD4, which tethers NSD3-NUT to chromatin, forming a complex that functions similarly to BRD4-NUT. In future studies, we plan to test this model by examining whether the interaction of NSD3-NUT with BRD4 is required for the oncogenic function of NSD3-NUT. In the context of BRD4-NUT-expressing NMC, we have shown that NSD3 is required for BRD4-NUT foci formation (Figure 5A), suggesting a role in the aggregation of large BRD4-NUT-containing complexes. However, it remains to be determined what function NSD3 serves in the formation of these foci.
Although its importance is not known in NSD3-NUT+ NMCs, NSD3 methyltransferase activity may be important to the function of BRD4-NUT NMC. The SET domain of NSD proteins is homologous to the Saccharomyces cerevisae histone 3 lysine 36 (H3K36) specific methyltransferase SET2 and is specific for H3K36 dimethylation (H3K36me2) (25). NSD3 has been reported to regulate H3K36 methylation and thereby active gene expression (8, 26). It is possible that akin to the aberrant activation of HoxA1 expression by NUP98-NSD1-mediated methylation of H3K36 in acute leukemia (14), NSD3 may contribute to the transcriptional activation of key targets of BRD4-NUT. Although the NUP98-NSD3 fusion oncogene has been described in acute leukemia as well (13), it has not been further characterized mechanistically. Proteomic analysis of NSD3 suggested that the protein also interacts with the histone protein variant, macroH2A1. MacroH2A1 replaces conventional H2A histones in a subset of nucleosomes, where it represses or activates transcription and participates in stable X chromosome inactivation (27–29). Moreover, macroH2A1 regulates cell growth and differentiation and is differentially expressed in cancer cells (30–32). Thus, NSD3 may regulate differentiation in BRD4-NUT expressing NMC cells by affecting differentiation specific genes via alterations of H3K36 or macroH2A1 levels.
BET and NSD3 Proteins in Cancer
NSD family proteins have been associated with other cancers (Reviewed in (33)). Chromosome translocation resulting in NSD2 (aka MMSET) overexpression leads to multiple myeloma (MM) whereas reduction in NSD2 levels suppresses cancer growth (34–36). Moreover, NSD3 is amplified in breast cancer cell lines and primary breast carcinomas (37, 38). NSD3 has been reported to contribute to the transformed phenotype and invasiveness of these breast cancer cells (39, 40). Most recently, mutations in NSD3 have been identified in pancreatic adenocarcinoma (41). Despite these associations, the mechanism by which NSD3 contributes to oncogenesis in these cancers remains poorly understood.
A key to understanding the role of NSD3 in cancer may be through its association with BRD4. The indispensability of the BRD4 chromatin-binding bromodomains in NMC (4, 6) and its ET domain’s role in recruiting NSD proteins demonstrates that BRD4 is a key player in BRD4-NUT chromatin-associated oncogenic complex formation. Moreover, recent studies with BET inhibitors have shown that BRD4 plays a role in other human cancers such as acute myeloid leukemia, multiple myeloma, and Burkitt lymphoma (42–44). In these cancers, as well as in NMC, BRD4 and BRD4-NUT, respectively, are required for the maintenance of MYC expression, and BET inhibitors repress expression of MYC, presumably through interference with BRD4-chromatin interaction (6, 43, 44). Our findings that siRNA knockdown or BET inhibitor blockade of BRD4 function induce differentiation in NSD3-NUT-expressing NMC cells indicates that NSD3 function depends on BRD4 and its interaction with chromatin (Figure 7). If NSD function is dependent upon BRD4 in other cancers, these NSD-associated cancers may be responsive to BET inhibitor therapy. Thus oncogenic NSD may be a biomarker of response to BET inhibitor therapy.
Drug Targeting of NSD3
Apart from BET inhibitors, this study highlights the importance of the therapeutic potential of targeting the NSD family of proteins. Histone modifying enzymes, including the NSD family, are often deregulated in cancer and aberrant histone modification profiles are intimately linked to carcinogenesis (45). Histone deacetylase (HDAC) inhibitors have been found to be effective in inhibiting NMC (17) and are already approved by the FDA for certain leukemias, while inhibitors to histone methyltransferases, including the NSD family, are under development (33, 45–47). Such therapies hold promise for NUT midline carcinoma as well as other cancers.
Methods
Fluorescence in situ hybridization
Dual-color fluorescence in situ hybridization (FISH) assays for NSD3 and NUT breakpoints were performed on formalin fixed, paraffin-embedded, 4 mm tissue sections as described (20). Probes used for the 15q14 breakpoint flanking NUT included the telomeric BAC clones RP11-1H8 and RP11-64o3, and the centromeric clones RP11-1084A12 and RP11-368L15. Probes used for the chromosome 8p11.23 NSD3 breakpoint were the flanking 5’ centromeric BAC clones CTD-2538P2 and RP11-957P17 and the 3’ telomeric BAC clones CTB-497A2 and RP11-90P5. Cytogenetic analysis and metaphase FISH was performed using standard methods (48) (49).
RNA-sequencing
RNA was extracted from live cultured 1221 cells using the RNeasy mini kit (Qiagen). Elim Biopharmaceuticals (Hayward, CA) performed the library preparation and sequencing. rRNA removal was performed using the Ribo-Zero kit (Epicentre, Madison, WI) following the manufacturer’s instructions. The library was prepared using standard Illumina protocols with proprietary modifications and sequenced using HiSeq2500 (Illumina, San Diego, CA). TopHat-Fusion (v2.0.8b bundled with TopHat2) was run with default parameters (as described at http://tophat.cbcb.umd.edu/fusion_tutorial.html#toph but with -r 50 and --max-intron-length 1000000) to identify novel fusion transcripts from paired-end 50 base reads (50) (51).
Reverse-transcriptase PCR (RT-PCR)
RT-PCR was as described (6) using the following primers: NSD3f1382 AAGAGCCACCGCCTGTTAAA, NUTr388 GCTGTCACAAATGGAGGTGC, GAPDH254f TCAAGTGGGGCGATGCTGGCGCT, GAPDH788rAGGGGGCCCTCCGACGCCTGCT.
Plasmids
BRD4 ET domain containing fragment (BRD4 444-722) was cloned into pcDNA5 FRT/TO-FLAG (Invitrogen) with an N-terminal SV40 NLS sequence to generate pcDNA5 FRT/TO-Flag-NLS-BRD4-ET (p6894). MSCV-CMV-Flag-HA-NSD3 (p6351) has been described previously (8). To make tetracycline-inducible N- and C-terminal BioTAP-tagged constructs, we transferred the gateway destination cassette from pHAGE-TRE (gift of Steven Elledge) to the pcDNA5 frt/to mammalian expression vector with N-terminal BioTAP tandem tag to make pcDNA5 frt/to-DEST-NBioTAP. NSD3-NUT was constructed by fusion PCR into pDONR223, then transferred into the pcDNA5 frt/to-DEST-NBioTAP vector by gateway cloning. Full length NSD3, NSD3Tr encoding amino acids 1–569 of NSD3, full length NUT, BRD4-NUT (derived from pcDNA5 frt/to FLAG-BRD4-NUT (17)) were PCR-cloned into pDONR223, then transferred by gateway cloning into the pHAGE-P CMVt N-HA GAW expression vector derived from the PHAGE lentiviral vector (52).
A tetracycline-inducible HA-tagged NSD3Tr was gateway cloned from pDONR223 into the tetracycline-inducible pHAGE-P CMVt N-HA GAW expression vector and into the tetracycline-inducible pHAGE-TRE-HA (gift of Steven Elledge).
Cell Culture
NMC cell lines, TC-797 (15) 10–15 (6), 8645(17), 293T, U2OS, and C33A cells were maintained as monolayer cultures in Dulbecco Modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% (v/v) Fetal Bovine Serum (FBS, SH3008803, Hyclone) and 1% pen-strep (GIBCO/Invitrogen). The 797TRex cell line was created using Flip-in technology as described (Invitrogen (6)) and maintained as above, but with the addition of Hygromycin (150 ug/ml, Sigma Aldrich, St. Louis, MO) and Blasticidin (7.5 ug/ml, Life Technologies, Grand Island, NY) to maintain selection of cDNA insert and tet repressor genes, respectively. 797Trex/Flag-NLS-ET and N-BioTAP-NSD3-NUT cell lines were generated by recombination with the plasmid pcDNA5 FRT/TO-FLAG-NLS-ET and – N-BioTAP-NSD3-NUT (above) using Flp-In technology (Invitrogen). The resulting cell line was maintained in Dulbecco Modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% (v/v) Fetal Bovine Serum (FBS, SH3008803, Hyclone), 1% pen-strep (GIBCO/Invitrogen), 7.5 ug Blasticidin/mL and 150 ug Hygromycin/mL. The 1221 cell line is derived from a lung metastasis from the index case of a 13 year old female with NSD3-NUT-positive NMC. The 1221 cells were grown and maintained in WIT media as described (17, 53). Tet-inducible C33A cells (C33A-6TR) cells were established by transfecting C33A cells with pcDNA6/TR vector (Invitrogen). Cells were selected and maintained with with 5 µg/ml Blasticidin and single cell clones were obtained. Single cell clones were then tested for leakiness using a Tet-inducible EGFP construct and clone 13 was chosen for further experiments due to a tight regulation of EGFP expression by the TR. The 1221, TC-797, 797TRex, 8645, and 10–15 cell lines were authenticated by FISH (above) demonstrating rearrangement of the NUT, BRD4, and/or NSD3 genes. The C33A cell line has been authenticated by documentation of p53 and pRb mutations (54). Neither the 293T or U2OS cell lines have been authenticated.
Luminescent cell viability assay
Cells were plated at a density of 3000 per well in a 96-well plate, and CellTiter-Glo (Promega, Madison, WI) was used to determine cell viability as a measure of ATP content according to the manufacturer’s instructions.
siRNA transfections
For TC-797, Per403, and 8645 cells, 7×106 cells were transfected with 50 nM siRNA using Nucleofector II (Lonza, Basel, Switzerland) and Amaxa solution R and plated in 100 mm cell culture dish. 1221 cells were transfected using RNAi-MAX (Invitrogen) using the manufacturer’s protocol. Briefly, reverse transfection procedure was used to deliver 50nM siRNA to 33×104 cells in a 6 well plate. Cells were analyzed for mRNA levels 24 h after transfection. Sequences of siRNA used were: siControl, ON-TARGET plus siRNA #1 (Dharmacon, Cat # D-001810-01-20), siNUT-1 (targeting coding sequence) AAACUCAGAACUUUAUCCUUAUU, siNUT-2 (targeting the 3’ UTR) UUACCUUUGGAAGGAGCUA, siBRD4 5’ siGENOME Human BRD4 (Dharmacon Cat # D-004937-02), siBRD4 3’ GGGAGAAAGAGGAGCGUGAUU, siNSD3-6 ON-TARGETplus Human WHSC1L1 (54904) (Dharmacon Cat # J-012875-06), siNSD3-7, ON-TARGETplus Human WHSC1L1 (54904) siRNA (Dharmacon Cat # J-012875-07), siNSD3 3’-1 CUGUAAACCUCUAAAGAAAUU, si NSD3 3’-2 GAAAGGUGCCAGCGAGAUUUU, siJMJD6-12 GGUAUAGGAUUUUGAAGCA (Dharmacon Cat # J-010363-12-0020), siJMJD6-13 GGAUAACGAUGGCUACUA (Dharmacon Cat # J-010363-13-0020), siNSD3-06 GAACGUGCUCAGUGGGAUA (Dharmacon Cat # J- J-012875-06-0020), siNSD3-07 GCUUGAGGUUCAUACUAAA (Dharmacon Cat # J-012875-07-0020), siGLTSCRI-05 GUAAUGAUCGACCGAAUGU (Dharmacon Cat # J-020751-05-0020), siGLTSCR1-08 CCACCACGUUCAAUGGGAA (Dharmacon Cat # J-020751-08-0020), siATAD-05 UAGCAGAAAUGUACAACUA (Dharmacon Cat # J-004738-05-0020), siATAD5-06 GCGCAAUAAUGUAUACUUU (Dharmacon Cat # J-004738-06-0020), siCHD4-07 (Dharmacon Cat # J-009774-07-0020), siCHD4-08(Dharmacon Cat # J-009774-08-0020).
Immunofluorescence
Immunofluorescence on TC-797 cells was performed as described (55) and nuclei were counterstained with ProLong® Gold antifade reagent with 4',6-diamidino-2-phenylindole (DAPI) (Life Technologies). Primary antibodies used were anti-NUT (1:1000, rabbit monoclonal clone C52, Cell Signaling Technology), and anti-HA (1:500, mouse monoclonal, Sigma-Aldrich). Secondary antibodies included goat anti-rabbit Alexa Fluor 594 and goat anti-mouse Alexa Fluor 488 (1:1000, Life Technologies, Grand Island, NY). Photos were taken on a Nikon Eclipse E600 fluorescent microscope (Melville, NY) using a Spot RTSlider camera (Diagnostic Instruments, Inc., Sterling Heights, MI), and Spot Advanced software (Diagnostic Instruments, Inc.).
Quantitation of BRD4-NUT foci was performed by analyzing immunofluorescent images with ImageJ software. BRD4-NUT foci were identified by equal threshold adjustment of all images. The Analyze Particles function in ImageJ was used to determine foci number for each cell. 40 cells were counted for each experimental condition and experiments were performed in triplicate. Statistics shown in all figures are from Student's t-Tests (two tailed).
High Throughput Immunofluorescence Analysis of NMC cells
Cells were transfected in 384 well format using 50 nM control siRNA (above), NSD3 siRNA (above), BRD4 siRNA, and NUT siRNA (above), as described (6), or a dose range of JQ1 treatment. Cells were stained with AE1/AE3 antibody (1:4, Dako, Carpinteria, CA) to measure keratin intensity, Ki-67 antibody (1:500, Cell Signalling Technology, Danvers, MA), and nuclei were stained with Hoechst 33342 (4 ug/ml, Molecular Probes). Imaging for keratin expression was performed using the ImageXpress high-throughput microscope with MetaXpress software (Molecular Devices, Sunnyvale, CA) as described (56). Shown are representative images taken at 40× magnification. Each condition was performed in triplicate in 384-well plate format in three separate experiments, where wells were analyzed using MetaXpress with Multi Wavelength Cell Scoring for cell number, average keratin fluorescence pixel intensity, and % Ki-67 per well. Statistics shown are from Student's T-tests.
Gene Expression Analysis using Quantitative Real-time PCR (qRT-PCR)
Total RNA was harvested at the indicated time points using TRizol (Invitrogen, Carlsbad, CA, USA) and the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and RNA (1 ug) was reversed transcribed into cDNA using the iScript cDNA synthesis kit (Bio-Rad). Quantitative RT-PCR was performed as described (6) using the reference gene Ribosomal protein L13a (RPL13a) mRNA levels as normalizing control. All qRT-PCR experiments are representative of triplicate qRT-PCRs from one of three independent experiments. qRT-PCR was performed in triplicate on a Bio-Rad iCycler in 96 well plate format with IQ SYBR Green supermix (Bio-Rad) and 1 ul of cDNA template per reaction. Amplification curves and Ct values were generated using MyiQ Single-Color Real-Time software (Bio-Rad). Primers used were 3' NSD3 fwd TTCTAGGAGTGCGGCCAAAG, 3' NSD3r CAGCTCTCCACCATCTCCAC, 5' NSD3f GCCCCAGTTCAGCCAATACT, 5' NSD3r ACCATACAAGGCCACCAAGG, RPL13Af CCTGGAGGAGAAGAGGAAAGAGA, RPL13Ar TTGAGGACCTCTGTGTATTTGTCAA, and using the TaqMan primers (Life Technologies, Grand Island, NY), JMJD6 (Hs00397095_m1), NSD3 (Hs00256555_m1), GLTSCR1 (Hs00185249_m1), and ATAD5 (Hs00227495_m1).
Immunoblotting
Cell lysis and immunoblotting was performed as described (17). Antibodies used were rabbit anti-NUT (1:500, AX.1 rabbit polyclonal antibody (57), anti-GAPDH (1:5000, mouse monoclonal 6C5, Life Technologies, Grand Island, NY), anti-BRD4 (1:1000, Bethyl Laboratories Inc., Montgomery, TX), anti-FLAG (1:1000, mouse monoclonal, Sigma-Aldrich, St Louis, MO), anti-involucrin (1:1000, mouse monoclonal, Sigma-Aldrich), anti-HA (1:1000, mouse monoclonal, Sigma-Aldrich), anti-NSD3 (1:500, mouse monoclonal clone 2E9, Lifespan Biosciences, Seattle, WA), anti-peroxidase anti-peroxidase complex / PAP antibody (1:5000, rabbit polyclonal, Sigma-Aldrich), anti-histone H3 (1:1000, mouse monoclonal ab10799, Abacam), anti-actin (1:1000, mouse monoclonal clone 4, Millipore) and anti-p300 (1:1000, mouse monoclonal clone RW128, EMD Millipore, Billerica, MA).
Immunoprecipitation
For co-immunoprecipitation experiments of HA-tagged proteins, C33A-6TR cells stably expressing HA-tagged constructs under a Tet-inducible promoter were used. Protein extracts were prepared 24 h after transfection or induction with Doxycycline (1 µg/ml). Cells were lysed in lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% Nonidet P-40) with freshly added protease inhibitors (Roche Complete, EDTA free protease inhibitor cocktail, Roche, Indianapolis, IN). Extracts were adjusted for protein concentration and 10% of extracts were used as Input. Immunoprecipitations were performed using 15 µl of HA-resin (Sigma A2095). Extracts were incubated overnight at 4°C, and precipitated proteins were detected by Western blot analysis.
Immunohistochemistry
Formalin-fixed, paraffin-embedded cell-blocks of cultured cells were prepared using Histogel (Richard-Allan Scientific) as described previously (2). Sections were stained with hematoxylin and eosin or by immunohistochemistry (IHC), which was performed on 5 mm sections prepared from formalin-fixed, paraffin-embedded cell-blocks. Immunohistochemical stains were performed using anti-NUT antibody (1:100, rabbit monoclonal clone C52, Cell Signaling Technology, Danvers, MA), anti-involucrin antibody (1:12000, Sigma-Aldrich, St. Louis), and Ki-67 (MIB-1 clone; DAKO USA, Carpinteria, CA (2). One hundred cells were counted per sample for Ki-67 percentage. Standard deviations for triplicate counts are shown in figures.
Supplementary Material
Significance.
The existence of a family of fusion oncogenes in squamous cell carcinoma is unprecedented, and should lead to key insights into aberrant differentiation in NMC and possibly other squamous cell carcinomas. The involvement of the NSD3 methyltransferase as a component of the NUT fusion protein oncogenic complex identifies a new potential therapeutic target.
Acknowledgments
We thank James E. Bradner (Dana Farber Cancer Institute) for sharing with us the BET inhibitor, JQ1. We are grateful to Jeffrey Galligan in the Howley laboratory who generated the C33A-6TR cell line used in these studies. This work was supported by grants from the National Institutes of Health: R01CA116720 (to P.M.H.), 2R01CA124633-06A1 (to C.A.F.), T32CA009361 (to S.R.), T32HL007627-27 (to E.W.) as well the Samuel Waxman Cancer Research Foundation (to C.A.F). We thank Ann Thomas and Michelle Speranza for their technical expertise that was very helpful for the cytogenetic analysis.
Footnotes
Disclosure of potential conflicts of interest: No potential conflicts of interest were disclosed.
References
- 1.French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer research. 2003;63(2):304–307. [PubMed] [Google Scholar]
- 2.French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, et al. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27(15):2237–2242. doi: 10.1038/sj.onc.1210852. Epub 2007/10/16. [DOI] [PubMed] [Google Scholar]
- 3.Bauer DE, Mitchell CM, Strait KM, Lathan CS, Stelow EB, Luer SC, et al. Clinicopathologic Features and Long-Term Outcomes of NUT Midline Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(20):5773–5779. doi: 10.1158/1078-0432.CCR-12-1153. Epub 2012/08/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. Epub 2010/09/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, et al. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150(4):673–684. doi: 10.1016/j.cell.2012.06.045. Epub 2012/08/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grayson ARWE, Cameron MJ, Godec J, Ashworth T, Ambrose JM, Aserlind AB, Wang H, Evan G, Kluk MJ, Bradner JE, Aster JC, French CA. MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene. 2013 doi: 10.1038/onc.2013.126. in press(in press):in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Molecular cell. 2005;19(4):535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 8.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Molecular and cellular biology. 2011;31(13):2641–2652. doi: 10.1128/MCB.01341-10. Epub 2011/05/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reynoird N, Schwartz BE, Delvecchio M, Sadoul K, Meyers D, Mukherjee C, et al. Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. The EMBO journal. 2010;29(17):2943–2952. doi: 10.1038/emboj.2010.176. Epub 2010/08/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haack H, Johnson LA, Fry CJ, Crosby K, Polakiewicz RD, Stelow EB, et al. Diagnosis of NUT Midline Carcinoma Using a NUT-specific Monoclonal Antibody. The American journal of surgical pathology. 2009;33(7):984–991. doi: 10.1097/PAS.0b013e318198d666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dang TP, Gazdar AF, Virmani AK, Sepetavec T, Hande KR, Minna JD, et al. Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. Journal of the National Cancer Institute. 2000;92(16):1355–1357. doi: 10.1093/jnci/92.16.1355. [DOI] [PubMed] [Google Scholar]
- 12.Haruki N, Kawaguchi KS, Eichenberger S, Massion PP, Gonzalez A, Gazdar AF, et al. Cloned fusion product from a rare t(15;19)(q13.2;p13.1) inhibit S phase in vitro. J Med Genet. 2005;42(7):558–564. doi: 10.1136/jmg.2004.029686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosati R, La Starza R, Veronese A, Aventin A, Schwienbacher C, Vallespi T, et al. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15) Blood. 2002;99(10):3857–3860. doi: 10.1182/blood.v99.10.3857. Epub 2002/05/03. [DOI] [PubMed] [Google Scholar]
- 14.Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nature cell biology. 2007;9(7):804–812. doi: 10.1038/ncb1608. Epub 2007/06/26. [DOI] [PubMed] [Google Scholar]
- 15.Toretsky JA, Jenson J, Sun CC, Eskenazi AE, Campbell A, Hunger SP, et al. Translocation (11;15;19): a highly specific chromosome rearrangement associated with poorly differentiated thymic carcinoma in young patients. Am J Clin Oncol. 2003;26(3):300–306. doi: 10.1097/01.COC.0000020960.98562.84. [DOI] [PubMed] [Google Scholar]
- 16.Kees UR, Mulcahy MT, Willoughby ML. Intrathoracic carcinoma in an 11-year-old girl showing a translocation t(15;19) Am J Pediatr Hematol Oncol. 1991;13(4):459–464. doi: 10.1097/00043426-199124000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz BE, Hofer MD, Lemieux ME, Bauer DE, Cameron MJ, West NH, et al. Differentiation of NUT midline carcinoma by epigenomic reprogramming. Cancer research. 2011;71(7):2686–2996. doi: 10.1158/0008-5472.CAN-10-3513. Epub 2011/03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson-Wicking K, Francis RW, Stirnweiss A, Ferrari E, Welch MD, Baker E, et al. Novel BRD4-NUT fusion isoforms increase the pathogenic complexity in NUT midline carcinoma. Oncogene. 2013;32(39):4664–4674. doi: 10.1038/onc.2012.487. Epub 2012/11/07. [DOI] [PubMed] [Google Scholar]
- 19.Engleson J, Soller M, Panagopoulos I, Dahlen A, Dictor M, Jerkeman M. Midline carcinoma with t(15;19) and BRD4-NUT fusion oncogene in a 30-year-old female with response to docetaxel and radiotherapy. BMC Cancer. 2006;6:69. doi: 10.1186/1471-2407-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan J, Diaz J, Jiao J, Wang R, You J. Perturbation of BRD4 protein function by BRD4-NUT protein abrogates cellular differentiation in NUT midline carcinoma. The Journal of biological chemistry. 2011;286(31):27663–27675. doi: 10.1074/jbc.M111.246975. Epub 2011/06/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annual review of pathology. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. Epub 2011/10/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ball A, Bromley A, Glaze S, French CA, Ghatage P, Kobel M. A rare case of NUT midline carcinoma. Gynecologic oncology case reports. 2012;3:1–3. doi: 10.1016/j.gynor.2012.09.004. Epub 2012/01/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mills AF, Lanfranchi M, Wein RO, Mukand-Cerro I, Pilichowska M, Cowan J, et al. NUT Midline Carcinoma: A Case Report with a Novel Translocation and Review of the Literature. Head and neck pathology. 2013 doi: 10.1007/s12105-013-0479-3. Epub 2013/08/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkler K. Surgical treatment of pulmonary metastases in childhood. The Thoracic and cardiovascular surgeon. 1986;34(Spec No 2):133–136. doi: 10.1055/s-2007-1022190. Epub 1986/11/01. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ, 3rd, et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. The Journal of biological chemistry. 2009;284(49):34283–34295. doi: 10.1074/jbc.M109.034462. Epub 2009/10/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell. 2010;39(2):222–233. doi: 10.1016/j.molcel.2010.07.008. Epub 2010/07/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costanzi C, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature. 1998;393(6685):599–601. doi: 10.1038/31275. Epub 1998/06/20. [DOI] [PubMed] [Google Scholar]
- 28.Gamble MJ, Frizzell KM, Yang C, Krishnakumar R, Kraus WL. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 2010;24(1):21–32. doi: 10.1101/gad.1876110. Epub 2009/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buschbeck M, Uribesalgo I, Wibowo I, Rue P, Martin D, Gutierrez A, et al. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat Struct Mol Biol. 2009;16(10):1074–1079. doi: 10.1038/nsmb.1665. Epub 2009/09/08. [DOI] [PubMed] [Google Scholar]
- 30.Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, Gamble MJ. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Molecular and cellular biology. 2011;31(20):4244–4255. doi: 10.1128/MCB.05244-11. Epub 2011/08/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sporn JC, Jung B. Differential Regulation and Predictive Potential of MacroH2A1 Isoforms in Colon Cancer. Am J Pathol. 2012 doi: 10.1016/j.ajpath.2012.02.027. Epub 2012/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Creppe C, Janich P, Cantarino N, Noguera M, Valero V, Musulen E, et al. MacroH2A1 regulates the balance between self-renewal and differentiation commitment in embryonic and adult stem cells. Molecular and cellular biology. 2012;32(8):1442–1452. doi: 10.1128/MCB.06323-11. Epub 2012/02/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morishita M, di Luccio E. Cancers and the NSD family of histone lysine methyltransferases. Biochim Biophys Acta. 2011;1816(2):158–163. doi: 10.1016/j.bbcan.2011.05.004. Epub 2011/06/15. [DOI] [PubMed] [Google Scholar]
- 34.Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111(6):3145–3154. doi: 10.1182/blood-2007-06-092122. Epub 2007/12/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stec I, Wright TJ, van Ommen GJ, de Boer PA, van Haeringen A, Moorman AF, et al. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Human molecular genetics. 1998;7(7):1071–1082. doi: 10.1093/hmg/7.7.1071. Epub 1998/06/09. [DOI] [PubMed] [Google Scholar]
- 36.Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92(9):3025–3034. Epub 1998/10/27. [PubMed] [Google Scholar]
- 37.Garcia MJ, Pole JC, Chin SF, Teschendorff A, Naderi A, Ozdag H, et al. A 1 Mb minimal amplicon at 8p11-12 in breast cancer identifies new candidate oncogenes. Oncogene. 2005;24(33):5235–5245. doi: 10.1038/sj.onc.1208741. Epub 2005/05/18. [DOI] [PubMed] [Google Scholar]
- 38.Angrand PO, Apiou F, Stewart AF, Dutrillaux B, Losson R, Chambon P. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics. 2001;74(1):79–88. doi: 10.1006/geno.2001.6524. Epub 2001/05/26. [DOI] [PubMed] [Google Scholar]
- 39.Yang ZQ, Liu G, Bollig-Fischer A, Giroux CN, Ethier SP. Transforming properties of 8p11-12 amplified genes in human breast cancer. Cancer research. 2010;70(21):8487–8497. doi: 10.1158/0008-5472.CAN-10-1013. Epub 2010/10/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Z, Thomsen R, Kahns S, Nielsen AL. The NSD3L histone methyltransferase regulates cell cycle and cell invasion in breast cancer cells. Biochemical and biophysical research communications. 2010;398(3):565–570. doi: 10.1016/j.bbrc.2010.06.119. Epub 2010/07/06. [DOI] [PubMed] [Google Scholar]
- 41.Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(16):5934–5941. doi: 10.1073/pnas.1202490109. Epub 2012/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(40):16669–16674. doi: 10.1073/pnas.1108190108. Epub 2011/09/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. Epub 2011/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. doi: 10.1038/nature10334. Epub 2011/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell research. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. Epub 2011/02/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. Epub 2009/09/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Best JD, Carey N. Epigenetic opportunities and challenges in cancer. Drug discovery today. 2010;15(1–2):65–70. doi: 10.1016/j.drudis.2009.10.010. Epub 2009/11/10. [DOI] [PubMed] [Google Scholar]
- 48.Marino-Enriquez A, Hornick JL, Dal Cin P, Cibas ES, Qian X. Dedifferentiated liposarcoma and pleomorphic liposarcoma: a comparative study of cytomorphology and MDM2/CDK4 expression on fine-needle aspiration. Cancer cytopathology. 2014;122(2):128–137. doi: 10.1002/cncy.21362. Epub 2013/11/15. [DOI] [PubMed] [Google Scholar]
- 49.La Starza R, Barba G, Nofrini V, Pierini T, Pierini V, Marcomigni L, et al. Multiple EWSR1-WT1 and WT1-EWSR1 copies in two cases of desmoplastic round cell tumor. Cancer genetics. 2013;206(11):387–392. doi: 10.1016/j.cancergen.2013.10.005. Epub 2014/01/07. [DOI] [PubMed] [Google Scholar]
- 50.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome biology. 2011;12(8):R72. doi: 10.1186/gb-2011-12-8-r72. Epub 2011/08/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. Epub 2009/03/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy GJ, Mostoslavsky G, Kotton DN, Mulligan RC. Exogenous control of mammalian gene expression via modulation of translational termination. Nature medicine. 2006;12(9):1093–1099. doi: 10.1038/nm1376. Epub 2006/08/08. [DOI] [PubMed] [Google Scholar]
- 53.Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer cell. 2007;12(2):160–170. doi: 10.1016/j.ccr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 54.Scheffner M, Munger K, Byrne JC, Howley PM. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(13):5523–5527. doi: 10.1073/pnas.88.13.5523. Epub 1991/07/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henikoff S, Ahmad K, Platero JS, van Steensel B. Heterochromatic deposition of centromeric histone H3-like proteins. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(2):716–721. doi: 10.1073/pnas.97.2.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Senapedis WT, Kennedy CJ, Boyle PM, Silver PA. Whole genome siRNA cell-based screen links mitochondria to Akt signaling network through uncoupling of electron transport chain. Molecular biology of the cell. 2011;22(10):1791–1805. doi: 10.1091/mbc.E10-10-0854. Epub 2011/04/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stelow EB, French CA. Carcinomas of the upper aerodigestive tract with rearrangement of the nuclear protein of the testis (NUT) gene (NUT midline carcinomas) Adv Anat Pathol. 2009;16(2):92–96. doi: 10.1097/PAP.0b013e31819923e4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.