

Abstract
In polycystic kidney disease (PKD), abnormal proliferation and genomic instability of renal epithelia have been associated with cyst formation and kidney enlargement. We recently showed that L-type calcium channel (Cav1.2) is localized to primary cilia of epithelial cells. Previous studies have also shown that low intracellular calcium level was associated with the hyperproliferation phenotype in the epithelial cells. However, the relationship between calcium channel and cystic kidney phenotype is largely unknown. In this study, we generated cells with somatic deficient Pkd1 or Pkd2 to examine ciliary CaV1.2 function via lentiviral knockdown or pharmacological verapamil inhibition. Although inhibition of CaV1.2 expression or function did not change division and growth patterns in wild-type epithelium, it led to hyperproliferation and polyploidy in mutant cells. Lack of CaV1.2 in Pkd mutant cells also decreased the intracellular calcium level. This contributed to a decrease in CaM kinase activity, which played a significant role in regulating Akt and Erk signaling pathways. Consistent with our in vitro results, CaV1.2 knockdown in zebrafish and Pkd1 heterozygous mice facilitated the formation of kidney cysts. Larger cysts were developed faster in Pkd1 heterozygous mice with CaV1.2 knockdown. Overall, our findings emphasized the importance of CaV1.2 expression in kidneys with somatic Pkd mutation. We further suggest that CaV1.2 could serve as a modifier gene to cystic kidney phenotype.
Introduction
Polycystic kidney disease (PKD) is one of the most common genetic diseases. It is characterized by formation and expansion of multiple fluid-filled cysts in the kidneys. Many previous studies have suggested that the abnormality of kidney cystic cells is contributed by interrupted calcium homeostasis [1–4], probably due to the abnormal calcium channel polycystin-2 (PC2) or its regulatory protein polycystin-1 (PC1)
Both PC1 and PC2 are localized to primary cilia of renal epithelia and are required for calcium signaling [5–7]. Cells isolated from the cyst lining of human patients and PKD animal models have been shown to lack normal flow-sensitive calcium signaling and have lower intracellular calcium concentration. This low calcium level has been thought to be critical for pathogenesis of PKD [2]. In cystic kidney-derived cells, the aberrant intracellular calcium level has been shown to cause cAMP-dependent activation of the MEK/ERK pathway and increased cell proliferation [4,8,9]. In contrast, cAMP often inhibits the MEK/ERK pathway in normal kidney cells. Calcium concentration has long been thought to be a major factor that differentiates the roles of cAMP between normal and cystic kidney cells. Furthermore, PKD phenotypes could be partially rescued by recovering intracellular calcium level, whereas reduction of that could reproduce PKD phenotypes [2–4].
One of the major complications of PKD is hypertension, occurring in 50 to 70 percent of cases before any significant reduction in glomerular filtration rate, with an average onset at 30 years of age [10]. Abnormalities in the renin-angiotensin system and vascular endothelial cilia have been proposed as the mechanisms of hypertension [11–13]. Because hypertension in PKD is still poorly understood, the hypertensive therapy has not been much different than therapy for hypertension in other chronic kidney diseases. L-type calcium channel blockers have been tested in PKD patients to control their blood pressure [14,15]. Considering the fact that calcium level is lower in cystic than normal epithelia [1–4], understanding the roles of L-type calcium channel in PKD is scientifically and clinically imperative. When heterozygous Han:SPRD Cy rats were treated with a serial of L-type calcium channel blockers to inhibit its largest subunit (CaV1.2), larger cysts were observed at a much earlier stage in treated- than in non-treated groups [1,16]. However, the molecular mechanism of CaV1.2 in the kidney remains largely unknown. In the present study, we examined the roles of CaV1.2 in the cilia and in cystic kidney formation in orthologous models of ADPKD. Our study suggested a potential role of CaV1.2 as a modifier gene in cystic renal epithelial cells.
Methods and Materials
Cell culture
Mouse kidney epithelial cells (wild-type, Pkd1−/− and Pkd2−/−) were isolated from embryonic day 15.5 kidneys from a cross of Pkd1+/− or Pkd2+/−mice that also carried a temperature-sensitive simian virus 40 (SV40) large T-antigen transgene. The heterozygous mice of Pkd1 or Pkd2 had normal characteristics as of wild-type mice. Because a mouse body temperature is about 39°C, the SV40 transgene remained inactivated. Of note is that the transgene was activated at 33°C and in the presence of IFN-γ. Therefore, the resulting cells from these mice were considered conditionally immortalized, because the expression of the SV40 large T-antigen could be regulated by temperature and IFN-γ in culture. All cell lines were further cloned by positive selection for renal epithelial marker (wheat germ agglutinin, WGA). Cell lines were grown at 33°C in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum supplemented with IFN-γ. In all experiments, a constant laminar flow generating a shear-stress of 1 dyne/cm2 was applied for 18 hours to mimics a physiological condition. Such force has been shown to be able to activate flow-sensitive cell signaling mediated by primary cilia [17].
Fluorescence automated cell sorting
Fully differentiated cells were rinsed with HBSS (Hank's balanced salt solution), and approximately 105 viable cells were analyzed for expression of epithelial marker. Cells were incubated with FITC-conjugated WGA antibody (10 μg/ml; Vector Laboratories) and subjected to a robust analysis using FACStar-PLUS with a laser excitation of 200 mm.
Lentivirus production and infection
HEK-293T cells were co-transfected with lentivirus packaging plasmids mix and one of four different constructs to block CaV1.2 expression. These constructs were labeled 1 (5’-GTC CAG CAC ACC TCC TTC AGG AAC CAT AT-3’), 2 (5’-TCA GAA GTG CCT CAC TGT TCT CGT GAC CT-3’), 3 (5’-TCA GAA GTG CCT CAC TGT TCT CGT GAC CT-3’) or scrambled (5’-AAA CCC ATG AGA GAC CTT TTA GAA GAT T-3’). Lentivirus was harvested at 48 and 72 hours after the initial transfection. The titration was determined to be ~107 with the method described previously [18,19]. One ml of each shRNA-lentivirus was added to cells grown on the 10-cm cell culture dishes. The efficiency of infection was determined with Accuri Flow Cytometer by monitoring the percentage of GFP positive cells. Gene silencing was further confirmed by Western Blot, and only the shRNA lentivirus with higher knockdown efficiency was selected for future use. Infected cell lines were maintained in cell growth medium described above and supplemented with 10 ug/ml of puromycin to maintain homogenous population. For in vitro applications, lentivirus was concentrated to a titration of 109–10 with an established method [20]. The verifications of CaV1.2 knockdown were examined after clonal expansion at passages 2, 3 and 5. Most of our studies on these cells were carried out at passages 3 to 6, except for those with verapamil at passage 8.
Immunoblot analysis
A total protein of 30 mg was analyzed with a standard Western blot. The following antibodies and dilutions were used in our analyses: phospho-ERK (Cell Signaling, 1:1000), anti-ERK (Cell Signaling, 1:1000), phosphor-AKT (Cell Signaling, 1:200), anti-AKT (Cell Signaling, 1:1000), anti-CaV1.2 (Alomone, 1:500) and GAPDH (Cell Signaling, 1:1,500). In some cases, wild-type cells were also treated with CaM kinase II inhibitor (10 μM; Sigma Aldrich, Inc) and KN-93 (2 μM; Sigma Aldrich, Inc) for 18 hours and 24 hours, respectively. Band intensity was quantified using NIH’s Image J software.
Immunofluorescence studies
Cells were fixed with 2% sucrose plus 3% paraformaldehyde, permeabilized with 0.5% (vol/vol) Triton-X, and incubated with rabbit Antibodies at 1:200 dilution. To stain CaV1.2, rabbit polyclonal anti-CaV1.2 (Alomone, 1:200) was applied to cells and incubated overnight at 4 °C. Mouse antibody to acetylated α-tubulin (1:10,000; Sigma Aldrich) was used as a ciliary marker, and cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) to label the nuclei. Respective secondary antibodies were used at a dilution of 1:500.
Chromosomal analysis
For flow cytometry analysis, cells were harvested and washed with cold phosphate buffered solution (PBS). Cells were then re-suspended at 1–2 × 106 cells/ml. One ml of cell aliquot was placed in a 15 ml polypropylene. Three ml of ethanol was added to the cell aliquot for fixation at −20 °C overnight. After two washes with PBS, solution containing propidium iodide and RNase A was added to cell pellet for three hours at 4 °C. Cells were analyzed with Accuri Flow Cytometer, as previously described [21,22].
For chromosomal counting, cells were plated for at least one day and grown to 80% confluence
They were then grown for an additional 2 h after incubating with 0.05 μg/ml colcemid solution for 30 min at 37 °C in the dark. Next, cells were collected and incubated with 0.56% KCl hypotonic solution for 45 min at 37 °C, followed by fixation with 3:1 methanol:acetic acid. The cells were dropped onto pre-cleaned glass slides and allowed to dry. Dried slides were counterstained with DAPI for microscopy analysis. At least 50 chromosome spreads were counted from each cell analysis, as previously described [23]. In some cases, Pkd1−/− and Pkd2−/− cells were treated with L-type calcium channel blocker verapamil (2 μM; Sigma Aldrich, Inc.) for 24 hours.
Calcium measurement
Cells were cultured on glass-bottom dishes and perfused 18 hours prior to experiment. After two washes with HBSS, cells were incubated with 5 μM of Fura-2 AM (Telf Labs) for 45 minutes in the dark. After incubation, cells were washed and left in HBSS for 15 min to allow Fura2-AM to hydrolyze. We have previously described details on experimental set-up and calcium calculation [17].
Animal models
Adult wild-type AB zebrafish were obtained from the Zebrafish International Resource Center (Eugene, OR) and used for breeding. Embryos were injected with 1 mM antisense translation blocking morpholino oligos (GeneTools) at the 1–2 cell stage. Zebrafish embryos were then cultured at 28.5°C in sterile egg water [24]. The following morpholino sequences were used: control MO: 5′-CCT CTT ACC TCA GTT ACA ATT TAT A-3′, cav1.2 MO: 5′-ACA TGT TTT TGC TTT CAT TTA CCA T-3′.
Previously generated Mx1Cre:Pkd1wt/flox mice at two months old were used in our studies [25]. In addition, only males were selected because they were shown to be more sensitive to CaV1.2 inhibition [16]. Polyinosine-polycytosine (pIpC, 62.5 μg) was injected intraperitoneally at seven days of age for five consecutive days. Pre-concentrated lentivirus (IFU=109–10/ml) was injected to animals at day 10 after birth subcutaneously. At two months of age, animals were euthanized and their kidneys were removed for analysis. Kidney samples were first cut in half. One half was used for immunoblotting, while the other half was for hematoxylin and eosin (H&E) staining.
Statistical analysis
All data are reported as mean±standard error of mean with statistical power greater than 0.8 at p<0.05. Data were then analyzed utilizing ANOVA test followed by Tukey’s post-test. Analysis of data was performed with Prism GraphPad 5 software.
Results
CaV1.2 was expressed in Pkd1 and Pkd2 cell lines
To confirm the presence of CaV1.2 in renal epithelial cells and determine whether its expression level was altered in different cell lines from cystic kidneys, we performed immunoblotting for CaV1.2 in wild type, Pkd1−/− and Pkd2−/− renal epithelial cells; all cell lines expressed renal epithelial marker, Wheat Germ Agglutinin (data not shown). We found that CaV1.2 was expressed in all cell lines but was two folds higher in mutant compared to wild-type cells (Figure 1a). To explore if PC1 or PC2 deficiency affected CaV1.2 distribution in renal epithelial cilia, we also studied CaV1.2 localization with immunofluorescence. CaV1.2 was mainly localized to primary cilia in wild-type cells but was found to be randomly distributed in the cytoplasm of Pkd cells (Figure 1b).
Figure 1. CaV1.2 was up-regulated in Pkd1−/− and Pkd2−/− cell lines.

a. Immunoblot analysis was performed to detect CaV1.2 expression in wild-type (WT), Pkd1−/− and Pkd2−/− cell lines. CaV1.2 was overexpressed in Pkd cells compared to wild-type cells. GAPDH was used as loading control. b. Overexpression of Cav1.2 was also confirmed by immunostaining. CaV1.2 was localized to cilia in wild-type cells. In Pkd cell lines, CaV1.2 was not detected in the cilia. Acetylated-α-tubulin (acet-a-tub) was used as cilia marker. N=3; * indicates significant difference from the control wild-type group.
CaV1.2 knockdown shortened cilia length in Pkd epithelial cells
To study the loss-of-function effects of CaF1.2, we transfected renal epithelial cells with lentivirus carrying different shRNA sequences specific for unique regions of mRNA coding for CaV1.2. Among the three sequences tested, shRNA sequence #3 produced the highest knockdown efficiency (Figure 2a). This sequence was thus selected for use in all of our future studies. To explore whether loss of CaV1.2 could play a role in cilia formation, we examined the cilia length in all cell groups (Figure 2b). In wild-type cells, we observed no change in cilia length between control and CaV1.2 knockdown. In contrast, knockdown of CaV1.2 significantly shortened cilia length in Pkd groups. This observation is intriguing because shorter cilia were often related to less sensitivity to extracellular cues, such as fluid-flow shear stress [26,27]. Also noteworthy is that Pkd1−/− and Pkd2−/− cell lines had longer cilia compared to wild-type cells.
Figure 2. CaV1.2 knockdown reduced cilia length in Pkd1−/− and Pkd2−/− cell lines.

a. Four different shRNA sequences were applied to wild-type (WT) cells via lentivirus delivery. The shRNA sequences specific for CaV1.2 were indicated as 1, 2, or 3, and scramble shRNA was used as a control (C). Additional non-infected control (NI) was also used. Immunobloting analysis was performed to determine CaV1.2 silencing efficiency. N=6. Thus, it was selected for all future applications. N=6. b. CaV1.2 knockdown had no effect on cilia length in wild-type cells but shortened cilia in Pkd1−/− and Pkd2−/− cells. N≥20 preparations for each group. # indicates significant difference from the control wild-type group; * indicates significant difference between scramble and CaV1.2 shRNA groups.
CaV1.2 regulated flow-induced cytosolic calcium level in Pkd epithelial cells
Despite its unclear physiological function in renal epithelial cells, CaV1.2 has been proposed to be a physiologically relevant calcium channel in the kidney [28]. To examine if CaV1.2 regulated cytosolic calcium concentration in physiological condition, we challenged both CaV1.2 knockdown and control groups of wild-type and Pkd cells with continuous fluid-flow at 0.7 dye/cm for 18 hours (Figure 3). There was no difference in calcium levels in wild-type cells between control and CaV1.2 knockdown. On the other hand, CaV1.2 knockdown decreased the already low calcium concentration in Pkd cell lines.
Figure 3. CaV1.2 knockdown decreased cytosolic calcium levels in Pkd1−/− and Pkd2−/− cell lines.
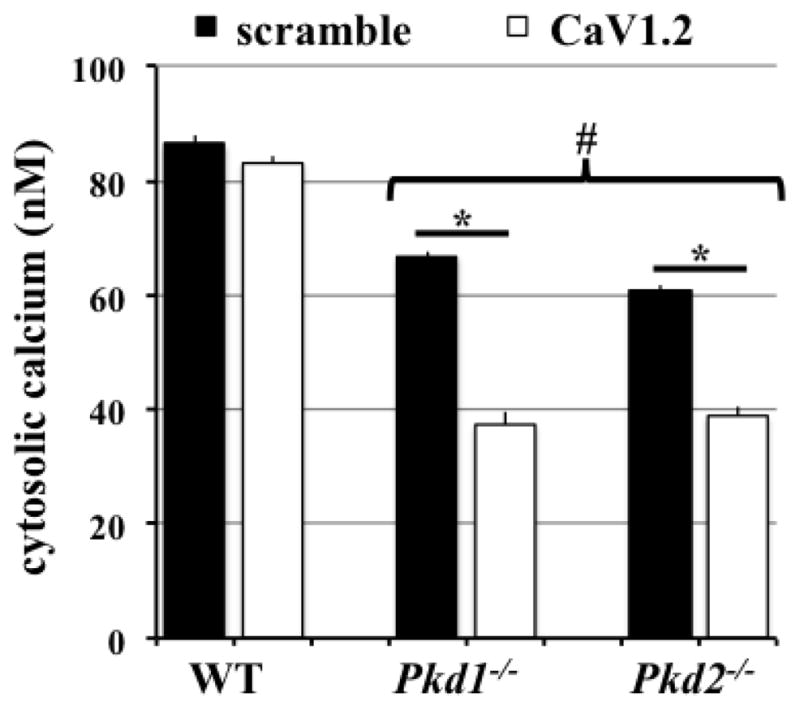
Wild-type (WT), Pkd1−/− and Pkd2−/− cells were perfused with fluid shear of 0.7 dye/cm2 for 18 hours. Cells were then loaded with Fura-2 for intracellular calcium measurement. Unlike wild-type cells in which CaV1.2 knockdown had no effect on intracellular calcium level, it significantly decreased the already lower calcium levels in Pkd cells. N=6 for each group. # indicates significant difference from wild-type; * indicates significant difference between scramble and CaV1.2 shRNA groups.
CaM kinase II activity was regulated by CaV1.2
Calcium ions serve as a very important second messenger in many signaling pathways. Therefore, we proposed that altered cytosolic calcium homeostasis could lead to changes in a signaling cascade. Because Akt has been shown to be abnormal in tissue samples isolated from PKD patients [21,22,29], we first examined phosphorylated Akt levels (Figure 4a). CaV1.2 knockdown did not change the phosphorylation level of Akt in wild-type cells, whereas it significantly reduced Akt phosphorylation in Pkd cells (Figure 4b). Phosphorylation ERK was consequently increased in CaV1.2 knockdown Pkd cells, consistent with the fact that inactivated Akt could not inhibit ERK phosphorylation [30]. Because calcium is an important regulator for CaM kinase II, which also phosphorylates and activates Akt [31], we treated wild-type cells with W7 or KN-93 (CaM kinase II inhibitors). As predicted, Akt phosphorylation was decreased in wild-type cells treated with the inhibitors. We thus propose that CaV1.2 played an important role in Akt and Erk activities through cytosolic calcium and CaM kinase II.
Figure 4. CaV1.2 knockdown altered Akt and Erk phosphorylation in Pkd1−/− and Pkd2−/− cells.


a. Immunoblotting was performed to detect Akt and Erk phosphorylation in wild-type (WT), Pkd1−/− and Pkd2−/− cells transfected with either scramble or CaV1.2 shRNA. Representative Western blots are shown, and W7 or KN-93 was used to block CaM Kinase II function. b. Decreased Akt phosphorylation was detected in Pkd cells with CaV1.2 knockdown, while Erk phosphorylation was elevated in these cells. Phosphorylations of Akt and Erk were not affected in wild-type cells with CaV1.2 silencing. N=3 for each blot; # indicates significant difference from the wild-type control; * indicates significant difference between scramble and CaV1.2 shRNA groups.
Effects of Cav1.2 knockdown on cell proliferation and polyploidy
PKD is characterized by abnormal cell proliferation and polyploidy [21,22]. Therefore, we explored if CaV1.2 knockdown affected these two factors. Our studies reinforced previous results that both Pkd1 and Pkd2 were involved in abnormal cell proliferation and polyploidy (Figure 5a). Importantly, our studies further indicated that although CaV1.2 knockdown (with shRNA) or CaV1.2 blockage (with verapamil) had no effects on wild-type cells, it significantly accelerated cell division and polyploidy formation in both Pkd1 and Pkd2 cells. To further confirm our flow cytometry analysis, we randomly selected 30 individual cells with large nuclei from each group to directly count the number of chromosomes (Figure 5b). Chromosome numbers remained relatively normal in wild-type cells without or with the CaV1.2 knockdown or CaV1.2 blockage. Although Pkd cells were polyploidy, CaV1.2 knockdown induced more polyploidy (≥4N). Of note, many Pkd cells with CaV1.2 knockdown had three times the normal number of chromosomes (≥6N).
Figure 5. CaV1.2 knockdown accelerated cell proliferation and polyploidy in Pkd1−/− and Pkd2−/− cells.
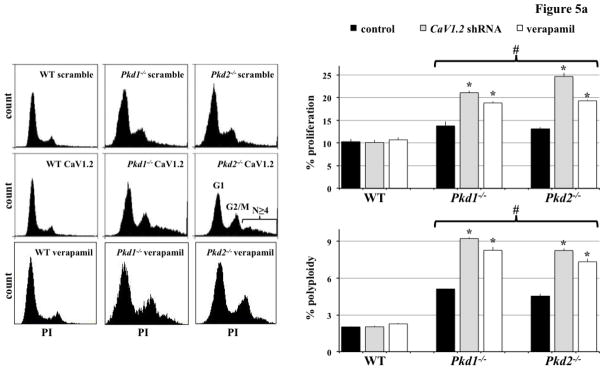

a. Flow cytometry analysis was carried out using propidium iodide (PI) to measure DNA content. Cell proliferation and polyploidy formation were more pronounced in Pkd cells with CaV1.2 knockdown (to block expression) or verapamil treatment (to block function). N=5 for each group; # indicates significant difference from the wild-type (WT); * indicates significant difference between scramble and CaV1.2 shRNA (or verapamil treated) groups. b. Polyploidy was further studied by directly counting the chromosome number from a single cell. A normal number of chromosomes (40) were observed in wild-type (WT) but not Pkd cells. Severity in polyploidy as depicted by numbers of chromosomes was more evident in Pkd cells with CaV1.2 knockdown (KD) or verapamil treatment (VA) compared to scramble control shRNA (C). N=30 cells for each group.
CaV1.2 modulated cystic kidney formation in zebrafish and Pkd1 mouse model
We have recently shown that abnormal cell division could induce polyploidy, which could further promote cystic kidney formation in zebrafish and mice [21,22]. To investigate the pathophysiological role of CaV1.2 in cyst formation, we utilized morpholino knockdown approach in zebrafish (Figure 6a). Supporting our hypothesis of the role of CaV1.2 in cyst formation, we consistently observed dilatated pronephric ducts in cav1.2 morpholino-knockdown fish. Unlike in fish, however, knockdown of CaV1.2 in the wild-type mice did not produce a cystic kidney (Figure 6b). In Pkd1+/− mice (Mx1Cre:Pkd1wt/flox ), we surprisingly observed very severe cystic kidneys in mice as young as two months old. Kidney to body weight ratio was significantly increased in Pkd1+/− mice with CaV1.2 knockdown (Figure 6c).
Figure 6. CaV1.2 modulates cystic kidney formation in zebrafish and Pkd1 heterozygous mice.


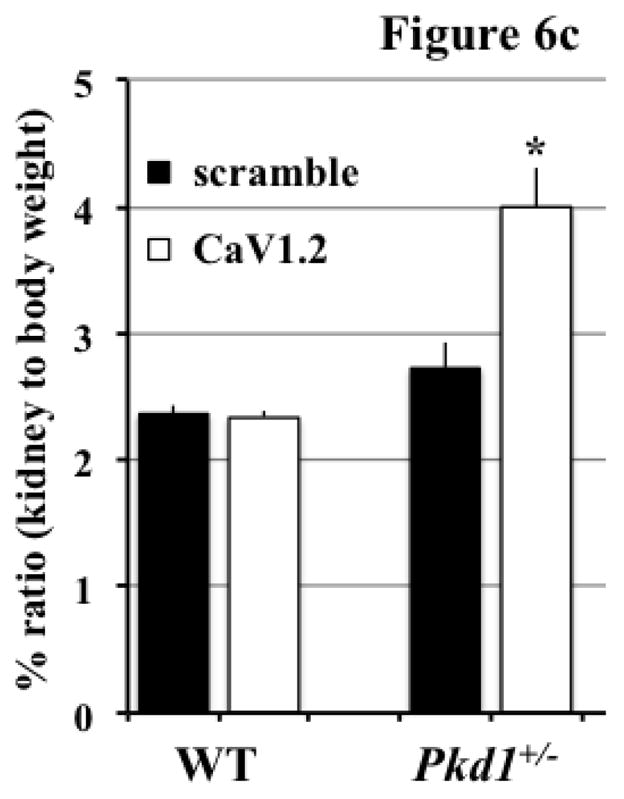
a. We used morpholino knockdown strategy to study the effect of CaV1.2 in zebrafish. Compared to scramble morpholino, cav1.2 morpholino resulted in pronephric duct dilations. Total count of the fish without (non-cystic) and with (cystic) pronephric duct dilation is tabulated in the bar graph. Arrows indicate pronephric ducts. b. Lentiviral infection carrying scramble or CaV1.2 shRNA was used in the wild-type (WT) or Pkd1 heterozygous mice. Cystic kidney phenotype could be easily observed from the isolated kidney. Further analysis indicated cystic-like phenotypes in Pkd1 mice with shRNA knockdown. A total observation from individual animals (n=5–12) without or with cystic phenotype is summarized in the bar graph. c. Kidney to body weight ratio was significantly increased in Pkd1 mice with shRNA knockdown.
Discussion
It has been well accepted that calcium ions play a significant role as a second messenger in translating extracellular stimulation into intracellular signaling [32]. The renal epithelium, one of the main tissues for sensing cues from the extracellular environment, is also dependent on calcium signaling to modulate cellular events such as mitosis, proliferation, fluid and ion transportation, cell volume regulation and many others. Although many calcium channels have been studied in the renal epithelium, not much effort was given to study voltage-dependent calcium channels in the kidney. This was probably due to the belief that the renal epithelium is not an excitable cell, unlike neuron, myocyte or muscle cell. Thus, any changes in epithelial membrane potential would be considered insignificant for voltage-operated channels.
Interestingly, independent research groups have identified voltage-dependent calcium channels, including L-type calcium channel (CaV1.2), in renal epithelial cells [28,33,34]. Based on their electrophysiological and pharmacological properties, voltage-dependent calcium channels are categorized into four groups (L, N, T, and P). The L-type Ca2+ channels are composed of multiple subunits (α1, α2, β, γ, and δ). Some of these subunits express ubiquitously within the kidney, including proximal tubules, medullary collecting ducts, cortical thick ascending limbs, distal convoluted tubules, and cortical collecting ducts [28]. Given the abundance of CaV1.2 within renal epithelial cells, it is intriguing to understand its functionality in the kidney.
In the present study, we reported for the first time that localization of CaV1.2 to primary cilia was disrupted in Pkd cell lines. This indicates that polycystins may regulate CaV1.2 ciliary localization. Furthermore, we observed a significantly higher expression level of CaV1.2 in Pkd cells. Because polycystin-2 calcium channel (PC2) was also malfunctioned in Pkd cells, we speculated that CaV1.2 could function in harmony with PC2. We thus hypothesized that PC2 deficiency would lead to compensatory up-regulation of CaV1.2. In accordance with this view, the loss of CaV1.2 did not have much effect on the intracellular calcium level in normal wild-type kidney cells. However, when CaV1.2 expression was suppressed in Pkd cells, a significant decrease in intracellular calcium level was observed. Because CaV1.2 knockdown did not induce cystic kidney formation in wild-type mice, we would speculate that effects of Pkd1 and Pkd2 on the CaV1.2 expression level would be less critical in the context of cyst formation. Nonetheless, we could not rule out the possibility of CaV1.2 overexpression in pathogenesis of renal diseases.
Considering that the CaV1.2 expression level was significantly higher in Pkd cells than in wild-type cells, it is logical to speculate that CaV1.2 would function as a secondary calcium-permeable channel to the polycystins complex. In the presence of polycystins, the expression of CaV1.2 was low, and so was its activity. When polycystins were not functioning, up-regulation of CaV1.2 expression might be necessary to compensate for the loss of the PC2 calcium channel. Depletion of CaV1.2 in Pkd cells could therefore shut down this compensatory mechanism and consequently reduce the already lower intracellular calcium concentration. Consistent with previous studies [3,16], such a discrepancy in cytosolic calcium levels leads to an increased Erk activity in PKD. Our studies also showed that the increased Erk activity could also be reproduced by inhibition of CaM kinase activity in wild-type cells. In wild-type cells, inhibiting CaM kinase II resulted in a low phosphorylated Akt level and consequently a higher Erk phosphorylation. We therefore hypothesize that CaM kinase II plays a central role in the association between intracellular calcium and downstream effectors, such as Akt and Erk (Figure 7).
Figure 7. Working model of renal CaV1.2.
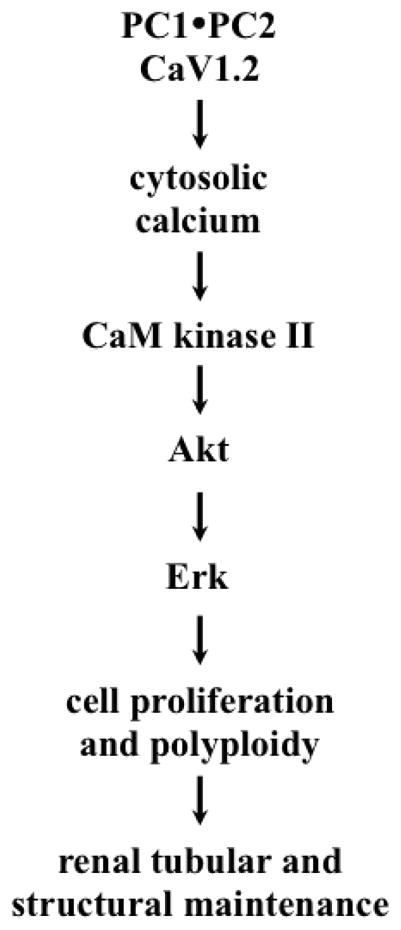
A schematic diagram depicts a hypothetical model of CaV1.2 in cystic kidney phenotypes. The functional role of CaV1.2 can be modulated by polycystins to regulate cytosolic calcium level. The calcium-signaling pathway involves CaM kinase II, Akt and Erk, and it also plays important roles in cellular proliferation and cell polyploidy. This in turn regulates structural maintenance of renal tubules.
On the cellular level, we observed that the length of primary cilia was longer in Pkd cells compared to control wild-type cells. We have recently shown that longer cilia tend to increase the sensitivity of renal epithelial cells to fluid-shear stress [26,27]. Thus, longer cilia in Pkd cells might be caused by a compensatory response to further extend the cilia length so the cells would be more sensitive to extracellular stimulation. Equally interesting is that reducing CaV1.2 expression in Pkd cells also resulted in shorter cilia. This might be a result of a disrupted ciliary assembly that requires intracellular calcium. It is also possible that CaV1.2 could interact with intraflagellar molecules to facilitate cilia length maintenance.
Our studies also indicated that CaV1.2 modulated cell proliferation and polyploidy formation in Pkd cells. By contrast, it did not have any effect on wild-type cells. The differential effects of CaV1.2 between wild-type and Pkd cells would very likely result from insufficient intracellular calcium in Pkd cells. This in turn leads to abnormal CaM kinase II, Akt and Erk pathways. Our in vivo studies confirmed the roles of CaV1.2 in kidney cyst formation. Renal cysts were consistently formed in wild-type zebrafish with cav1.2 morpholino knockdown, unlike in wild-type mice with CaV1.2 shRNA knockdown. This is probably due to less redundancy in genetic makeup for zebrafish compared to mice. Regardless, our studies offer a unique correlation between the expression of CaV1.2 and cystic kidneys. Taken together, we propose that CaV1.2 may function as a genetic modifier for Pkd, the loss of which aggravates the cystic kidney phenotype.
Highlights.
CaV1.2 inhibition causes abnormal cellular events in Pkd1 or Pkd2 deficient cells.
Inhibition of Cav1.2 leads to cystic kidney phenotype in wild-type zebrafish.
Inhibition of Cav1.2 leads to cystic kidney phenotype in heterozygous PKD mouse.
Acknowledgments
This work was funded by NIH DK080640 to SMN and DK51050 to JZ. The completion of this work by Xingjian Jin and Brian S. Muntean partially fulfilled the requirements for their respective graduate programs in medicine and pharmaceutical sciences. Authors also thank Maki Takahashi for technical support and Charisse Montgomery for editing assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nagao S, Morita M, Kugita M, Yoshihara D, Yamaguchi T, et al. Polycystic kidney disease in Han:SPRD Cy rats is associated with elevated expression and mislocalization of SamCystin. Am J Physiol Renal Physiol. 2010;299:F1078–1086. doi: 10.1152/ajprenal.00504.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, et al. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci U S A. 2007;104:4389–4394. doi: 10.1073/pnas.0700499104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17:178–187. doi: 10.1681/ASN.2005060645. [DOI] [PubMed] [Google Scholar]
- 4.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 5.Jin X, Mohieldin AM, Muntean BS, Green JA, Shah JV, et al. Cilioplasm is a cellular compartment for calcium signaling in response to mechanical and chemical stimuli. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-013-1483-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, et al. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol. 2006;17:1015–1025. doi: 10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- 7.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 8.Sweeney WE, Jr, von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19:1331–1341. doi: 10.1681/ASN.2007060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, et al. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004;66:964–973. doi: 10.1111/j.1523-1755.2004.00843.x. [DOI] [PubMed] [Google Scholar]
- 10.Kelleher CL, McFann KK, Johnson AM, Schrier RW. Characteristics of hypertension in young adults with autosomal dominant polycystic kidney disease compared with the general U.S. population. Am J Hypertens. 2004;17:1029–1034. doi: 10.1016/j.amjhyper.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 11.Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med. 1990;323:1091–1096. doi: 10.1056/NEJM199010183231602. [DOI] [PubMed] [Google Scholar]
- 12.AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104:860–869. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, et al. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitobe M, Yoshida T, Sugiura H, Shiohira S, Shimada K, et al. Clinical effects of calcium channel blockers and renin-angiotensin-aldosterone system inhibitors on changes in the estimated glomerular filtration rate in patients with polycystic kidney disease. Clin Exp Nephrol. 2010;14:573–577. doi: 10.1007/s10157-010-0329-5. [DOI] [PubMed] [Google Scholar]
- 15.Kanno Y, Suzuki H, Okada H, Takenaka T, Saruta T. Calcium channel blockers versus ACE inhibitors as antihypertensives in polycystic kidney disease. QJM. 1996;89:65–70. doi: 10.1093/oxfordjournals.qjmed.a030139. [DOI] [PubMed] [Google Scholar]
- 16.Nagao S, Nishii K, Yoshihara D, Kurahashi H, Nagaoka K, et al. Calcium channel inhibition accelerates polycystic kidney disease progression in the Cy/+ rat. Kidney Int. 2008;73:269–277. doi: 10.1038/sj.ki.5002629. [DOI] [PubMed] [Google Scholar]
- 17.Nauli SM, Jin X, AbouAlaiwi WA, El-Jouni W, Su X, et al. Non-motile primary cilia as fluid shear stress mechanosensors. Methods Enzymol. 2013;525:1–20. doi: 10.1016/B978-0-12-397944-5.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding B, Kilpatrick DL. Lentiviral vector production, titration, and transduction of primary neurons. Methods Mol Biol. 2013;1018:119–131. doi: 10.1007/978-1-62703-444-9_12. [DOI] [PubMed] [Google Scholar]
- 19.Muntean BS, Jin X, Williams FE, Nauli SM. Primary cilium regulates CaV1.2 expression through Wnt signaling. J Cell Physiol. 2014 doi: 10.1002/jcp.24642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merten OW, Charrier S, Laroudie N, Fauchille S, Dugue C, et al. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther. 2011;22:343–356. doi: 10.1089/hum.2010.060. [DOI] [PubMed] [Google Scholar]
- 21.Aboualaiwi WA, Muntean BS, Ratnam S, Joe B, Liu L, et al. Survivin-Induced Abnormal Ploidy Contributes to Cystic Kidney and Aneurysm Formation. Circulation. 2014;129:660–672. doi: 10.1161/CIRCULATIONAHA.113.005746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.AbouAlaiwi WA, Ratnam S, Booth RL, Shah JV, Nauli SM. Endothelial cells from humans and mice with polycystic kidney disease are characterized by polyploidy and chromosome segregation defects through survivin down-regulation. Hum Mol Genet. 2011;20:354–367. doi: 10.1093/hmg/ddq470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.AbouAlaiwi WA, Rodriguez I, Nauli SM. Spectral karyotyping to study chromosome abnormalities in humans and mice with polycystic kidney disease. J Vis Exp. 2012:3887. doi: 10.3791/3887. pii. doi: 3810.3791/3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muntean BS, Horvat CM, Behler JH, Aboualaiwi WA, Nauli AM, et al. A Comparative Study of Embedded and Anesthetized Zebrafish in vivo on Myocardiac Calcium Oscillation and Heart Muscle Contraction. Front Pharmacol. 2010;1:139. doi: 10.3389/fphar.2010.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takakura A, Contrino L, Beck AW, Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19:2351–2363. doi: 10.1681/ASN.2007101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdul-Majeed S, Nauli SM. Dopamine receptor type 5 in the primary cilia has dual chemo- and mechano-sensory roles. Hypertension. 2011;58:325–331. doi: 10.1161/HYPERTENSIONAHA.111.172080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdul-Majeed S, Moloney BC, Nauli SM. Mechanisms regulating cilia growth and cilia function in endothelial cells. Cell Mol Life Sci. 2012;69:165–173. doi: 10.1007/s00018-011-0744-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao PL, Wang XT, Zhang XM, Cebotaru V, Cebotaru L, et al. Tubular and cellular localization of the cardiac L-type calcium channel in rat kidney. Kidney Int. 2002;61:1393–1406. doi: 10.1046/j.1523-1755.2002.00267.x. [DOI] [PubMed] [Google Scholar]
- 29.Fischer DC, Jacoby U, Pape L, Ward CJ, Kuwertz-Broeking E, et al. Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD) Nephrol Dial Transplant. 2009;24:1819–1827. doi: 10.1093/ndt/gfn744. [DOI] [PubMed] [Google Scholar]
- 30.Chen JY, Lin JR, Cimprich KA, Meyer T. A two-dimensional ERK-AKT signaling code for an NGF-triggered cell-fate decision. Mol Cell. 2012;45:196–209. doi: 10.1016/j.molcel.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong B, Valencia CA, Liu R. Ca(2+)/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J Biol Chem. 2007;282:25131–25140. doi: 10.1074/jbc.M702123200. [DOI] [PubMed] [Google Scholar]
- 32.Jones TJ, Nauli SM. Mechanosensory calcium signaling. Adv Exp Med Biol. 2012;740:1001–1015. doi: 10.1007/978-94-007-2888-2_46. [DOI] [PubMed] [Google Scholar]
- 33.Andreasen D, Jensen BL, Hansen PB, Kwon TH, Nielsen S, et al. The alpha(1G)-subunit of a voltage-dependent Ca(2+) channel is localized in rat distal nephron and collecting duct. Am J Physiol Renal Physiol. 2000;279:F997–1005. doi: 10.1152/ajprenal.2000.279.6.F997. [DOI] [PubMed] [Google Scholar]
- 34.Monteil A, Chemin J, Bourinet E, Mennessier G, Lory P, et al. Molecular and functional properties of the human alpha(1G) subunit that forms T-type calcium channels. J Biol Chem. 2000;275:6090–6100. doi: 10.1074/jbc.275.9.6090. [DOI] [PubMed] [Google Scholar]