

Abstract
Polo-like kinase 4 (Plk4) is a key regulator of centriole duplication, an event critical for the maintenance of genomic integrity. Here we showed that Plk4 relocalizes from the inner Cep192 ring to the outer Cep152 ring as newly recruited Cep152 assembles around the Cep192-encircled daughter centriole. Crystal structure analyses revealed that Cep192 - and Cep152-derived peptides bind the cryptic polo box (CPB) of Plk4 in opposite orientations and in a mutually exclusive manner. The Cep152-peptide bound to the CPB markedly better than the Cep192-peptide and effectively snatched the CPB away from a preformed CPB–Cep192-peptide complex. A cancer-associated Cep152 mutation impairing the Plk4 interaction induced defects in procentriole assembly and chromosome segregation. Thus, Plk4 is intricately regulated in time and space through ordered interactions with two distinct scaffolds, Cep192 and Cep152, and a failure in this process may lead to human cancer.
As the principal microtubule-organizing center in mammalian cells, the centrosome plays a central role inspindle formation and chromosome segregation during mitosis. Centrosomes contain two orthogonally arranged centrioles, which duplicate precisely once per cell cycle 1–3. A failure in this process can result in abnormal centrosome numbers, improper spindle formation and chromosome missegregation that ultimately leads to genomic instability and human disorders 4–6. Therefore, accurate control of centriole duplication is fundamentally required for normal cell division and proliferation.
Centriole duplication begins by assembling a procentriole in G1/S phase. Proper recruitment of a member of the polo kinase subfamily, Plk4, to centrosomes appears to be a key event that triggers the entire duplication process 7–9. Interestingly, while nematodes and flies utilize distinct centrosomal scaffolds, Spd-2 and Asterless (Asl), respectively, to recruit their Plk4 orthologs 10–12, humans require both Cep192 (Spd-2 ortholog) and Cep152 (Asl ortholog) for this event 13,14. Whether Cep192 and Cep152 function cooperatively 14 or hierarchically 13 to recruit Plk4 to centrosomes remains controversial.
In this study, we set out to investigate how the two dissimilar scaffold proteins, Cep192 and Cep152, regulate Plk4 localization at subcentrosomal structures. We showed that they interact with Plk4 in a temporally and spatially regulated manner, thus ensuring timely formation of distinct Plk4 complexes at different subcentrosomal structures. Our data suggest that ordered binding of these two scaffolds to Plk4 is critical to promote Plk4-mediated centriole biogenesis and to maintain genomic integrity.
RESULTS
Plk4 ring enlarges as Cep152 localizes to centrioles
To understand the functional relationships between Plk4 and the two Plk4-binding scaffold proteins, Cep192 and Cep152, at the initial stage of centriole biogenesis, we first examined the subcentriolar localization patterns of these proteins in U2OS cells by performing three-dimensional structured illumination microscopy (3D-SIM) analysis (Fig. 1 and Supplementary Fig. 1).
Figure 1.

Two distinct sizes of Plk4 ring structures in the absence or presence of the Cep152 ring around Cep192-decorated centrioles. (a) 3D -SIM images showing asynchronously growing U2OS cells co-immunostained with anti-Plk4 (red), anti-Sas6 (blue; pseudo-colored in gray), Alexa 647 (magenta)-conjugated anti-Cep192 N-terminal (N), and Alexa 488 (green)-conjugated anti-Cep152 middle region (M) antibodies. Arrows in the 1st panel indicate the outer diameters of two Plk4 rings—one from a daughter (D) and the other from a mother (M) centriole before and after Cep152 recruitment, respectively. A bracket on the 2ndpanel indicates Plk4 signals colocalized with a nascent Cep152 toroid assembling at a daughter centriole. Arrowheads on the 3rd and 4th panels indicate dot -like Plk4 (red) signals colocalized with Sas6 (gray) on Cep152 toroids. Scale bars, 0.5 μm. (b) Quantification of the outer diameters of Cep192, Cep152, and Plk4 ring signals for the samples in Figure 1a. G1 centrioles prior to Cep152 recruitment (n=25) or after Cep152 recruitment (n=69), or S centrioles without discernable Plk4 ring signals (n=41) were measured. Error bars, s.d. (c) An immunoblot showing U2OS cells silenced for control Luciferase (GL siRNA) or CEP152 (CEP152 siRNA). Asterisk, cross-reacting protein. (d,e) Immunostaining (d) of the cells in c and subsequent quantification (e) from three independent experiments (n=105 for GL siRNA, n=116 for CEP152 siRNA). Arrows in d, the outer diameters of Cep192 and Plk4 rings; scale bars in d, 0.5 μm; error bars in e, s.d. An uncropped blot image for c is shown in Supplementary Figure 8a.
Early in G1, Plk4 assumed a ring-like structure surrounding the proximal part of cylindrical centrioles. Interestingly, 3D-SIM images showed that the centrioles decorated with Cep192 but not Cep152 (i.e., daughter centrioles prior to Cep152 recruitment) exhibited a Plk4 ring with an outer diameter of ~443 ± 34 nm, while the centrioles surrounded with both Cep192 and Cep152 (i.e., mother and late-stage daughter centrioles) displayed a significantly larger Plk4 ring (590 ± 48 nm outer diameter) (Fig. 1a, 1st and 2nd panels; Fig. 1band Supplementary Fig. 1b). The sizes of these rings were in good agreement with the findings of Lawo et al 15. Consistent with a report that the N-terminus of Cep152 is outwardly oriented 16, Plk4, which binds to the N-terminal region of Cep15213,14, was detected at the outskirts of the Cep152 middle region (M), where the antibody epitope is located (Fig. 1a,b and Supplementary Fig. 1b). Thus, as Cep152 is recruited around the Cep192-encircled daughter centrioles, Plk4 reassembles into a bigger ring at the boundary of the newly forming Cep152 toroid. Consistent with this view, the morphology of the Plk4 ring structure closely mirrored that of Cep152 during the early stages of Cep152 toroid formation (Fig. 1a, 2nd panel; bracket). Furthermore, depletion of Cep152 allowed Plk4 to reassociate with the inner Cep192 ring (Fig. 1c–e). In lateG1/S, Plk4 fluorescence became a dot-like signal and colocalized with Sas6 (Fig. 1a), a component whose recruitment is critical for procentriole assembly 17,18.
Interaction of human CPB with Cep192 and Cep152
Since Plk4 colocalized with Cep192 and Cep152 at subcentriolar structures (Fig. 1), we examined whether it interacts with these proteins in vivo. We observed that immunoprecipitation of Cep192 or Cep152 coprecipitated Plk4 at the endogenous level (Fig. 2a). However, Cep192 and Cep152 did not interact with each other under the same conditions (Fig. 2a). These findings suggest that Plk4 forms two distinct complexes with Cep192 or Cep152.
Figure 2.

Biochemical and structural analysis of Plk4 CPB in complex with Cep192-58mer or Cep152-60mer. (a) Interaction of Plk4 with Cep192 and Cep152 at endogenous levels. HeLa cell lysates were subjected to immunoprecipitation (IP) analysis with the indicated antibodies cross-linked to agarose beads. GL siRNA, Luciferase siRNA; IgG, control rabbit IgG. (b) Immunoprecipitation (IP) analysis was performed with 293T cells cotransfected with the indicated constructs. White asterisks, remaining FLAG-Plk4 CPB signals from the second panel immunoblot; Black asterisks, non-specific bands from IgG light chain. (c) Reverse alignment of Cep152-60mer and Cep192-58mer. Identical (vertical line) and conserved (colon) residues are indicated. (d) MBP pull-downs were performed using purified proteins. CBB, Coomassie-stained gel; asterisks, contaminated proteins; numbers, the relative amount of Cep192-58mer or Cep152-60mer bound to a CPB dimer (2.0). (e) Immunoprecipitation (IP) analysis with 293T cells cotransfected with the indicated constructs. To achieve stable overexpression, a kinase-inactive Plk4 K41M mutant was used. CBB, Coomassie-stained gel; arrows, coimmunoprecipitated FLAG-Plk4; asterisk, cross-reacting protein; H.C. and L.C., IgG heavy and light chains. (f,g) Overall structures of the CPB–Cep192-58mer and the CPB–Cep152-60mer complexes. Each subunit (blue or orange) of a homodimeric CPB consists of PB1 and PB2 folds. N, N-terminus; C, C-terminus; D-rich, D-rich motif. Uncropped blot images for a and b are shown in Supplementary Figure 8b,c.
Understanding the molecular basis of how Plk4 interacts with Cep192 and Cep152 may shed light on the mechanism of how the Plk4 ring reassembles concurrently with the recruitment of Cep152 to Cep192-associated centrioles. Cryptic polo box (CPB; composed of PB1 and PB2 motifs) 19 is centrally required for targeting Plk4 to centrosomes 9,20. However, how CPB interacts with its binding targets is unknown. Notably, unlike Plk1–3, which bind to phosphorylated epitopes 21, Plk4 formed a tight homomeric complex that binds to its targets in a phospho-independent manner (Supplementary Fig. 2a –c).
Next, we sought to structurally characterize how Plk4 CPB interacts with Cep192 and Cep15213,14,22,23. We found that a minimal CPB fragment containing residues 580–808 (referred to hereafter as CPB) was sufficient for homomerization (Supplementary Fig. 2d; also, see details below) and that it exhibited the full capacity to interact with the two previously defined motifs, Cep192 (201–280) and Cep152 (1–217) 13 (Supplementary Fig. 2e). However, in disagreement with the Drosophila CPB model 20, the PB1 of CPB did not interact with another PB1, while the PB2 interacted with another PB2 efficiently (Fig. 2b).
Our initial attempt to co-crystallize CPB with Cep192 (201–280) or Cep152 (1–217) failed due to the instability of bacterially expressed proteins. Systematic deletion analysis showed that the Cep152 (1–60) fragment (hereafter, Cep152-60mer) bound to CPB as efficiently as Cep152 (1–217) and coprecipitated the full-length Plk4 at a degree somewhat comparable to that of the full-length Cep152 (Supplementary Fig. 2f,g). The Cep152-60mer exhibited no apparent homology with Cep192 (201–280). However, upon reverse alignment of these two sequences, we were able to identify a Cep192 (201 –258) fragment (hereafter, Cep192-58mer) that contains analogously positioned, predicted α-helices and D-rich motifs (Fig. 2c), suggesting that they may bind to CPB in opposite orientations. Deletion of the 58-mer region completely abolished the Cep192 -Plk4 interaction, and the Cep192-58mer precipitated the full-length Plk4 efficiently (Supplementary Fig. 2h,i). These observations suggest that the Cep192-58mer is necessary and sufficient to bind to Plk4.
In vitro binding analyses showed that maltose-binding protein (MBP)-Cep192-58mer bound to a CPB dimer with ~2:2 stoichiometry, whereas MBP-Cep152-60mer bound to the latter with ~1:2 stoichiometry (Fig. 2d). In line with these fin dings, Cep192 was less effective than Cep152 in coprecipitating Plk4 from transfected 293T cells (Fig. 2e) (Assessment of the binding stoichiometry for this result was not possible because of the different molecular sizes of Cep192 and Cep152).
Structure of apo-CPB, CPB–Cep192-58mer and CPB –Cep152-60mer
Next, we determined the crystal structures of human apo-CPB and CPB complexed with Cep192-58mer or Cep152-60mer by molecular replacement (see the Methods) (Table 1). Like the Drosophila CPB 20, the structure of human CPB was composed of two PB domains, PB1 (580–700) and PB2 (701–808), each containing an α-helix and a six-or seven-stranded antiparallel β-sheet (Supplementary Fig. 3a). The PB1 and PB2 domains were connected by a two-residue linker (S700 and P701). Distinct from the Drosophila CPB proposed to form a “donut-shaped” homodimer with both PB1-PB1 and PB2-PB2 contacts, all human CPB structures presented here contained an “X-shaped” homodimer with a symmetrical PB2-PB2 interface, although their crystal lattices differed from one another. This dimeric interaction was verified by a coimmunoprecipitation assay (Fig. 2b). Furthermore, the PB2-PB2 interface was also observed in the Drosophila CPB structure (PDB: 4G7N), but a different symmetry mate was chosen to form the donut-shaped CPB dimer 20. It was impossible to fit the human CPB structure into the Drosophila CPB model 20 (Supplementary Fig. 3b).
Table 1.
Data collection and refinement statistics
| Apo-CPB | CPB–Cep192-58mer | CPB–Cep152-60mer | |
|---|---|---|---|
| Data collection | |||
| Space group | P6122 | P6122 | P21212 |
| Cell dimensions | |||
| a, b, c (Å) | 128.1, 128.1, 168.4 | 67.3, 67.3, 287.6 | 126.6, 63.9, 79.5 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 50.0–2.60 (2.64–2.60) | 30.0–2.85 (2.90–2.85) | 50.0–2.76 (2.80–2.76) |
| Rmerge | 6.8 (27.6) | 11.5 (57.4) | 6.8 (30.3) |
| I/σI | 48.0 (5.7) | 10.9 (3.1) | 11.3 (2.6) |
| Completeness (%) | 98.7 (96.7) | 99.7 (99.0) | 92.0 (71.4) |
| Redundancy | 21.3 (8.5) | 9.5 (8.2) | 3.8 (2.0) |
| Refinement | |||
| Resolution (Å) | 36.1–2.60 | 29.1–2.85 | 37.3–2.76 |
| No. reflections | 25,386 | 9,773 | 15,858 |
| Rwork/Rfree | 0.202/0.249 | 0.235/0.260 | 0.258 / 0.288 |
| No. atoms | |||
| Protein | 3,622 | 2,033 | 3,904 |
| Ligand/ion | 8 | 0 | 0 |
| Water | 113 | 45 | 16 |
| B factors | |||
| Protein | 58.3 | 45.5 | 50.0 |
| Ligand/ion | 74.8 | – | – |
| Water | 40.3 | 38.2 | 23.4 |
| r.m.s. deviations | |||
| Bond lengths (Å) | 0.009 | 0.015 | 0.004 |
| Bond angles (°) | 1.29 | 1.47 | 0.92 |
Values in parentheses are for highest-resolution shell. All diffraction data were obtained from a single crystal.
The X-shaped CPB dimer was generated by a non-crystallographic dyad axis and fastened by four hydrogen bonds between two antiparallel β13-strands, one from each subunit, resulting in the formation of a continuous 14-stranded antiparallel β-sheet. The dimer interface was strengthened by multiple hydrophobic interactions between the two β13 strands and also the two α2 helices. PB1 domains formed two wings that do not directly interact with each other (Supplementary Fig. 3a).
In the complexes, both Cep192-58mer and Cep152-60mer bound to the concave surface of CPB along the α1 of PB1 and over the α1–β7 junction (Fig. 2f,g), inducing slight alterations in the CPB structure (Supplementary Fig. 3c). Remarkably, the 58mer and 60mer peptides were oriented in opposite directions, which reflect the reverse sequence alignment of the two peptides (Fig. 2c). In addition, one Cep192-58mer bound to each subunit of a CPB dimer, generating a symmetrical 2:2 heterotetrameric complex (Fig. 2f), whereas one Cep152-60mer bound to each CPB dimer, forming a 1:2 heterotrimeric complex (Fig. 2g; also, see details below). Unlike Plk1 PBD, which interacts with a phosphorylated peptide through the β1 of PB1 and also partly the β8 and β9 of PB224, Plk4 CPB interacted with its target primarily through the α1 and β1 of PB1 and the β7 of PB2 (Supplementary Fig. 3d), requiring no phosphorylated epitope.
Structural of CPB–Cep192-58mer versus CPB –Cep152-60mer
Cep192-58mer and Cep152-60mer formed three major contacts with CPB. Although the centrally located α-helical sequences from the 58mer and the 60mer exhibit no detectable sequence homology (Fig. 2c), they bound to an overlapped region of CPB by engaging in multiple hydrophobic and hydrogen bond interactions with several common residues along the α1 and β1 of PB1 (Fig. 3a,b, left; see details in Supplementary Fig. 3e). The 58mer α-helix was skewed at an oblique angle to the axis of the CPB α1, whereas the 60mer α-helix was antiparallel to the CPB α1, forming tightly packed hydrophobic interactions between the two α-helices. The coiled-coil interface was reinforced by salt bridges between the D20 and D23 of the 60mer and the K711 (rectangled) of the CPB (Fig. 3b, left).
Figure 3.

The binding modes of Cep192-58mer and Cep152-60mer to Plk4 CPB and mutational analyses of residues critical for the interaction. (a,b) Electrostatic surface representation of the CPB–58mer (a) and the CPB –60mer (b) complexes. The K711 and K/R crater residues of CPB and the E21 of Cep152-60mer are highlighted in rectangles. See Supplementary Figure 3e – h for details. (c–e) Immunoprecipitation (IP) analyses of 293T cells cotransfected with the indicated constructs. –, control vector; numbers, relative signal intensities. (f, g) Characterization of a cancer-associated CEP152 E21K mutation. U2OS cells stably expressing either control vector or siRNA-resistant CEP152-silor CEP152-silE21K mutant were silenced for either control Luciferase (GL siRNA) or CEP152 (CEP152 siRNA) (see Supplementary Table 2), and then immunostained (see Supplementary Figure 4a–c). The numbers of centrosomal Sas6 signals (from 0 to ≥3) among interphase cells (f) and the percentage of mitotic cells with missegregating chromosomes (chr.) (g) were quantified from three independent experiments (≥200 cells/cell line/experiment). Error bars, s.d. Uncropped blot images for c–e are shown in Supplementary Figure 8c–e.
In addition, the D-rich motif at the N-terminus of Cep192-58mer or the C-terminus of Cep152-60mer interacted with a K/R-enriched crater formed by one arginine and five lysine residues (rectangled) at the tip of the PB1 domain (Fig. 3a,b, right; see details in Supplementary Fig. 3f,g). Among the crater-forming residues, the side chain of R684 shifted in different directions upon binding either Cep192-58mer or Cep152-60mer (Supplementary Fig. 3c). The K685 residue, centrally located in the crater, appeared to play a key role in binding the D-rich motif (Fig. 3a,b, right). Notably, the D-rich motifs exhibited a weak electron density, suggesting that their charge-charge interactions with the K/R crater were diffused cloud-like instead of one-to-one correspondence (Supplementary Fig. 3g). A hydrophobic residue present in the middle of the D-stretch of the 58mer and 60mer (I217 and L42, respectively) fit well in the hydrophobic pocket at the center of the K/R crater (Fig. 3a,b, right, and Supplementary Fig. 3f for details). The D-rich motif of the 58mer efficiently neutralized the charge potential of the K/R crater, while that of the 60mer, which contains fewer acidic residues, negated the charge potential less effectively (Supplementary Fig. 3g).
Finally, a unique acidic sequence at the N-terminus of theCep152 -60mer (from E15 to E21) appeared to efficiently counteract the positive charge potential near the CPB dimeric interface (i.e., PB2 interface basic patch). This charge–charge attraction again occurred through group interactions with poorly defined electron densities for individual side chains. In the CPB–Cep192-58mer complex, the neutralizing effect was achieved by the negative helical dipoles of two 58mer peptides coming together near the CPB dimeric interface (Supplementary Fig. 3h).
Consistent with the structural results above, mutational analyses showed that the R684 in the K/R crater was vital for Cep192-58mer binding but not for Cep152-60mer binding, whereas the K711, located near the dimeric interface, was crucial for Cep152-60mer binding, but much less so for Cep192-58mer binding (Fig. 3c,d). Interestingly, the K685 of PB1 was required for binding to both Cep192-58mer and Cep152-60mer (Fig. 3c,d). This may be due to its dual roles—one in the intramolecular interaction with the E635 of PB1 and the other in the intermolecular electrostatic interaction with the D-rich motif residues of Cep192-58mer and Cep152-60mer (Supplementary Fig. 3f).
Significance of the CEP152 E21K mutation in human cancers
Plk4 haploinsufficiency has been shown to cause mitotic abnormalities, including chromosome missegregation that ultimately leads to spontaneous generation of cancers in mice 25. Proper interaction of Plk4 with Cep192 and Cep152 is likely critical for regulating Plk4 function in centriole duplication and mitotic progression 13,14. A search of human mutation databases found the CEP152 E21K mutation identified in two out of 212 cases of colon and rectum adenocarcinoma [Catalogue of Somatic Mutations in Cancer (http://www.sanger.ac.uk/cosmic)]; however, the same searches did not uncover any cases involving the CEP192 mutation. The Cep152-60mer E21 (rectangled) located near the N-terminus of its α-helix may neutralize the positively charged potential of both its own helical dipole moment and the CPB surface (Fig. 3b, left). We predicted that reversal of charges in the E-to-K mutation would destabilize the formation of α-helix and therefore the association of Cep152 with CPB. Indeed, the E21K mutation greatly impaired the Cep152–Plk4 interaction (Fig. 3e). Consequently, CEP152 RNAi cells expressing a siRNA-insensitive CEP152-silE21K mutant exhibited a severe defect in the recruitment of Sas6 (Fig. 3f and Supplementary Fig. 4a,b), an event critical for procentriole assembly. These cells also exhibited significantly increased levels of misaligned and lagging chromosomes (Fig. 3g and Supplementary Fig. 4c).
Disruption of the CPB–Cep192-58mer complex by Cep152-60mer
Discrete enlargement of Plk4 ring structures on Cep152-recruited centrioles (Fig. 1a) and similar binding modes of Cep192-58mer and Cep152-60mer to CPB (Fig. 2f,g) suggest that Cep192–Plk4 and Cep152–Plk4 complexes may not coexist in vivo. Indeed, superposition of CPB-bound Cep192-58mer and Cep152-60mer (root-mean-square deviation [r.m.s.d.] of 2.2 Å across all Cα atoms of the CPB dimer) showed that Cep152 -60mer not only directly interfered with the binding of Cep192-58mer to the same CPB subunit, but also clashed with the second Cep192-58mer already bound to the other CPB subunit (Fig. 4a), suggesting that binding of Cep192-58mer and Cep152-60mer to CPB is mutually exclusive.
Figure 4.

Mechanistic basis of repositioning Cep192-bound Plk4 with incoming Cep152 at daughter centrioles. (a) Superposition of the structures of Cep192-58mer and Cep152-60mer individually bound to Plk4 CPB. Arrowed bracket indicates the region of steric clash between the 60mer and the 58mer binding to two different subunits of a CPB dimer. N, N-terminus; C, C-terminus. (b) Fluorescence polarization (FP)-based binding assays were performed using Alexa 488 -conjugated (*) MBP or MBP -fused ligand and increasing concentrations of unconjugated Plk4 CPB. (c) FP inhibition assays were performed by providing increasing concentrations of MBP -Cep152-60mer or MBP-Cep192-58mer to preformed MBP-Cep192-58mer*–CPB complex or MBP-Cep152-60mer*–CPB complex, respectively. The data obtained from three independent experiments. Error bars, s.d. (d) A biochemical assay demonstrating the ability of Cep152-60mer to snatch CPB from the Cep192-58mer–CPB complex. Proteins eluted after the addition of increasing concentrations of MBP-Cep152-60mer were analyzed. See detailed procedures in Methods. Slanted arrows, the first fraction containing the Coomassie-stainable amount of co-eluted MBP-Cep152-60mer and CPB; Vertical arrows, the fraction with the highest levels of MBP-Cep152-60mer and CPB; Asterisk, a non-specific protein co-migrating with the 60mer; Numbers, relative amounts of CPB coeluted with the 60mer.
To directly investigate the incompatible nature of these bindings to CPB, we performed a fluorescence polarization (FP) binding assay using Alexa 488-conjugated MBP-Cep192-58mer and MBP-Cep152-60mer. Results showed that the CPB-binding affinity of Cep152-60mer (Kd= 32 ± 4 nM) was ~5.5 -fold higher than that of Cep192-58mer (Kd= 177 ± 23 nM) (Fig. 4b). Consistent with these results, Cep152-60mer efficiently disrupted a preformed complex between Alexa 488-conjugated MBP-Cep192-58mer and CPB (IC50= 64 ± 4 nM). In a reverse experiment, provision of up to an order of magnitude higher concentration of Cep192 -58mer failed to noticeably dissociate Alexa 488-conjugated MBP-Cep152-60mer from CPB (Fig. 4c; see Methods).
In an experiment using an Ni-NTA–immobilized His6-MBP-Cep192-58mer–CPB complex, MBP-Cep152-60mer efficiently snatched the CPB away from the preformed complex with ~1:2 stoichiometry (Fig. 4d; see the CPB/MBP-60mer ratios prior to MBP -60mer saturation). Consistent with this finding, MBP-Cep152-60mer also outcompeted MBPL-Cep192-58mer in binding to CPB in a size-exclusion chromatography (Supplementary Fig. 5a). A similar result was obtained when Cep192 (201–280) and Cep152 (1–217) containing their entire Plk4 -binding motifs 13 were incubated with a Plk4 C-terminal (580–970) fragment (Supplementary Fig. 5b). These in vitro binding results were corroborated by the observation that, in transfected 293T cells, Cep152 was able to efficiently out compete Cep192 in binding to Plk4, but not vice versa (Supplementary Fig. 5c,d). Collectively, these data demonstrate that Cep192 and Cep152 are not compatible in binding to Plk4, and suggest that a free form of Cep152 is able to take Plk4 away from the preformed Cep192–Plk4 complex at daughter centrioles.
DISCUSSION
Characteristics of Plk4 CPB binding
Our results demonstrated that CPB utilizesits PB1 α1 flanking region, K/R crater, and the dimeric interface basic patch to interact with Cep192-58mer and Cep152-60mer, suggesting that the molecular architecture underlying the formation of these complexes is largely similar. However, the opposite orientations of the bound peptides and the differential usages of these binding sites suggest that CPB may function as a versatile binding module that can interact with diverse cellular targets and mediate distinct cellular processes. Moreover, each target’s unique binding mode may serve as the molecular basis for establishing hierarchical interactions between targets functioning in the same pathway.
It should be noted that, unlike the PBDs from Plk1–3, Plk4 CPB did not require a phosphorylated epitope for binding. Moreover, the CPB failed to interact with its C-terminal PB3 (residues 854–970), which appeared to have only a weak capacity to forma homodimeric PB3–PB3 complex 19(Supplementary Fig. 2d). WhetherPlk4 PB3 binds to its own target and how it functions in conjunction with its CPB have yet to be studied.
Molecular basis of unidirectional scaffold switching
We showed that Cep152 recruitment induces Plk4 relocation from the outer edge of the Cep192 ring to that of the Cep152 ring (Fig. 1), suggesting that Plk4 translocates unidirectionally from the Cep192 scaffold to the Cep152 scaffold. In support of this interpretation, Cep152-60mer was able to efficiently snatch the CPB from the Cep192-58mer–CPB complex (Figs. 4d and 5).
Figure 5.
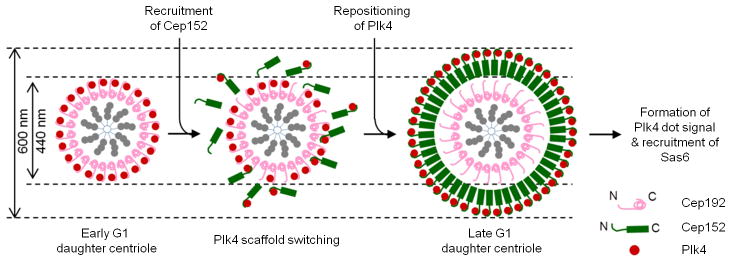
A schematic diagram illustrating the mechanism of how Cep152 snatches Plk4 away from the Cep192–Plk4 complex and repositions Plk4 at the outer boundary of a newly formed Cep152 ring structure. Early G1 daughter centrioles are decorated with a cylindrical Cep192 structure, in which the N-terminus of Cep192 points outward (Supplementary Figure 1a). At this stage, Plk4 forms a smaller (~440 nm outer diameter) ring through the interaction with the N-terminal region of Cep192 (see Supplementary Figure 1). As Cep152 is being recruited to this location in late G1, Cep152 stochastically interacts with the Cep192-bound Plk4 and snatches the Plk4 away from the Cep192 scaffold (scaffold switching). Subsequent assembly of a Cep152 ring around the Cep192 toroid in an N-terminus outward fashion prompts the repositioning of Plk4 to the outer edge of the Cep152 ring, thus resulting in a much larger Plk4 ring structure in diameter (~600 nm). Around the time of procentriole formation, Plk4 assumes a dot-like signal at the future procentriole assembly site and recruits Sas6 to this site to induce centriole biogenesis.
The mechanism underlying this unidirectional cellular process appears to stem from the different binding natures of Cep192-58mer versus Cep152-60mer to Plk4 CPB. Although the physiological binding stoichiometry for endogenous proteins is yet to be determined, Cep192-58mer bound to the CPB with 2:2 stoichiometry, whereas Cep152-60mer bound to the latter with 1:2 stoichiometry (Fig. 2d). This is because the C-termini of two Cep192-58mers engaged in heterotetrameric junctional interactions (Supplementary Fig. 5e), whereas the N-terminal Cα backbone (e.g., C α of E15–D20) of Cep152-60mer that is bound to one of the two subunits extended beyond the PB2-PB2 dimeric junction and interacted with the basic patch residues in the other subunit (Supplementary Fig. 5f). This extended binding would prevent another Cep152-60mer from binding to the second subunit (Supplementary Fig. 5g).
So then, how can the binding of one Cep152-60mer dislodge two Cep192-58mers interacting with both subunits of the CPB dimer? First, in vitro biochemical analysis showed that the 60mer binds to CPB with ~5.5-fold higher affinity than the 58mer. Second, superimposing CPB-bound Cep192-58mer with Cep152-60mer revealed that the extended N-terminus of the 60mer directly interferes with the binding of the C-terminal helix region of the 58mer to the other CPB subunit (Fig. 4a, arrowed bracket). Third, deletion of a portion of the 58mer α-helix extending toward the CPB β7 and β13 eliminated the ability of the 58mer to bind to CPB (Supplementary Fig. 5h), suggesting that the steric clash between the N-terminal region of the 60mer and the C-terminal α-helix of the 58mer is sufficient to dislodge the 58mer from the CPB dimer. Intriguingly, the binding of the 58mer and 60mer to CPB occurs in opposite orientations. This reverse binding mode may help avoid the steric hindrance that could arise if the two bulky Cep192 and Cep152 proteins attempt to bind to Plk4 from the same direction.
Significance of ordered Plk4 binding to Cep192 or Cep152
Our earlier studies have shown that two human centrosomal scaffolds, Cep192 and Cep152, do not appreciably interact with each other, and that the impairment of either one of the Cep192- and Cep152-dependent Plk4 interactions is sufficient to induce a defect in Sas6 recruitment to the procentriole assembly site 13. These findings suggest that Cep192 and Cep152 form two biochemically distinct and functionally nonredundant complexes with Plk4and that both complexes are required to promote Plk4 -mediated centriole biogenesis. In support of this view, depletion of Cep6326 disrupted Cep152 localization to centrioles, but did not appear to alter Cep192 localization to this site (Supplementary Fig. 6a–c).
Here we elucidated the molecular mechanism showing how Cep152 snatches Plk4 away from the Cep192–Plk4 complex and, as a result, repositions Plk4 at the outer edge of a newly forming Cep152 ring (Fig. 5). As expected if the recruitment of the high affinity Cep152 scaffold caused Plk4 to relocate to the outer rim of the Cep152 ring, depletion of Cep152 permitted Plk4 to reassociate with the inner Cep192 ring structure (Fig. 1c–e). Furthermore, when Plk4 was overexpressed, excess Plk4 was additionally localized along the entire length of the Cep192 structure (Supplementary Fig. 6d–g). These data suggest that Cep192 and Cep152 intricately regulate subcentrosomal localization of Plk4.
It should be noted, however, that unlike humans, nematodes utilize Spd-2 (Cep192 ortholog) to recruit Zyg-1 (Plk4 homolog) to centrosomes 7, while flies resort to Asl (Cep152 ortholog) to recruit its Plk412,27. These apparent differences hint that nematodes and flies have lost the ability of one or the other of the two scaffolds to promote Plk4 functions when evolved from their most recent common ancestor, while humans have kept the function of both scaffolds to cope with complex centriolar assembly processes.
Then, what advantage is gained in humans by having two scaffolds, Cep192 and Cep152, binding to Plk4 in an ordered manner? One possibility is that, by binding to Plk4, these two scaffolds modulate Plk4 function differently at distinct stages of early G1 phase. We found that Plk4-bound Cep152, but not Cep192, efficiently interacted with Cep135 (Supplementary Fig. 7a), a downstream component critical for procentriole assembly 28,29. Thus, the binding of Cep152 to Plk4 is likely to allow Plk4 to form a biologically active complex with downstream effectors, such as Cep135, and to induce procentriole assembly with other components critical for this process. On the other hand, since overexpression of Plk4 induces centriole over duplication 9,30, the binding of Cep192 to Plk4 may be important in sequestering Plk4 and preventing it from inducing unscheduled centriole duplication until Cep152 is recruited to early G1 centrioles. Notably, binding of Cep192 or Cep152 to Plk4 did not appreciably influence the Plk4 kinase activity (Supplementary Fig. 7b). Thus, these two scaffold proteins modulate Plk4-dependent centriole biogenesis by altering its bound states rather than its catalytic activity.
Here we showed that two centrosomal scaffold proteins, Cep192 and Cep152, interact with Plk4 in a temporally and spatially regulated manner, thus ensuring timely formation of distinct Plk4 complexes at different subcentrosomal structures. In this regard, it is notable that the CEP152 E21K mutation found in human cancers crippled the Cep152-Plk4 interaction that led to a defect in procentriole assembly and chromosome segregation (Fig. 3e–g, and Supplementary Fig. 4a–c). Similar defects were also observed with another cancer -associated, Plk4-binding defective, CEP152 V8A mutation (Supplementary Fig. 4d–h). These findings highlight the importance of converting Plk4 from a Cep192-tethered state (sequestered state) to a Cep152-bound state (procentriole assembly state) to properly regulate centriole duplication and mitotic progression.
Scaffold proteins play diverse roles in various biological processes, such as insulating or tethering signaling components. However, unlike these somewhat passive roles, the two functionally distinct scaffolds that we described here operate in an opposite manner to shift a common binding target from one state to another and modulate its biological function. Incompatible binding modes and differential binding affinities are the basis of unidirectional scaffold switching that may serve as a new paradigm for understanding complex assembly processes involving multiple scaffold proteins.
ONLINE METHODS
Plasmid construction
Detalis for all constructs used in this work are provided in Suppl. Note.
Cell culture and transfection
For U2OS and 293T cells were cultured as recommended by American Type Culture Collection. Transfection was carried out using either Lipofectamine 2000 (Invitrogen) for protein overexpression or Lipofectamine RNAiMAX (invitrogen) for siRNA transfection. U2OS cells were transfected with siRNA twice against control Luciferase (GL siRNA) or CEP152 (CEP152 siRNA) for 96 h. To arrest cells in S or M phase, cells were treated with thymidine (Thy) or nocodazole (Noc) for 20 h, respectively.
Lentivirus production and generation of stable cell lines
Lentiviruses expressing siRNA-insensitive CEP152-sil, CEP152-sil (E21K), or CEP152-sil (V8A) were generated by cotransfecting 293T cells with pHR′-CMVΔR8.2Δvpr, pHR′-CMV-VSV-G (protein G of vesicular stomatitis virus), and pHR′-CMV-SV-puro-based CEP152-sil WT, E21K, or V8A mutant, using the CaCl2 transfection method as described previously 31. Stable U2OS cell lines expressing the gene of interest were generated by lentivirus infection followed by selection with 2 μg/ml of puromycin (Sigma). The resulting cells were then transfected two times with the indicated siRNA to deplete the RNAi-sensitive endogenous protein (see Supplementary Figure 4a,e for detailed information). All the siRNAs used in this study are listed in Supplementary Table 2.
Antibodies
All the antibodies used in this study are summarized in Supplementary Table 1. A rabbit polyclonal Cep192 (2240–2538) antibody was generated against bacterially expressed His6-Cep192 (2240–2538) at Young In Frontier Co., Ltd (Seoul, South Korea) and then affinity-purified using GST-Cep192 (2240–2538) immobilized to Affi-gel 15 (Bio-Rad Laboratories). Antibodies for Cep192 N (1–500), Cep152 M (491–810), and Plk4 (580–970) are previously described 13. The polyclonal Plk4 p-S305 antibody was generated by immunizing rabbits with a synthetic peptide, NH2-SSSTSISGpSLFDKRRLL-NH2 (amino acids 297–313 in human Plk4; bold pS indicates the phospho-S305 residue) (Young In Frontier Co., Ltd, Seoul, South Korea). The p-S305 antibody was then affinity purified before use. Other primary antibodies were purchased from outside sources. See Supplementary Table 1 for additional information about the antibodies used in this study. Validations for commercial and newly generated antibodies are available on the manufactures’ websites and Supplementary Figure 8.
Immunostaining
Asynchronously growing U2OS cells were fixed with 4% paraformaldehyde and immunostained as described previously 32 using the indicated antibodies. Alexa Fluor-conjugated rabbit Cep192 and Cep152 antibodies were generated using Alexa Fluor labeling kits (Invitrogen). Where indicated, these antibodies were used to avoid cross-reactivities among more than two rabbit antibodies. Stained cover slips were mounted with a non-polymerizing mounting media from Vector Laboratories.
For images in Figure 1a, cells were first stained with rabbit anti-Plk4 and mouse anti-Sas6 antibodies. After completing these stainings, the resulting samples were then additionally stained with Alexa 647-conjugated Cep192 N-terminal (N) and Alexa 488-conjugated Cep152 middle region (M) antibodies. For images in Supplementary Figure 1a, Alexa 488-conjugated Cep192 N-terminal (N) and Alexa 647-conjugated Cep192 C-terminal (C) antibodies were used.
Three-dimensional structured illumination microscopy (3D-SIM)
Structured Illumination Microscopy (SIM) was performed on an ELYRA PS.1 microscope from Carl Zeiss Microscopy. Images were observed through a 63x/1.4NA objective and recorded using an Andor iXon 885 EMCCD (1024 × 1024 pixels, 8 × 8 μm pixel size, 65% QE), for a maximum field of view of 80 × 80 μm of the sample. Structured Illumination raw datasets were acquired by projecting grids onto the sample generated from the interference of the 0th and ±1st diffraction orders from a phase grating. For the 405, 488, 561 and 642 nm excitation, phase gratings of spacing 23, 28, 34 and 34 μm (respectively) were used to generate illumination grids for maximum resolution improvement of each color. Each super-resolved image required five grid shifts (phases) and three grid rotations for a total of 15 images per super-resolved z-plane per color. The ELYRA PS.1 system’s maximum laser output was 50, 200, 200, 150 mW (respectively), with a dedicated ND filter wheel for each laser for fine power control. During acquisition, laser power, camera exposure time and camera gain were adjusted so that high contrast images (50% camera dynamic range, 16 bit) were acquired. For most images, a camera exposure time of 50 ms was used. To measure the diameter of ring signals, acquired images were analyzed using a Zeiss Zen software.
Immunoprecipitation and immunoblotting analyses
Immunoprecipitation was performed essentially as described previously 32 in TBSN buffer [20 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.5% Nonidet P-40, 5 mM EGTA, 1.5 mM EDTA, 20 mM p-nitrophenylphosphate and protease inhibitor cocktail (Roche)]. Immunoblotting was performed following standard procedures, and immobilized proteins were detected by enhanced chemiluminescence (ECL) substrate (Pierce). Original images of immunoblots used in this study can be found in Supplementary Figure 8.
To detectPlk4 coimmunoprecipitated with Cep192 or Cep152 at endogenous levels, anti-Cep192 or anti-Cep152 antibodies were first cross-linked to protein A/G agarose beads using Pierce Crosslink IP Kit (Thermo Scientific), and then used for immunoprecipitation analysis with HeLa lysates prepared in the TBSN buffer.
For lambda phosphatase (λ-PPase)-treated experiments, cell lysates were prepared in a phosphatase lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.5% Nonidet P-40, 1 mM EDTA, 1 mM MgCl2, 2 mM MnCl2], incubated with 2000 units of λ-Phosphatase (New England Biolabs) for 1 h at 30°C, and then subjected to coimmunoprecipitation analysis with the indicated antibodies.
In vitro kinase reaction
For Plk4 kinase assay in Supplementary Figure 7b, 293T cells individually transfected with control vector (−), FLAG-Plk4 (WT or K41M mutant), GFP-Cep192, or GFP-Cep152 were lysed and the resulting lysates were mixed prior to immunoprecipitation with anti-FLAG-M2 bead (Sigma). Kinase assays were performed essentially as described previously 32 in a buffer containing 100 μM ATP (5 μCi of [γ-32P] ATP; 1 Ci = 37 GBq) for 30 min at 30°C. Bacterially expressed His6-Cep192 (201–310) fragment was included as an in vitro substrate. A half of the samples was separated by 10% SDS -PAGE, stained with coomassie (CBB), and then auto radiographed. The other half of the samples was transferred to PVDF and immunoblotted with anti-p-S305 antibody 33.
Protein expression and purification
All the proteins used in this study were expressed in an E. coli Rosetta strain (Novagen). Cells were cultured in LB medium containing 50 μg/ml kanamycin at 37°C. The cells were then treated with 0.1mM IPTG and cultured for additional 10 h at 18 °C before harvest. The cell pellets were lysed in an ice-cold buffer [20 mM Tris–HCl (pH 7.5), 500 mM NaCl and 10% glycerol] by ultrasonication. His6-MBP-fused human Plk4 CPB (residues 580–808) was purified using a 5-mL HisTrap chelating column (GE Healthcare), and the bound protein was eluted with the lysis buffer plus a step gradient of 25 mM, 50 mM, or 500 mM imidazole. The resulting protein was digested with a recombinant tobacco etch virus protease (TEV) to remove the His6-MBP tag, and subjected to further purification by cation exchange chromatography using a 5-mL HiTrap SP (GE Healthcare).
The His6-MBP-fused human Cep192-58mer (residues 201–258) or Cep152-60mer (residues 1–60) proteins were purified using a 5-mL HisTrap chelating column (GE Healthcare) similarly as above, and then further purified by ion exchange chromatography using a 5-mL HiTrap Q (GE Healthcare).
To generate complexes between Plk4 CPB and His6-MBP-Cep192-58mer or His6-MBP-Cep152-60mer, the CPB was incubated with 5 molar excess of either the MBP-58mer or the MBP-60mer in 20 mM Tris–HCl (pH 7.5) and 150 mM NaCl at 4 °C for 3 h. The resulting complexes were digested with TEV to remove the His6-MBP tag, and then sequentially purified by Amylose resin (Qiagen), 5-ml HisTrap (GE Healthcare), and HiLoad 16/60 Superdex 200 (GE Healthcare) size-exclusion chromatography. All the purification steps were performed in the same buffer used for the complex formation. The purified CPB–Cep192-58mer or CPB–Cep152-60mer complexes exhibited greater than 95% purity.
The MBPL-Cep192-58mer and MBP-Cep152-60mer lacking the N-terminal His6 tag were purified sequentially using a 5-mL MBPTrap (GE Healthcare) and a 5-mL HiTrap Q (GE Healthcare). To separate the molecular size between MBP-fused Cep192-58mer and Cep152-60mer, the Cep192-58mer was fused with a variant of MBP (MBPL), containing functionally-inert 30 additional residues. All the purifications were carried out using AKTA Explorer (GE healthcare).
Crystallization and data collection
For crystallization, purified apo-CPB was concentrated to 20 mg/mL in a buffer containing 20 mM HEPES (pH 7.0), 500 mM NaCl, 10% (v/v) glycerol and 10 mM dithiothreitol, while purified CPB–Cep192-58mer or CPB–Cep152-60mer complexes were concentrated to 5 mg/ml in a buffer containing 20 mM Tris–HCl (pH 7.5) and 150 mM NaCl. Since MBP-fused Cep192-58mer and Cep152-60mer form more stable complexes with Plk4 CPB than their respective unfused forms, the CPB complexes were generated in the presence of MBP. The resulting complexes were then digested with TEV to cleave off MBP prior to placing them under different crystallization conditions.
For the human apo-CPB crystals, initial crystallization hits were found using the Hampton Index screen. Using a sitting-drop vapor-diffusion method at 22°C, crystals were optimized by mixing 1 μL of protein solution with 1 μL of reservoir solution containing 100 mM Bis-Tris (pH 5.5), 2.0 M ammonium sulfate and 1% (v/v) methanol and equilibrating the mixture against 100 μL of reservoir solution. The crystals suitable for diffraction analysis were obtained within 2 days. For the CPB–Cep192-58mer and CPB–Cep152-60mer complexes, initial crystallization hits were found using Qiagen Classics and JCSG+ screens. Using a hanging-drop vapor-diffusion method at 22°C, crystals were optimized by mixing 0.8 μl of protein solution with 1 μl of reservoir solution [20% (w/v) Polyethylene Glycol 1500] and equilibrating the mixture against 200 μl of reservoir solution. The CPB–Cep192-58mer crystals suitable for diffraction analysis were obtained within 3 days. To obtain the CPB–Cep152-60mer crystals suitable for diffraction analysis, 2 μl of the protein and reservoir solution (1 μl each) were mixed and equilibrated against 200 μl of reservoir solution containing22% (w/v) Polyethylene Glycol 4000, 3% Iso-propanol, 0.1 M Tris–HCl (pH 8.5), and 0.2 M Lithium Sulfate. Under these conditions, crystals were reproducibly obtained within 5 days.
Crystals were flash frozen in liquid nitrogen after stepwise soaking in 6%, 12.5%, and 18.5% (v/v) glycerol plus mother liquor. The diffraction data for the apo-CPB crystals were collected at the wavelength of 0.97949 Å, at 100 K, on the 5C beamline of the Pohang Accelerator Laboratory, Kyungbuk, South Korea, and the data for the CPB–Cep192-58mer and CPB–Cep152-60mer crystals were collected at the wavelength of 1.0 Å, at 100 K, on the 22-BM beamline of the Advanced Photon Source, Argonne, IL, U.S.A.. The data were then indexed, integrated, and scaled with the HKL-2000 program 34.
Structure determination
The crystal structures of apo-CPB (PDB: 4N9J), CPB–Cep192-58mer (PDB: 4N7Z), and CPB–Cep152 60mer (PDB: 4N7V) were solved by a molecular replacement program, Phaser-1.335. The Drosophila CPB structure (PDB: 4G7N) 20 was used as a n initial search model. Further model building was performed manually using WinCoot 36. Refinement was performed with PHENIX 37 and CNS 38. Ramachandran statistics of the refined apo-CPB, CPB–Cep192-58mer and CPB–Cep152-60mer models using MolProbity 39 showed that 95.0%, 94.2% and 91.7% of all the residues in each model, respectively, lied in favored region, while5.0%, 5.8% and 8.3% of them, respectively, lied in allowed region with no outliers. In all the CPB structures, six N-terminal residues (W580–R585) were disordered and the residues 625-632, 653-656, and 753-766 in loop regions exhibited weak electron density maps, and therefore were poorly defined. In addition, the C-terminal G808 residue in both apo-CPB and the CPB–60mercomplex, and the C-terminal P807 and G808 residues in the CPB–58mercomplex could not be traced. The D 668 and N669 residues of the CPB–58mer complex were also not registered.
The N-termini and C-termini of both Cep192 -58mer (201–214 and 242–258) and Cep152-60mer (1–15 and 47–60) were disordered, suggesting that these regions are highly flexible upon binding to CPB.
The statistics for the collected data and refinement are summarized in Table 1. The quality of the model was analyzed by using MolProbity 39. Electrostatic surface potentials were calculated using the PyMol Molecular graphics program (Delano Scientific).
In vitro binding and quantification
Peptide-based pull-down assay was carried out as described previously 24. Briefly, the indicated peptides bearing an N-terminal Cys-(CH2)6 linker (1 mM stock) were cross -linked to the beads using SulfoLink coupling gel (Pierce). The resulting bead-immobilized peptides were incubated with 293T cell lysates expressing FLAG-Plk4 and the resulting precipitates were analyzed by immunoblotting.
In vitro protein -protein interaction analysis was performed using bacterially expressed purified proteins. His6-MBP-TEV-Plk4 CPB, His6-MBP-TEV-Cep192 full-length (1-2537), His6-MBP-TEV-Cep192 (201–280), His6-MBP-TEV-Cep192 58mer (201–258), His6-MBP-TEV-Cep152 full-length (1-1654), His6-MBP-TEV-Cep152 (1–217), and His6-MBP-TEV-Cep152 60mer (1–60) were expressed in an E. coli Rosetta strain and purified with amylose resin (New England Biolabs). GST-Plk4 C (580–970) was purified with glutathione-agarose beads (Sigma). MBP pull-down assay was carried out as described previously 40. Bead-immobilized His6-MBP control, His6-MBP-TEV-Cep192 58mer, or His6-MBP-TEV-Cep152 60mer was incubated with Plk4 CPB in an in vitro binding buffer [50 mM Tris -HCl (pH 7.4), 150 mM NaCl, 1 m MEDTA, and 0.1% Nonidet P-40]. After incubation for 2 h at 4°C, beads were washed four times with binding buffer and analyzed by 12% SDS-PAGE. The resulting gel was stained with Coomassie and the amounts of bound proteins were quantified using TINA software 2.0 program. For the competition binding assay, bead-immobilized GST-Plk4 C was incubated with either His6-MBP-Cep192 (201–280) or His6-MBP-Cep152 (1–217) alone, or with both in the in vitro binding buffer for 1 h at 20°C. Samples were prepared and analyzed as described above.
Fluorescence polarization assays
To carry out fluorescence polarization (FP)-based binding assays, purified MBP control, MBP-Cep192-58mer, and MBP-Cep152-60mer were first conjugated with Alexa Fluor 488 fluorescent dye using a labeling kit from Invitrogen. To determine the binding affinity of Alexa 488-MBP alone, Alexa 488-MBP-Cep192-58mer, or Alexa 488-MBP-Cep152-60mer to Plk4 CPB, 100 nM of each of these conjugates was mixed with different concentrations (ranging from 0.015 nM to 8,000 nM) of Plk4 CPB in a buffer [50 mM NaCl, 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.01% Nonidet P-40] for 10 min. Samples were then analyzed in the 384-well format using a SpectraMax Paradigm Multi-Mode Microplate Detection Platform (Molecular Device). The data obtained from three independent experiments were analyzed using the GraphPad Prism 6 program, and the results were provided in Figure 4b.
FP-based inhibition assays for Plk4 CPB binding were performed similarly as described previously 41. Briefly, 100 nM of Alexa 488-conjugated MBP-Cep192-58mer or MBP-Cep152-60mer was preincubated for 10 min with Plk4 CPB at the final concentration of 518 nM (EC75% of MBP-Cep192-58mer binding) or 67 nM (EC75% of MBP-Cep152-60mer binding), respectively. The resulting preformed complexes were challenged with increasing concentrations (from 0.31 nM to 1250 nM) of unconjugated MBP-Cep152-60mer or MBP-Cep192-58mer, respectively. Samples were analyzed using the same microplate detection platform described above. Experiments were performed in triplicates, and the results were analyzed using the GraphPad Prism 6 program. The data were provided in Figure 4c.
Ni-NTA-based CPB switching assay between Cep192-58mer and Cep152-60mer
The entire switching assay was performed using AKTA Explorer (GE healthcare). Partially purified1.5 μ of the His6-MBP-Cep192-58mer–CPB complex (a total volume of 8 ml) was loaded onto 1 ml HisTrap column (GE Healthcare) in a loading buffer [20 mM Tris-HCl (pH 7.5) and 150 mM NaCl] supplemented with 50 mM Imidazole (buffer A) to eliminate nonspecific binding of the proteins to the column. The elution buffer (buffer B) was composed of buffer A plus 3.9 μ of MBP-Cep152-60mer. The elution was performed by applying a linear gradient [after equilibration from 0 to 5 ml, a linear gradient (0 to 100% buffer B) from 5 to 20 ml, 100% buffer B from 20 to 25 ml, and 0% buffer B from 25 to 35ml were followed; see the gradient profile in Figure 4d]. The flow was maintained at 0.2 ml/min and each fraction was collected at 1 ml. The indicated fractions (20 μl per each fraction) in Figure 4d were mixed with sample buffer and analyzed by 12% SDS-PAGE.
Size-exclusion chromatography
The experiment was performed using AKTA Explorer (GE healthcare). Partially purified 2.1 μ MBP -CPB, 8.5 μM MBPL-Cep192-58mer, and 8.0 μM MBP-Cep152-60mer were mixed (a total volume of 1 ml) in a buffer containing 20 mM Tris-HCl (pH 7.5) and 150 mM NaCl, and then incubated overnight at 4°Cto provide sufficient time to form a complex(es). The resulting sample was loaded onto the 16/60 Superdex 200 column (GE healthcare). The gel filtration was carried out at 1 ml/min in the same buffer. Samples were collected at 4 ml per fraction and 20 μl of each fraction was analyzed by 8% SDS-PAGE.
Statistical Analysis
P values were calculated by unpaired two-tailed t-test from the mean data of each group. All values are given as mean of n ± s.d.
Supplementary Material
Acknowledgments
We are grateful to V. Barrand S. Garfield for critical reading of the manuscript and B. Chhun and E. Shumsky for technical assistance on 3D-SIM. This work was supported in part by US National Cancer Institute (K.S.L) and National Institute of Diabetes and Digestive and Kidney diseases (W.Y.) intramural grants, the GRL Program grant (K20815000001) from the National Research Foundation of Korea (B.H.O.), the National Research Foundation of Korea (NRF; 2011-0030027) (S.J.K.), the Korea Basic Science Institute’s research grant T33418 (J.K.B), the World Class Institute (WCI 2009-002) program (B.Y.K.) funded by the Ministry of Science, ICT, and Future Planning of Korea, and the Next-Generation BioGreen 21 Program (#PJ009594), Rural Development Administration, Republic of Korea.
Footnotes
ACCESSION CODES
The crystal structures of apo-CPB, the CPB–Cep192-58mer complex and the CPB–Cep152-60mer complex are deposited in the Protein Data Bank under accession codes 4N9J, 4N7Z and 4N7V, respectively.
AUTHOR CONTRIBUTION
Experiments were designed and data interpreted by S.-Y.P., J.-E.P., T.-S.K, J.-H.K, M.-J.K., B.K., L.T. R.N.M., M.A., and S.K.. Crystal structures of apo-CPB structure (J.-H.K., and B.K.), the CPB–Cep192-58mer complex (S.-Y.P., L.T., J.-H.K., B.K., and J.-E.P.), and the CPB–Cep152-60mer complex (S.-Y.P., L.T., M.-J.K., B.K., and J.-E.P.) were determined independently. FP, 3D-SIM, and cell-based analyses were performed by J.-E.P., while all the biochemical analyses were performed by T.-S.K.. In addition, B.Y.K., J.K.B., H.H., and K.W.L. provided reagents and resources, and R.L.E., S.J.K., B.H.O., W.Y., and K.S.L. designed the experiments, interpreted the data, and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Gönczy P. Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol. 2012;13:425–435. doi: 10.1038/nrm3373. [DOI] [PubMed] [Google Scholar]
- 2.Brito DA, Gouveia SM, Bettencourt-Dias M. Deconstructing the centriole: structure and number control. Curr Opin Cell Biol. 2012;24:4–13. doi: 10.1016/j.ceb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Nigg EA, Stearns T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13:1154–1160. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep. 2012;13:501–514. doi: 10.1038/embor.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 7.O’Connell KF, et al. The C. eleganszyg -1 gene encodes a regulator of centrosome duplication with distinct maternal and paternal roles in the embryo. Cell. 2001;105:547–558. doi: 10.1016/s0092-8674(01)00338-5. [DOI] [PubMed] [Google Scholar]
- 8.Bettencourt-Dias M, et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol. 2005;15:2199–2207. doi: 10.1016/j.cub.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 9.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 10.Delattre M, Canard C, Gönczy P. Sequential protein recruitment in C. elegans centriole formation. Curr Biol. 2006;16:1844–1849. doi: 10.1016/j.cub.2006.07.059. [DOI] [PubMed] [Google Scholar]
- 11.Pelletier L, O’Toole E, Schwager A, Hyman AA, Müller-Reichert T. Centriole assembly in Caenorhabditis elegans. Nature. 2006;444:619–623. doi: 10.1038/nature05318. [DOI] [PubMed] [Google Scholar]
- 12.Dzhindzhev NS, et al. Asterless is a scaffold for the onset of centriole assembly. Nature. 2010;467:714–718. doi: 10.1038/nature09445. [DOI] [PubMed] [Google Scholar]
- 13.Kim TS, et al. Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc Natl Acad Sci USA. 2013;110:E4849–4857. doi: 10.1073/pnas.1319656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonnen KF, Gabryjonczyk AM, Anselm E, Stierhof YD, Nigg EA. Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J Cell Sci. 2013;126:3223–3233. doi: 10.1242/jcs.129502. [DOI] [PubMed] [Google Scholar]
- 15.Lawo S, Hasegan M, Gupta GD, Pelletier L. Subdiffraction imaging of centrosomes reveals higher-order organizational features of pericentriolar material. Nat Cell Biol. 2012;14:1148–1158. doi: 10.1038/ncb2591. [DOI] [PubMed] [Google Scholar]
- 16.Sonnen KF, Schermelleh L, Leonhardt H, Nigg EA. 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open. 2012;1:965–976. doi: 10.1242/bio.20122337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitagawa D, et al. Structural basis of the 9 -fold symmetry of centrioles. Cell. 2011;144:364–375. doi: 10.1016/j.cell.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Breugel M, et al. Structures of SAS -6 suggest its organization in centrioles. Science. 2011;331:1196–1199. doi: 10.1126/science.1199325. [DOI] [PubMed] [Google Scholar]
- 19.Leung GC, et al. The Sak polo -box comprises a structural domain sufficient for mitotic subcellular localization. Nat Struct Biol. 2002;9:719–724. doi: 10.1038/nsb848. [DOI] [PubMed] [Google Scholar]
- 20.Slevin LK, et al. The structure of the plk4 cryptic polo box reveals two tandem polo boxes required for centriole duplication. Structure. 2012;20:1905–1917. doi: 10.1016/j.str.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elia AE, et al. The molecular basis for phospho -dependent substrate targeting and regulation of Plks by the polo-box domain. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 22.Cizmecioglu O, et al. Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to the centrosome. J Cell Biol. 2010;191:731–739. doi: 10.1083/jcb.201007107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatch EM, Kulukian A, Holland AJ, Cleveland DW, Stearns T. Cep152 interacts with Plk4 and is required for centriole duplication. J Cell Biol. 2010;191:721–729. doi: 10.1083/jcb.201006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yun SM, et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ko MA, et al. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat Genet. 2005;37:883–888. doi: 10.1038/ng1605. [DOI] [PubMed] [Google Scholar]
- 26.Zhao H, et al. The Cep63 paralogue Deup1 enables massive de novo centriole biogenesis for vertebrate multiciliogenesis. Nat Cell Biol. 2013;15:1434–1444. doi: 10.1038/ncb2880. [DOI] [PubMed] [Google Scholar]
- 27.Dix CI, Raff JW. Drosophila Spd-2 recruits PCM to the sperm centriole, but is dispensable for centriole duplication. Curr Biol. 2007;17:1759–1764. doi: 10.1016/j.cub.2007.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin YC, et al. Human microcephaly protein CEP135 binds to hSAS -6 and CPAP, and is required for centriole assembly. EMBO J. 2013;32:1141–1154. doi: 10.1038/emboj.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inanç B, et al. Abnormal centrosomal structure and duplication in Cep135 -deficient vertebrate cells. Mol Biol Cell. 2013;24:2645–2654. doi: 10.1091/mbc.E13-03-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleylein-Sohn J, et al. Plk4 -induced centriole biogenesis in human cells. Dev Cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 31.Johmura Y, et al. Regulation of microtubule -based microtubule nucleation by mammalian polo-like kinase 1. Proc Natl Acad Sci USA. 2011;108:11446–11451. doi: 10.1073/pnas.1106223108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee KS, Yuan YL, Kuriyama R, Erikson RL. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol Cell Biol. 1995;15:7143–7151. doi: 10.1128/mcb.15.12.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sillibourne JE, et al. Autophosphorylation of polo -like kinase 4 and its role in centriole duplication. Mol Biol Cell. 2010;21:547–561. doi: 10.1091/mbc.E09-06-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, et al. PHENIX: a comprehensive Python -based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brünger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Chen VB, et al. MolProbity: all -atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang YH, et al. Mammalian polo -like kinase 1-dependent regulation of the PBIP1–CENP-Q complex at kinetochores. J Biol Chem. 2011;286:19744–19757. doi: 10.1074/jbc.M111.224105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, et al. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat Chem Biol. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.