

Abstract
Objectives
Perry syndrome consists of autosomal dominant Parkinsonism, depression, weight loss, and central hypoventilation. Eight mutations in 16 families have been reported: p.F52L, p.G67D, p.G71R, p.G71E, p.G71A, p.T72P, p.Q74P, and p.Y78C located in exon 2 of the dynactin 1 (DCTN1) gene on chromosome 2p13.1.
Methods
Genealogical, clinical, genetic, and functional studies were performed in three kindreds from New Zealand, the United States, and Colombia. A diaphragmatic pacemaker was implanted in the proband from the Colombian family to treat her respiratory insufficiency. Dopaminergic therapy was initiated in probands from two families.
Results
Besides the probands, 17 symptomatic relatives from all families were identified. The cardinal signs of Perry syndrome were present in all three probands with symptomatic disease onset in their fifth or sixth decade of life. Parkinsonism was moderate with a partial response to dopaminergic treatment. All affected persons but two died of respiratory insufficiency. The proband from the Colombian family is alive most likely due to early diagnosis and implantation of a diaphragmatic pacemaker. Two-and-a-half-year follow-up examination has revealed that the diaphragmatic pacemaker is optimally functioning without any major complications. In the Colombian and US families, the DCTN1 p.G71R and in the New Zealand family the DCTN1 p.Y78C mutations were identified. In functional assays, both mutations altered microtubule binding consistent with a pathogenic role.
Conclusions
Perry syndrome is a rare condition, but new cases are expected to be diagnosed worldwide. Early diagnosis prevents life-threatening acute respiratory failure. Diaphragmatic pacemakers should be considered as an effective symptomatic treatment option.
INTRODUCTION
Perry syndrome is a rare autosomal dominant disorder first described in 1975 in a Canadian family [1]. To date, 15 additional families with genetically confirmed Perry syndrome have been found in Japan, France, the United Kingdom, the United States, Turkey, South Korea, and Colombia [1– 9]. The clinical presentation of the syndrome comprises four cardinal signs: central hypoventilation, weight loss, depression, and Parkinsonism [1, 10]. The mean age at symptom onset is 48 years and the mean disease duration is five years. Neuropathological changes include neuronal loss and gliosis in the substantia nigra, and transactive response DNA-binding protein 43 (TDP-43) positive inclusions and no Lewy bodies [2]. Etiopathologically, Perry syndrome is most likely a TDP-43 proteinopathy, but without evidence of mutations in the transactive response DNA-binding protein 43 (TARDBP) gene [2]. Although TDP-43 and ubiquitin immunopositive inclusions are similar to those found in frontotemporal dementia and amyotrophic lateral sclerosis, they affect the extrapyramidal system and spare both motor neurons and cortical regions in this syndrome [2]. To date eight pathogenic mutations in exon 2 in the dynactin 1 (DCTN1) gene on chromosome 2p13.1 have been identified: p.F52L, p.G67D, p.G71R, p.G71E, p.G71A, p.T72P, p.Q74P, and p.Y78C. The clinical and pathological presentations are similar, except for carriers of the p.G71A mutation, which display neither hypoventilation, nor weight loss [4], and p.F52L mutation carriers have do not develop depression, or if they do develop depression, it occurs late in the disease course [9].
DCTN1 encodes p150Glued, the subunit of the dynactin complex that links intracellular cargo to the dynein motor complex for retrograde transport [11]. p150Glued also independently interacts with microtubules via its N-terminal cytoskeleton-associated protein glycine-rich (CAP-Gly) domain. Herein, we report three kindreds with Perry syndrome, including family 1 from New Zealand, family 2 from the United States and family 3 from Colombia. All families present with mutations in the highly conserved CAP-Gly domain which show functional deficits in binding to microtubules, underscoring their pathogenic roles.
METHODS
GENEALOGICAL AND CLINICAL INVESTIGATIONS
Genealogical and clinical evaluations were performed by means of medical chart reviews, interviews of the patients and their relatives, and neurological examinations. All aspects of this study were approved by the Institutional Review Boards of Mayo Clinic, Pontificia Universidad Javeriana, and Christchurch Hospital.
MOLECULAR GENETIC AND FUNCTIONAL STUDIES
Sequence analysis of DCTN1 exon 2 was performed. To test the pathogenicity of the identified mutations a microtubule binding assay was performed. HEK293E cells (Invitrogen, CA) were grown in Dulbecco’s Modified Eagle Medium (Invitrogen, CA) supplemented with 10% Fetal Bovine Serum (PAA Laboratories, PA) at 37°C under humidified conditions. pLenti6.3-DCTN1-V5 wild-type (wt), p.G59S, p.G71R, and p.Q74P were previously described [4]. The p.Y78C mutation was introduced using DCTN1 wt as a template for standard PCR based mutagenesis followed by restriction digest and ligation via EcoRI and the DCTN1 internal restriction site AccIII. The generated construct was sequence verified using BigDye Terminator v.3.1 and an ABI 3100 Genetic Analyzer (Applied Biosystems, CA). To perform a microtubule binding assay, HEK293E cells were transiently transfected using Xtremegene 9 (Roche, Germany) with wt or mutant pLenti-DCTN1-V5 constructs for 72 hours or left untransfected. Cells were harvested and lysed in microtubule binding assay buffer (80mM HEPES, pH 7.5, 10mM NaCl, 5mM MgCl2, 0.2% NP-40) in the presence of protease inhibitors (Roche, Germany). After measuring the protein concentration using bicinchoninic acid (Pearce Pharmaceuticals, Australia), 100 μg were used for microtubule binding assay (Microtubule Binding Protein Spin-Down Assay Kit, Cytoskeleton) according to manufacturer instructions. 10 μl of the supernatant and the resuspended pellet fraction or 25 μg of total protein were loaded onto 4–12% Bis-Tris gradient gels (Invitrogen, CA) and blotted onto polyvinylidene fluoride (PVDF) membrane. After blocking in 5% milk/Tris Buffered Saline with Tween-20 (TBST), membranes were incubated with anti-V5 (1:10,000, Invitrogen, CA) and anti- β-tubulin (1:200,000, clone 2-28-33, Sigma, MO) or anti-GAPDH (1:100,000, Biodesign International) antibodies overnight followed by anti-mouse secondary antibodies coupled to horseradish peroxidase (Jackson Immunoresearch, PA). Immunoreactive signals were visualized using Immobilon Western HRP substrate (Millipore, MA) on X-ray films. Band densitometry was analyzed using ImageJ software, version 1.46. Statistical analysis was performed using unpaired, two-sided student’s t-test.
RESULTS
FAMILY 1 (New Zealand)
GENEALOGICAL AND CLINICAL INVESTIGATIONS
A 58-year-old Caucasian man presented with a four-year history of increasing somnolence, fatigue, and personality changes with marked apathy. He also reported a weight loss of 8 kg over three months despite an increased appetite and intake of food. He had mild depression and slight cognitive impairment. Upon clinical examination, the patient showed tachypnea and mild Parkinsonian signs, which included intermittent resting tremor of the upper extremities and hypomimia, which were not treated with dopaminergic therapy. The patient responded well to bi-level positive airway pressure (BiPAP) therapy. He continued to work, albeit part-time and in a less demanding role and was able to cycle recreationally until just before his death 20 months after the diagnosis was established. He died of a sudden worsening of his respiratory insufficiency.
The patient had three asymptomatic children aged 25–30 years. The patient’s father suffered from depression and apathy, and he died of respiratory failure at the age of 66 years. The father’s sister also had depression and apathy, and she died of respiratory failure after electroconvulsive therapy (ECT) when she was 51 years old. The father’s brother also died of respiratory failure, pneumonia, and brain stem degeneration when he was aged 52 years (pedigree structure is shown in Figure 1a).
Figure 1. Pedigree structures of three kindreds with Perry syndrome.

Panel 1a) includes the pedigree with the DCTN1 p.Y78C (c.233A>G) mutation, and 1b and 1c) pedigrees with the DCTN1 p.G71R (c.211G>A) mutation. Standard symbols were used. Round symbols indicate females, squares males, diagonal lines indicate the individual is deceased. Diamonds were used to disguise gender. The solid arrowhead indicates the proband. Black full-filled symbols indicate individuals who suffered from Perry syndrome. Asterisks indicate that genetic testing for DCTN1 gene mutation was performed.
The diagnostic procedures included arterial blood gas (ABG) tests. They confirmed compensated hypercapnic respiratory failure. An overnight polysomnography showed severe central sleep apnea and unusual rapid shallow breathing during the rapid eye movement sleep. Spirometry, electroencephalography (EEG), computed tomography (CT), and magnetic resonance imaging (MRI) of the brain were normal.
MOLECULAR GENETIC AND FUNCTIONAL STUDIES
The c.233A>G (p.Y78C) mutation in the DCTN1 gene was identified in the proband. Subsequently, the corresponding mutant cDNA was cloned, expressed in human cell culture and examined in functional assays to study its pathogenicity. To uncover putative deficits associated with p150Glued p.Y78C, we performed microtubule binding assays [4]. In brief, cell lysates containing comparable amounts of overexpressed DCTN1 wt protein or p.Y78C mutant were incubated with preassembled microtubules in vitro (Figure 2a). Other mutant DCTN1 proteins (p.G59S, p.G71R, and p.Q74P) had previously been analyzed and served as pathogenic controls in the current assay. To assess binding to microtubules, reactions were placed on a glycerol cushion and separated by high-speed centrifugation. p150Glued wt completely co-sedimented with microtubules in the pellet fraction indicative of its highly efficient binding. However, the binding of Perry syndrome mutants was significantly reduced, as demonstrated by their increased presence in the supernatant fraction (Figure 2b). The p.G59S mutation showed reduced binding to microtubules, however, did not reach statistical significance, in agreement with our previous study [4]. In addition, we performed immunostaining of overexpressed DCTN1 in human HEK293 cells to evaluate their aggregation propensities. Neither wt nor any of the analyzed mutants resulted in overt aggregation or appreciable cell death (data not shown).
Figure 2. Microtubule Binding assay for p.Y78C, p.G71R, p.G59S, p.Q74P DCTN1 mutants.
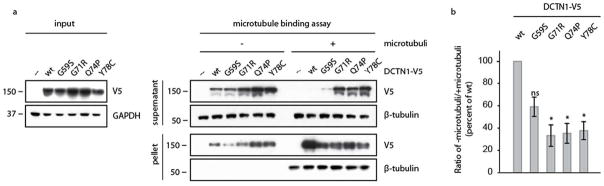
HEK293E cells were transfected with either wild-type or mutant DCTN1-V5, as indicated. To test the affinity to microtubules, equal amounts of lysates were incubated with assembled microtubules in vitro, placed on a glycerol cushion and separated via centrifugation at 100,000g. Supernatant and pellet were subjected to SDS-PAGE, blotted and analyzed with antibodies against V5 and β-tubulin. (a) Shown is a representative Western Blot of supernatant (middle, top) and pellet (middle, bottom). Input samples are indicated to the left. Equal loading is demonstrated by GAPDH probing. (b) Shown is the densitometric ratio of supernatant without microtubules to supernatant with microtubules as the mean value of five independent experiments. In contrast to wild-type DCTN1-V5, mutant DCTN1-V5 show considerable less microtubule binding, being significant for p.G71R, p.Q74 and p.Y78C. *: p<0.05, ns: not significant.
FAMILY 2 (United States)
GENEALOGICAL AND CLINICAL INVESTIGATIONS
A 58-year-old Caucasian man from the United States was admitted to the hospital with a nine-year-history of respiratory insufficiency, fatigue, apathy, and unexplained weight loss. Tachypnea and predominantly akinetic-rigid Parkinsonism with intermittent resting tremor were observed on clinical examination. The tremor slightly improved with a total daily dose of 400 mg of levodopa. The patient died as a result of rapid deterioration of his respiratory insufficiency. Both his mother and sister were reported to have had a similar disease course and fate. The sister was diagnosed as having mitochondriopathy based only on the presence of ragged red fibres on muscle biopsy. Her 33-year-old son and the patient’s 28-year–old son were reported to be asymptomatic. The patient had two twin brothers who were 13 months older than him; one died in an accident at the age of 18 years and the other one was reported to be asymptomatic (Pedigree structure is presented in Figure 1b).
ABG results showed hypercarbia in the patient. His initial response to BiPAP and oxygen was positive by day and at night. Muscle biopsy revealed chronic myopathic features. No deletions of mtDNA were found. On polysomnography, sleep apnea and a central hypoventilation syndrome were documented.
MOLECULAR GENETIC AND FUNCTIONAL STUDIES
Genetic testing revealed the c.211G>A (p.G71R) mutation in exon 2 of the DCTN1 gene in the proband. As described above, DCTN1 p.G71R was similar to all other Perry syndrome mutations analyzed; it equally and significantly disrupted microtubule binding (Figure 2), which confirmed our previous findings [4].
FAMILY 3 (Colombia)
GENEALOGICAL AND CLINICAL INVESTIGATIONS
A 56-year-old Colombian woman of Caucasian descent was admitted in 2011 to the hospital with her first episode of acute respiratory failure. Her case has recently been reported [5]. She had a two-year history of fatigue, Parkinsonism, apathy, and anxiety. Therapy with oral levodopa at a maximum daily dosage of 1000 mg slightly improved her tremor. The patient also reported a weight loss of 15 kg in 6 months without dietary changes. Following diagnostic tests of the respiratory system [5], the patient underwent an implantation surgery of a diaphragmatic pacemaker. At her two-and-a-half-year follow-up examination after the implantation surgery, her diaphragmatic pacemaker has been observed to be optimally functioning. The patient has been highly independent in her everyday life.
Similar symptoms were observed in the patient’s mother, three maternal aunts, one male and two female cousins, and her brother and sister. All except her sister died of respiratory insufficiency between the ages of 60–80 years. The sister’s first complaints manifested at the age of 47 years and included bradykinesia and hypomimia. She refused genetic tests for DCTN1 gene mutations. The patient has three asymptomatic daughters aged 35– 38 years (Pedigree structure is presented in Figure 1c).
MOLECULAR GENETIC AND FUNCTIONAL STUDIES
Genetic testing revealed the c.211G>A (p.G71R) mutation in exon 2 of the DCTN1 gene in the patient and her middle daughter. No mutation was found in her other children.
DISCUSSION
The probands from the three kindreds with Perry syndrome presented all of the obligatory clinical signs. The tentative diagnosis was not established until the respiratory insufficiency manifested. The symptom onset in our cases was typical and occurred at the beginning of the sixth decade of their lives. Mechanisms that explain the late onset of clinical manifestations in Perry syndrome remain unknown.
Parkinsonian signs were moderate and seen in all patients, but only two probands required dopaminergic treatment, and in these two, only a partial responsiveness was observed; however, the maximal daily dose varied between 400mg/day and 1000mg/day, but typically, high doses of levodopa (2000mg/day or higher) have been observed to be more beneficial for alleviating Parkinsonian signs, especially in the treatment of tremor in the early stages of the disease [8]. In one family, marked improvement of bradykinesia and depression due to a levodopa therapy was initially observed [9]. However, the authors did not specify the dosage.
All probands revealed signs of mild depressive disorders. In a more severe form, these can lead to suicide or suicidal thoughts in roughly one third of the patients [10]. Weight loss without dietary changes is a poorly understood phenomenon, which presumably results from involvement of brainstem nuclei: increased caloric intake may be of some help.
Caroppo et al. [7] have recently described a French family with the DCTN1 p.G71E mutation that is claimed to show a high intrafamilial variability with different phenotypes, including behavioral variant of frontotemporal dementia, progressive supranuclear palsy-like phenotype, and isolated Parkinsonism. Another family from Japan with the DCTN1 p.F52L mutation has recently been reported to have the initial signs of isolated Parkinsonism. Later in the disease course, weight loss and respiratory insufficiency were observed. However, only some affected family members developed depression and frontotemporal atrophy on MRI [9]. Although none of our patients was identified to have these mutations, we did not observe any atypical clinical presentations in our cases. Affected individuals from the three families, including two probands, died of hypoventilation. This is due to the neuronal loss in the pre-Bötzinger complex of the medulla oblongata and mostly occurs at night [10].
The DCTN1 p.G71R mutation identified in two of our families is known as being causative for Perry syndrome and so far has been described in four families from Canada, Turkey, England and South Korea [4, 8, 6]. Affected individuals with this mutation have presented all cardinal signs of Perry syndrome. Moreover, the patients from England and South Korea also showed limitation of both upward and downward vertical saccades. In contrast, the DCTN1 p.Y78C mutation described in our New Zealand family has been published only once in a Korean patient [6]. Interestingly, neither respiratory failure, nor weight loss were reported in this patient [6], whereas our carrier presented all cardinal signs of Perry syndrome. Brain imaging studies revealed no relevant pathological changes in our patients, although midbrain and frontotemporal atrophy has already been reported [7, 9].
ABG showed respiratory acidosis and hypercapnia. Nocturnal BIPAP was the method of choice to prolong survival and improve quality of life. The implantation of a diaphragmatic pacemaker, which was performed in the proband from the Colombian family seems to be a good and safe alternative to provide a patient with a much wider autonomy in his or her everyday life.
Using a microtubule-binding assay as a functional read-out for pathogenicity, we could show a decrease in binding microtubules in the p150Glued p.G71R, p.Q74P, and p.Y78C mutations. This provides evidence of a destabilized dynactin complex and thus evidence of their pathogenicity. All these mutations reside within the N-terminal cytoskeleton-associated protein–glycine-rich (CAP-Gly) domain of p150Glued and affect evolutionary conserved residues. However, the p.G59S mutation that also locates to the CAP-Gly domain of p150Glued has been identified in hereditary motor neuropathy 7B (HMN7B), also known as distal spinal and bulbar muscular atrophy. Importantly, two recent reports have uncovered differential effects of Perry syndrome and HMN7B mutations in DCTN1, thus providing a molecular explanation for the pathogenesis of these distinct neurodegenerative disorders [12, 13]. Perry syndrome mutations lie on the surface, in or close to the microtubule-binding site, and thus locally affect p150Glued without change of protein stability. However, the HMN7B mutation likely affects overall folding of the dynactin subunit, which causes protein instability and aggregation. This results not only in diminished microtubule association, but additionally affects the binding to the dynein motor complex. As a result, Perry syndrome mutations that preserve other dynactin functions specifically reduced the flux of cargo from the neurite tip, whereas the HMN7B mutant in the CAP-Gly domain disrupts axonal transport [12, 13].
Given that pathogenic DCTN1 mutations are most commonly missense mutations, it is possible that the mutant protein may competitively inhibit the function of the wild type protein, which is a process that may be progressive and may lead to increasing dysfunction of intracellular transport. Such a scenario would be in line with findings that Perry syndrome mutations neither interfere with the dimerization of p150Glued nor with its integration into the dynactin complex, but rather result in compromised CAP-Gly domain function [12, 13].
In conclusion, three families with Perry syndrome indicate that this syndrome is more common than previously thought and can be found with a global distribution. An early diagnosis can significantly improve the quality of life and can prevent life-threatening episodes of acute respiratory failure. Implantation of a diaphragmatic pacemaker should be considered as a safe and effective symptomatic treatment option of respiratory insufficiency in patients with Perry syndrome. In these cases, Parkinsonism, although always present, may not be disabling. Further functional studies will hopefully provide insights into the underlying pathogenic mechanisms and will nominate a therapeutic intervention strategy that will halt the progression of this fulminant disorder.
Acknowledgments
We thank the patients and their relatives for their assistance in creating this manuscript. We also thank Mrs. Kelly E. Viola, ELS for her editorial support.
Author contributions
Pawel Tacik: Drafting and revising the manuscript, acquisition of patient data, analysis and interpretation of the data.
Fabienne Fiesel: Functional analysis, analysis and interpretation of the data drafting manuscript.
Shinsuke Fujioka: Revising the manuscript, analysis and interpretation of the data.
Owen A. Ross: Revising the manuscript; analysis and interpretation of the data.
Felipe Pretelt: Revising the manuscript, acquisition of patient data.
Camilo Castañeda Cardona: Revising the manuscript, acquisition of patient data.
Alexa Kidd: Revising the manuscript, acquisition of patient data.
Michael Hlavac: Revising the manuscript, acquisition of patient data.
Tony Raizis: Revising the manuscript, acquisition of patient data.
Michael S. Okun: Revising the manuscript, acquisition of patient data.
Sharleen Traynor: Revising the manuscript, acquisition of patient data.
Audrey J. Strongosky: Revising the manuscript, acquisition of patient data.
Wolfdieter Springer: Design, analysis and interpretation of functional data, revising the manuscript.
Zbigniew K. Wszolek: Design and conceptualization of the study, acquisition of patient data, revising the manuscript.
Financial Disclosure/Conflict of Interest
| Pawel Tacik | is supported by the Max Kade Foundation. |
| Fabienne C. Fiesel | reports no disclosure |
| Shinsuke Fujioka | is partially supported by the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch. |
| Owen A. Ross | is partially supported by NIH/NINDS R01 NS078086 and supported by the NIH/NINDS P50 NS072187 |
| Felipe Pretelt | reports no disclosure |
| Camilo Castañeda Cardona | reports no disclosure |
| Alexa Kidd | reports no disclosure |
| Michael Hlavac | reports no disclosure |
| Tony Raizis | reports no disclosure |
| Michael S. Okun | serves as a consultant for the National Parkinson Foundation, and has received research grants from NIH, NPF, the Michael J. Fox Foundation, the Parkinson Alliance, Smallwood Foundation, the Bachmann-Strauss Foundation, the Tourette Syndrome Association, and the UF Foundation. Dr. Okun has previously received honoraria, but in the past >36 months has received no support from industry. Dr. Okun has received royalties for publications with Demos, Manson, Amazon, and Cambridge (movement disorders books). Dr. Okun is an associate editor for New England Journal of Medicine Journal Watch Neurology. Dr. Okun has participated in CME activities on movement disorders in the last 36 months sponsored by PeerView, Prime, and by Vanderbilt University. The institution and not Dr. Okun receives grants from Medtronic and ANS/St. Jude, and the PI has no financial interest in these grants. Dr. Okun has participated as a site PI and/or co-I for several NIH, foundation, and industry sponsored trials over the years but has not received honoraria. |
| Sharleen Traynor | reports no disclosure |
| Audrey J. Strongosky | reports no disclosure |
| Wolfdieter Springer | is partially supported by Mayo Clinic Center for Individualized Medicine, the Marriott Family Foundation and a Gerstner Family Career Development Award. |
| Zbigniew K. Wszolek | is partially supported by the NIH/NINDS P50 NS072187, the Michael J. Fox Foundation for Parkinson’s Research, and the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch. |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Pawel Tacik, Email: Tacik.Pawel@mayo.edu.
Fabienne C. Fiesel, Email: Fiesel.Fabienne@mayo.edu.
Shinsuke Fujioka, Email: Fujioka.Shinsuke@mayo.edu.
Owen A. Ross, Email: Ross.Owen@mayo.edu.
Felipe Pretelt, Email: feferpre@hotmail.com.
Camilo Castañeda Cardona, Email: camilomeister@gmail.com.
Alexa Kidd, Email: Alexa.Kidd@cdhb.health.nz.
Michael Hlavac, Email: Michael.Hlavac@cdhb.health.nz.
Anthony Raizis, Email: Anthony.Raizis@cdhb.health.nz.
Michael S. Okun, Email: okun@neurology.ufl.edu.
Sharleen Traynor, Email: smtraynor@gmail.com.
Audrey J. Strongosky, Email: Strongosky.Audrey2@mayo.edu.
Wolfdieter Springer, Email: Springer.Wolfdieter@mayo.edu.
Zbigniew K. Wszolek, Email: Wszolek.Zbigniew@mayo.edu.
References
- 1.Perry TL, Bratty PJ, Hansen S, Kennedy J, Urquhart N, Dolman CL. Hereditary mental depression and Parkinsonism with taurine deficiency. Arch Neurol. 1975;32:108–13. doi: 10.1001/archneur.1975.00490440058009. [DOI] [PubMed] [Google Scholar]
- 2.Wider C, Dachsel JC, Farrer MJ, Dickson DW, Tsuboi Y, Wszolek ZK. Elucidating the genetics and pathology of Perry syndrome. J Neurol Sci. 2010;15;289:149–54. doi: 10.1016/j.jns.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aji BM, Medley G, O’Driscoll K, Larner AJ, Alusi SH. Perry syndrome: a disorder to consider in the differential diagnosis of Parkinsonism. J Neurol Sci. 2013;330:117–8. doi: 10.1016/j.jns.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Farrer MJ, Hulihan MM, Kachergus JM, Dächsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier B, Chapon F, Tsuboi Y, Yamada T, Gutmann L, Elibol B, Bhatia KP, Wider C, Vilariño-Güell C, Ross OA, Brown LA, Castanedes-Casey M, Dickson DW, Wszolek ZK. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;412:163–5. doi: 10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pretelt F, Castañeda Cardona C, Tacik P, Ross OA, Zbigniew Wszolek ZK. Latin America’s first case of Perry syndrome and a new treatment option for respiratory insufficiency. J Neurol. 2014;26:620–1. doi: 10.1007/s00415-014-7262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung EJ, Hwang JH, Lee MJ, Hong J-H, Ji KH, Yoo W-K, Kim SJ, Song HK, Lee CS, Lee M-S, Kim YJ. Expansion of the clinicopathological and mutational spectrum of Perry syndrome. Parkinsonism Relat Disord. 2014 doi: 10.1016/j.parkreldis.2014.01.010. In Press. [DOI] [PubMed] [Google Scholar]
- 7.Caroppo P, Le Ber I, Clot F, Rivaud-Péchoux S, Camuzat A, De Septenville A, Boutoleau-Bretonnière C, Mourlon V, Sauvée M, Lebouvier T, Bonnet AM, Levy R, Vercelletto M, Brice A for the French Clinical and Genetic Research Network on Frontotemporal Dementia/Frontotemporal Dementia–Amyotrophic Lateral Sclerosis. DCTN1 Mutation Analysis in Families With Progressive Supranuclear Palsy-Like Phenotypes. JAMA Neurol. 2014;71:208–15. doi: 10.1001/jamaneurol.2013.5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newsway V, Fish M, Rohrer JD, Majounie E, Williams N, Hack M, Warren JD, Morris HR. Perry syndrome due to the DCTN1 G71R mutation: a distinctive levodopa responsive disorder with behavioral syndrome, vertical gaze palsy, and respiratory failure. Mov Disord. 2010;25:767–770. doi: 10.1002/mds.22950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Araki E, Tsuboi Y, Daechsel J, Milnerwood A, Vilarino-Guell C, Fujii N, Mishima T, Oka T, Hara H, Fukae J, Farrer MJ. A Novel DCTN1 mutation with late-onset parkinsonism and frontotemporal atrophy. Mov Disord. 2014 doi: 10.1002/mds.25833. [DOI] [PubMed] [Google Scholar]
- 10.Wider C, Wszolek ZK. Rapidly progressive familial Parkinsonism with central hypoventilation, depression and weight loss (Perry syndrome) - a literature review. Parkinsonism Relat Disord. 2008;14:1–7. doi: 10.1016/j.parkreldis.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 11.Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–79. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- 12.Lloyd TE1, Machamer J, O’Hara K, Kim JH, Collins SE, Wong MY, Sahin B, Imlach W, Yang Y, Levitan ES, McCabe BD, Kolodkin AL. The p150(Glued) CAP-Gly domain regulates initiation of retrograde transport at synaptic termini. Neuron. 2012;74:344–60. doi: 10.1016/j.neuron.2012.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moughamian AJ1, Holzbaur EL. Dynactin is required for transport initiation from the distal axon. Neuron. 2012;74:331–43. doi: 10.1016/j.neuron.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]