

SUMMARY
We identified a novel gain of function mutation in STAT1 in a patient with disseminated Apophysomyces trapeziformis infection who had never had mucocutaneous candidiasis or autoimmunity. To our knowledge this is the first report of a genetic predisposition associated with mucormycosis.
Keywords: STAT1, Immunodeficiency, Mucormycosis, Apophysomyces trapeziformis, Interferon gamma
TO THE EDITOR
Signal transduction and activator of transcription (STAT) 1 belongs to a family of transcription factors that mediate growth factor and cytokine signaling. Complete recessive mutations in STAT1 cause susceptibility to viral, mycobacterial and bacterial infections and are fatal early in life. In contrast, heterozygous dominant negative mutations in STAT1 cause mild disseminated mycobacterial infections.1 Recently, heterozygous dominant gain of function (GOF) mutations in STAT1 were described as causing impaired STAT1 dephosphorylation, diminished IL-17 producing T cells and chronic mucocutaneous candidiasis (CMC).2 These GOF mutations have also been associated with intracellular dimorphic fungal infections, disseminated coccidioidomycosis, histoplasmosis,3 and cases of FOXP3-wild type IPEX-like syndrome.4 Progressive multifocal leukencephalopathy, autoimmunity, arterial aneurysms, and squamous cell cancers have also been reported.2–5 We identified a novel gain of function STAT1 mutation in a patient with disseminated mucormycosis caused by Apophysomyces trapeziformis. Previous reports of A. trapeziformis infection were associated with penetrating trauma in immunocompetent hosts.6 This is the first description of a primary immunodeficiency linked to disseminated mucormycosis.
A 24-year-old man of mixed African and Asian descent presented with 3 weeks of fever (104°F), myalgia, and night sweats, two weeks of headache associated with right-sided facial numbness and swelling, pleuritic chest pain and shortness of breath. Tender, enlarged, mobile bilateral inguinal and axillary lymphadenopathy was present along with discrete subcutaneous nodules in the right deltoid, right scapula and in the right paraspinal muscles. Facial swelling and numbness were associated with weakness of the right muscles of mastication. He had bibasilar crackles and a triphasic pericardial friction rub without murmurs. The WBC count was 27×109/L with 28% eosinophils; troponinI was 2.4ng/ml. Chest CT showed a 2 cm nodule in the right lower lobe (Fig 1A, upper panel) and lesions at the right shoulder, supraspinatus (Fig 1A, middle panel) and deltoid (Fig 1A, lower panel). MRI of the brain detected a 1.7 × 1.1 cm thick-walled rim-enhancing lesion in the anterior temporal lobe and enhancement of the right masticatory muscles (Fig 1B). Cardiac MRI showed myopericarditis with pericardial effusion and tamponade (Fig 1C). Biopsy of deltoid muscle and lymph node showed an eosinophilic infiltrate surrounding aseptate fungal hyphae with right angle branching (Fig 1D), consistent with mucormycosis.7 Cultures from the right deltoid muscle biopsy and blood grew Apophysomyces trapeziformis, identified by colony and microscopic morphology and confirmed by sequencing. The isolate was susceptible to amphotericin B, itraconazole, and posaconazole. Serum galactomannan, urine Histoplasma antigen, serum cryptococcal antigen, serum coccidiodes antigen, trichinella and neurocysticercosis serologies, mycobacterial staining and culture from multiple sites of infection were all negative.
Figure 1.
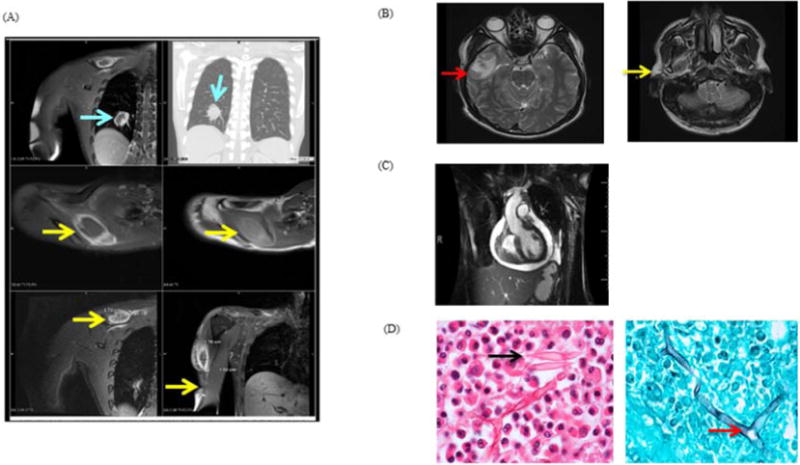
A, Chest MRI. Lesions are visible in the right lung (top panels), right supraspinatus (arrows, middle panels) and deltoid (lower panels); B, Brain MRI T2W images. Thick-walled right temporal lobe lesion (left); enhancement of the right muscles of mastication (right); C, Cardiac MRI: Pericardial effusion with mild RV collapse; D, H&E inguinal lymph node biopsy (left) with intense eosinophilic infiltrate, Charcot-Leyden crystals (arrow); silver stain of deltoid muscle (right). Aseptate fungal elements with 90° branching hyphae were observed (arrow).
He was from northern Florida, had not travelled outside the southern United States, had no history of previous infections, sick contacts, penetrating trauma or CMC. Other conditions predisposing to mucormycosis, such as diabetes and hemochromatosis were excluded. The patient was formerly healthy with the exception of episodes of “prostatitis”; no further records were available. The family history was not significant.
The patient was treated with liposomal amphotericin B (7.5mg/kg/day) and posaconazole. Blood cultures cleared in 36 hours after initiation of treatment. However, his old lesions continued to worsen and he developed new ones. Liposomal amphotericin was increased to 10mg/kg and he was started on micafungin (150mg/day).6 After 2 weeks, IFN-γ subcutaneously (50mcg/m2) three times weekly was added to augment cellular immune response. He received this combination for 8 weeks followed by oral posaconazole. Follow-up imaging studies showed progressive decrease and disappearance of lesions along with improvement in symptoms. The patient was discharged on long-term therapy with posaconazole. He has been well at 3, 6 and 12 months follow-up.
The identification of a disseminated fungal infection in an otherwise healthy patient, let alone an unusual mold, initiated a search for an immune predisposition. No identified genetic predispositions to disseminated Mucorales infections are known. The recent description of several cases of fatal A. trapeziformis associated with trauma and diabetes did not apply in this case.6,8 Mutations in CARD9 were not found and NADPH oxidase activity was normal, excluding chronic granulomatous disease. The association of cases of disseminated fungal infections in patients with GOF mutations in STAT1, including some with adult onset, prompted us to sequence STAT1. Full length sequencing of STAT1 genomic and cDNA identified the novel heterozygous mutation c.1110G>C; p.E370D, which resides in the DNA binding domain, and is predicted to be deleterious. The mutation was not found in dbSNP 138, or the 1000 Genomes Project. The patient’s parents were not available for mutation screening.
To investigate its function, peripheral blood mononuclear cells (PBMCs) and transformed EBV-B cells from normal donors, the E370D patient, a patient with GOF STAT1 mutation (E353K),3 and a patient with a loss of function (LOF) mutation (M654K) were stimulated with IFN-γ (400 IU/ml, 30min). STAT1 phosphorylation (pSTAT1) was examined by immunoblotting and flow cytometry. pSTAT1 was increased in GOF patient cells (Fig 2A, 2B) with characteristic delay in STAT1 dephosphorylation (Fig 2C) and was reduced in LOF patient cells (Fig 2A–2C). Transcriptional activity of the mutants was evaluated in the STAT1-deficient U3A cell line by luciferase activity of a reporter gene under the control of the GAS promoter. Transfection of the STAT1-E370D construct into the U3A cells led to enhanced IFN-γ stimulation compared to WT (Fig 2D), similar to the E353K GOF mutant and strikingly different from the dominant negative STAT1 mutants L706S and M654K. Consistent with these observations, expression of IFN-γ-induced chemokine genes (CXCL9, CXCL10) was enhanced in transfected U3A cells, PBMCs and EBV-B cells (not shown). IFN-γ up-regulation of LPS (200ng/ml) induced TNF-α (18h cultures) was higher in patient PBMCs (16-fold induction) vs. normal (4-fold) or LOF cells (Fig 2E). When PBMCs were stimulated with phorbol 12-myristate 13-acetate (PMA)/ionomycin, IL-17 producing T cells were mildly reduced: E370D, 1.07%; E353K, 1.27%; controls, 2.0 ± 0.87%. Overall, these laboratory findings are characteristic of dominant GOF STAT1 mutation leading to hyper-responsiveness to IFN-γ.
Figure 2.
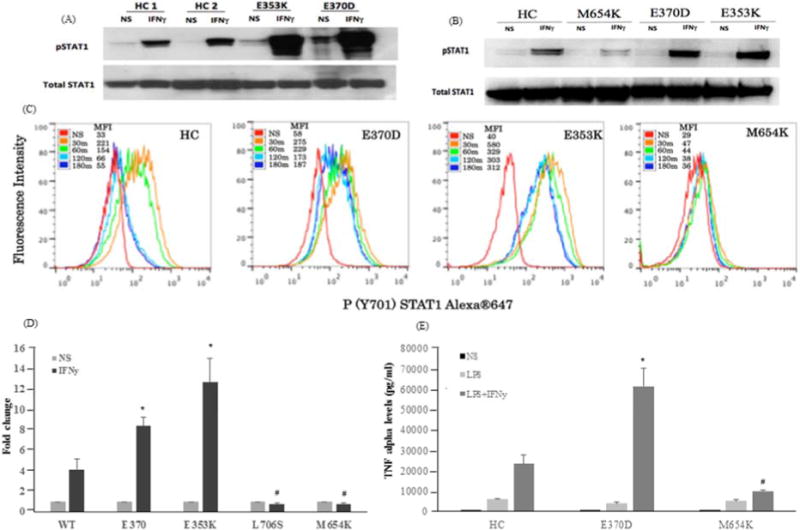
A, Immunoblotting (pSTAT1) of PBMCs and B, EBV-B cells from patients (E370-current patient, E353K -GOF control, M654K-LOF control) and healthy controls (HC) were stimulated or not (NS) with IFN-γ; C, pSTAT1 dephosphorylation (30–180 min) in EBV-B cells by flow cytometry. MFI, mean fluorescence intensity; D, Luciferase activity on U3A cells transfected with WT or mutant STAT1 (E370D, E353K, L706S and M654K-LOF controls). Data are mean (± SEM; n=3); E, Evaluation of TNF-α production (pg/ml) using bead cytokine assay from PBMCs of patients and HC (n=4) in response to LPS and IFN-γ. *, # p < 0.05 vs. WT.
Apophysomyces trapeziformis is a recently described cause of mucormycosis, which rarely infects humans. It causes environmentally acquired infections predominantly in the immunocompromised host.8 Infections with Apophysomyces spp. have been identified in traumatically inoculated immunocompetent hosts and it may also result from inhalation of spores into the sinuses.8,9 In 2011, 15 cases of A. trapeziformis infection were identified in individuals injured in an Enhanced Fujita Scale 5 tornado in Joplin, Missouri.6 Treatment descriptions are limited to case reports, including extensive tissue damage and debridement and use of liposomal amphotericin B in combination with posaconazole or micafungin.6
This novel GOF mutation in STAT1 in a patient with disseminated Apophysomyces trapeziformis infection who had never had CMC or autoimmunity represents the first genetic predisposition to A. trapeziformis. STAT1 GOF mutations have not previously been associated with filamentous molds, let alone members of the Mucorales.
The finding of disseminated A. trapeziformis occurring in a patient with STAT1 GOF mutation without associated trauma or any other predisposing clinical conditions suggests that GOF STAT1 mutations can also underlie disseminated mucormycosis.
Acknowledgments
We are grateful to Nick Adamo and Debra A. Long Priel for technical assistance. Thanks are also due to Ingrid Portillo for administrative assistance.
Financial support: This study was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors have no conflicts of interest.
References
- 1.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515–28. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208:1635–48. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias DL, Paulson ML, et al. STAT1 gain of function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. 2013;131:1624–34. doi: 10.1016/j.jaci.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant gain-of-function STAT1 mutations in Foxp3+ Ipex-like syndrome. J Allergy Clin Immunol. 2013;131:1611–23. doi: 10.1016/j.jaci.2012.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hori T, Ohnishi H, Teramoto T, Tsubouchi K, Naiki T, Hirose Y, et al. Autosomal-dominant chronic mucocutaneous candidiasis with STAT1-mutation can be complicated with chronic active hepatitis and hypothyroidism. J Clin Immunol. 2012;32:1213–20. doi: 10.1007/s10875-012-9744-6. [DOI] [PubMed] [Google Scholar]
- 6.Neblett Fanfair R, Benedict K, Bos J, Bennett SD, Lo YC, Adebanjo T, et al. Necrotizing cutaneous mucormycosis after a tornado in Joplin, Missouri, in 2011. N Engl J Med. 2012;367:2214–25. doi: 10.1056/NEJMoa1204781. [DOI] [PubMed] [Google Scholar]
- 7.Alvarez E, Stchigel AM, Cano J, Sutton DA, Fothergill AW, Chander J, et al. Molecular phylogenetic diversity of the emerging mucoralean fungus Apophysomyces: proposal of three new species. Rev Iberoam Micol. 2010;27:80–9. doi: 10.1016/j.riam.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Walsh TJ, Gamaletsou MN, McGinnis MR, Hayden RT, Kontoyiannis DP. Early clinical and laboratory diagnosis of invasive pulmonary, extrapulmonary, and disseminated mucormycosis (zygomycosis) Clin Infect Dis. 2012;54(Suppl 1):S55–60. doi: 10.1093/cid/cir868. [DOI] [PubMed] [Google Scholar]
- 9.Liang KP, Tleyjeh IM, Wilson WR, Roberts GD, Temesgen Z. Rhino-orbitocerebral mucormycosis caused by Apophysomyces elegans. J Clin Microbiol. 2006;44:892–8. doi: 10.1128/JCM.44.3.892-898.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]