

Abstract
Ku-dependent C-NHEJ (classic non-homologous end joining) is the primary DNA EJing (end joining) repair pathway in mammals. Recently, an additional EJing repair pathway (A-NHEJ; alternative-NHEJ) has been described. Currently, the mechanism of A-NHEJ is obscure although a dependency on LIGIII (DNA ligase III) is often implicated. To test the requirement for LIGIII in A-NHEJ we constructed a LIGIII conditionally-null human cell line using gene targeting. Nuclear EJing activity appeared unaffected by a deficiency in LIGIII as, surprisingly, so were random gene targeting integration events. In contrast, LIGIII was required for mitochondrial function and this defined the gene’s essential activity. Human Ku:LIGIII and Ku:LIGIV (DNA ligase IV) double knockout cell lines, however, demonstrated that LIGIII is required for the enhanced A-NHEJ activity that is observed in Ku-deficient cells. Most unexpectedly, however, the majority of EJing events remained LIGIV-dependent. In conclusion, although human LIGIII has an essential function in mitochondrial maintenance, it is dispensable for most types of nuclear DSB repair, except for the A-NHEJ events that are normally suppressed by Ku. Moreover, we describe that a robust Ku-independent, LIGIV-dependent repair pathway exists in human somatic cells.
1. Introduction
A serious challenge to genomic integrity is the occurrence of a DNA DSB (double-strand break) [1]. To avoid the pathological outcomes that result from even a single unrepaired DNA DSB, all cells have developed efficient DSB repair pathways. In most organisms, there are two major pathways: HR (homologous recombination) and C-NHEJ (classic-non-homologous end joining) [2, 3]. HR is preferentially used in lower organisms, however in mammals — and particularly in human cells — the majority of DSBs are repaired via C-NHEJ.
C-NHEJ facilitates the direct ligation of the broken ends of a DSB. Since the DNA termini formed at DSBs are, however, often complex and can contain non-ligatable end groups, the repair of such DNA lesions may require the processing of the ends prior to ligation [1, 4]. This requirement often leads to the loss or addition of nucleotides from either side of the DSB, making C-NHEJ “error-prone”. The mechanism of C-NHEJ-mediated DSB repair postulates that Ku (the Ku70/Ku86 heterodimer) binds to the DSB ends, where it recruits downstream C-NHEJ factors that facilitate processing [5]. Finally LIGIV (DNA ligase IV), in association with XRCC4 (X-ray-cross-complementation gene 4) and XLF (Cernunnos/XRCC4-like factor), performs the end ligation reaction [1]. This linear, stepwise model for C-NHEJ may be oversimplified as there is evidence that LIGIV, XRCC4 and XLF may perform roles both upstream and downstream in the repair process [6-8].
There is an additional EJing pathway present in higher eukaryotes. It has interchangeably been referred to as MMEJ (micro-homology-mediated end joining) [9], B-NHEJ (backup-NHEJ) [10] and A-NHEJ (alternative-NHEJ) [11], {hereafter, A-NHEJ}. Unlike the HR and C-NHEJ pathways, which are conserved from bacteria to man, the A-NHEJ pathway has evolved in a somewhat checkered manner and can only be detected in about a third of eukaryotic genomes [12]. It is presumed that an end-binding factor besides Ku is required to bind onto the broken DNA ends, stabilize them, protect them from random nuclease degradation and finally funnel the ends into the A-NHEJ pathway [13]. Then, because microhomology is frequently used to mediate the repair event, some end resection is required [14]. Alignment activities to bring the microhomologies into register are also needed, followed by the action of a flap-like nuclease to trim non-base paired regions and finally a ligation complex to covalently link the ends back together [15]. Because the pathway uses microhomology to mediate the repair event, deletions always accompany the repair event, as does loss of one of the blocks of microhomology [4].
Several laboratories have made dedicated attempts to identify A-NHEJ factors. In particular, a brute-force nuclear extract fractionation protocol identified LIGIII {DNA ligase III; [12]}, heretofore known only for its role in BER (base excision repair), as the candidate ligase required for A-NHEJ [16]. Using guilt-by-association as a scientific rationale, PARP1 {poly (ADP-ribose) polymerase 1} and XRCC1 (X-ray cross complementing gene 1), two proteins known to interact with LIGIII during BER, were subsequently identified as also being involved in A-NHEJ [13, 17, 18]. PARP1 is presumed to compete with Ku for binding to broken DNA ends thereby dictating pathway choice [13, 18] whereas XRCC1 appears to act as a chaperone for LIGIII [19]. Additional factors have also been implicated in A-NHEJ. Thus, CtIP (C-terminal interacting protein) and the MRN (Mre11:Rad50:Nbs1) complex — factors known to be involved in the end resection events required for HR — have also been implicated in the end resection steps of A-NHEJ [20-24].
If the factors needed for A-NHEJ are not completely defined and the A-NHEJ reaction mechanism nebulous, it is also fair to say that the biological role(s) of A-NHEJ is even more poorly understood. Most of the current interest in A-NHEJ, however, stems from its implicated use in the chromosomal translocations that are present in cancer cells. Sequencing of human cancer genomes has revealed that many [25-27], albeit not all [28] chromosomal translocations have microhomology at their breakpoint junctions, which implicates A-NHEJ in their genesis. This hypothesis gained support from work in which LIGIII conditionally-null murine cells showed decreased translocation frequencies and reduced microhomology usage [29-32]. An additional biological process where A-NHEJ has been implicated is in the random insertion events associated with rAAV (recombinant adeno-associated virus)-mediated gene targeting. Although rAAV can mediate high frequency gene targeting, the majority of the viral integration events still occur randomly [33]. Moreover, our laboratory has reported that a reduction in the C-NHEJ genes Ku70 [34] and LIGIV [35] had almost no impact on the random rAAV integration rate — implying that these events may be mediated instead by A-NHEJ. In summary, although A-NHEJ was a neglected subject for many years, in the past decade it has proven itself to be an increasingly interesting and biologically relevant topic.
A key feature of A-NHEJ is its dependence on LIGIII [16]. Unlike the other ligases, LIGIII is molecularly heterogeneous [12, 36, 37]. Thus, alternative translation initiation generates mitochondrial and nuclear forms of LIGIII, which either contain or lack a MLS (mitochondrial localizing sequence), respectively [36]. The existence of LIGIII isoforms implies diverse functional roles for LIGIII. One experimental approach to unraveling the complexity of LIGIII is to generate a LIGIII-deficient model system, which has already been accomplished in the chicken cell line, DT40 [38], and in the mouse [30, 32, 39]. In these systems the gene is essential due to its presumed requirement for mitochondrial DNA replication. Moreover, in LIGIII conditionally-null mice no obvious nuclear DNA repair phenotypes could be detected [30, 32]. The extrapolation of these studies to humans is unfulfilled as neither LIGIII patients nor LIGIII-deficient human cell systems have been described.
In this study, we conditionally inactivated the LIGIII gene in the HCT116 human cell line and confirmed that the loss of LIGIII results in death due to mitochondrial dysfunction. We also constructed a cell line that exclusively expressed a mitochondrial-only form of LIGIII. The nuclear LIGIII deficiency in this cell line caused a growth retardation, but it did not affect the overall NHEJ repair activity nor did it result in hypersensitivity to DNA damaging agents. Unexpectedly, we also demonstrate that LIGIII-dependent A-NHEJ does not mediate rAAV random integration events. These findings were extended by constructing human cell lines that were doubly deficient for either Ku and LIGIII or Ku and LIGIV. These experiments demonstrated that LIGIII is required for the enhanced A-NHEJ activity that is observed in Ku-deficient cells and that the vast majority of repair events in a Ku-deficient cell are still LIGIV-dependent. In conclusion, human LIGIII has an essential function in mitochondrial maintenance, however it is dispensable for most nuclear DSB repair, except for the A-NHEJ that is normally suppressed by Ku. In addition, we demonstrate that human cells have a robust Ku-independent, but LIGIV-dependent EJing activity.
2. Material and methods
2.1. Primers
The sequences for all primers referenced in the Materials and Methods section can be found in Supplemental Table 1.
2.2. Construction of LIGIII-null cells
Conditional and non-conditional knockout LIGIII targeting vectors were constructed as described, with a few modifications [33, 40]. For the conditional knockout vector, the left and right homology arms, the latter of which contained the floxed exon 5, were generated by PCR. For the left homology arm, Exon5_LARM_F1 and Exon5_LARM_SacR1 primers were used. For the right homology arm, the PCR products generated from Exon5_RARM_XhoF1 × Exon5_RARM_R1 primers and Exon5_KpnF1 × Exon5_XhoR1 primers were ligated after XhoI restriction enzyme digestion. The relevant homology arms and a NEO (neomycin-resistance) gene cassette were assembled together through ligations followed by unique restriction enzyme digestions, and then cloned into the pAAV-MCS vector. A non-conditional knockout targeting vector was generated in a similar way, but it did not include the floxed exon 5 sequences. To select for productively infected cells, the rAAV-infected cells were incubated in 1 mg/ml G418-containing media for approximately 2 weeks. At this time, genomic DNA was purified from all G418-resistant clones and PCR was used to screen for the subset of those in which correct targeting had taken place. Targeted clones were screened with Exon5_SC_F2 × NeoR2 primers, and retargeted clones were confirmed by LIG3_LArm_F3 × LIG3_RArm_R2 primers.
2.3. Construction of a Ku86flox/−:LIGIIImito/− cell line
Conditional and knock-in LIGIII targeting vectors were constructed as described above. In the first round of targeting, Ku86flox/− cells [41] were infected with a conditional knockout (i.e., “flox:NEO”) vector, as described in the creation of the LIGIIIflox/− cell line. After confirming correct targeting, clones were Cre-treated, and screened for the loss of the drug selection cassette (NEO), but retention of the conditional (floxed) exon, thus yielding a Ku86flox/−:LIGIII+/flox cell line. In the second round of targeting, these cells were infected with a rAAV knock-in vector, which introduced ATC point mutations into the two closely spaced ATGs that enable nuclear LIGIII expression. For the rAAV LIGIII knock-in targeting vector construction, the left homology arm was PCR-amplified from wild-type HCT116 genomic DNA using Lig3Mut_LArmF and Lig3Mut_LArmR primers. The right homology arm was amplified using Lig3Mut_RArmF and Lig3Mut_RArm3R primers.
For the screening of the correctly targeted clones, Lig3pNeDaKI-EF1 × PGK-R2 and ZeoF1 × Lig3pNeDaKI-ER3 primer sets were used to ascertain the integrity of the left and right homology arms, respectively. After confirmation of a Ku86flox/−:LIGIIImito:NEO/flox cell line, clones were transiently treated with Cre recombinase and subcloned to identify for Ku86flox/−:LIGIIImito/− clones. Screening of the Cre recombinase-treated clones was performed with LArmF × Lig3pNeDaKI-ER primers.
2.4. Construction of a Ku86flox/−:LIGIV−/− cell line
LIGIV targeting vectors were used as described [35] in two rounds of gene targeting with Ku86flox/− cells [41] to generate a Ku:LIGIV doubly-mutant cell line.
2.5. LIGIII complementation
To construct the mitochondrial-only LIGIII cDNA, the second and third ATGs in the ORF (open reading frame) of the LIGIII cDNA were mutated to ATC. This mitochondrial-only LIGIII cDNA was cloned into the pcDNA3.1(+) vector with a C-terminal HA epitope tag. For the nuclear-only LIGIII, the N-terminal ORF that encodes the MLS was deleted and a FLAG-epitope tag was added to the C-terminus. This modified, nuclear-only LIGIII cDNA was also cloned into a modified pcDNA3.1(+) vector, where the NEO gene had been replaced with a puromycin-resistance gene. Complementation constructs were linearized by PvuI restriction enzyme digestion and stably transfected into the relevant cell lines with Lipofectamine 2000. For selection, 1 mg/ml G418 and 2 μg/ml puromycin, respectively, were used.
2.6. Use of an inducible Cre system
A PiggyBac transposon system [42] was used with slight modification. Cells were subcultured into 24-well plates a day before transfection. A vector expressing PiggyBac-transposase (0.4 μg) and a vector containing PiggyBac-CreERt2-transposon lacking a GFP marker (0.4 μg) were transfected using Lipofectamine 2000 according to the manufacturer’s protocol. The transfected cells were subcultured 48 hr after transfection into a 96-well plate with 2 μg/ml of puromycin in the media for selection. Clones stably expressing CreERt2 were subsequently identified by immunoblotting using an antibody directed against Cre recombinase (data not shown). For CreERt2 induction, 10 nM of 4-hydroxytamoxifen (4-OHT), which was dissolved in ethanol, was used.
2.7. Immunoblotting
Whole cell extracts were prepared with RIPA buffer and 30 μg of protein was electrophoresed on a 4% to 20% gradient SDS (sodium dodecyl sulfate)-polyacrylamide gel and a rabbit, anti-human LIGIII monoclonal antibody (Gene Tex) was used at a 1:1000 dilution. HA (Covance) and FLAG (Sigma) antibodies were also used at a 1:1000 dilution. The actin antibody (Santa Cruz), which was used for the loading control, was diluted 1:250.
2.8. Immunocytochemistry (ICC)
Cells were plated on multi-chamber slides a day before analysis and subsequently fixed with 4% (v/v) paraformaldehyde in phosphate-buffered saline for 10 min. Slides were incubated in antigen retrieval buffer {100 mM Tris, 5% (v/v) urea, pH 9.5} at 95°C for 10 min. Permeabilization was performed with 0.1% Triton X-100. The LIGIII antibody was used at a 1:1000 dilution and an Alexa Fluor 488 goat, anti-mouse IgG antibody (Invitrogen) was used to visualize LIGIII. DAPI (0.2 μg/ml) was used to stain the nucleus.
2.9. Etoposide and MMS (methyl methane sulfonate) sensitivity
An etoposide sensitivity assay was performed as described [35] with slight modifications. The cells were plated on a 6-well cell culture plate approximately 17 to 19 hr prior to drug treatment. Etoposide was dissolved in dimethyl sulfoxide to give a 10 mM stock solution. The cells were then incubated in etoposide-containing medium for 7 to 10 days, fixed and stained with crystal violet. For the MMS sensitivity test, cells were incubated in MMS-containing media for 1 hr and then maintained in drug-free media for 7 to 10 days. In all survival experiments the wild-type cells were grown for 7 days and the LIGIII mutant lines were grown for 10 days to accommodate the latter’s slower growth phenotype.
2.10. DNA EJing assays and plasmid rescue
The in vivo EJing reporter plasmid pEGFP-Pem1-Ad2 [43] and the total A-NHEJ reporter pEJ2 [44] were used as described. HindIII- or I-SceI-digested pEGFP-Pem1-Ad2 or I-SceI-digested pEJ2 plasmid was co-transfected with a pCherry plasmid — used as a control of transfection efficiency — into the relevant cell line using Lipofectamine 2000. Green (EGFP) and red (Cherry) fluorescence were measured by FACS (fluorescence-activated cell sorting) 24 or 48 hr later. Repair efficiency was presented as a ratio of cells that were doubly positive for red and green over the number of cells that were only positive for red fluorescence.
For plasmid rescue experiments [43], HindIII- or I-SceI-digested pEGFP-Pem1-Ad2 plasmid was transfected into the desired cell lines without the pCherry plasmid. Plasmids were rescued 24 hr after transfection using a mini-preparation protocol, transformed into E. coli and repaired plasmids were selected on LB plates containing 30 μg/ml of kanamycin. All the repair products were analyzed by sequencing, using a variety of primers located upstream and downstream of the Ad2 exon sequence.
2.11. Microhomology assay
A DNA microhomology assay was performed as described [43]. Cells were subcultured into 6-well plates a day before transfection. pDVG94 plasmid (2.5 μg) digested with EcoRV and AfeI was transfected using Lipofectamine 2000. Plasmid DNA was recovered using a modified mini-preparation protocol at 24 to 48 hr after transfection. Repaired DNA junctions were PCR amplified using FM30 and 5′-radiolabeled DAR5 primers. PCR products were then digested with BstXI. Digested PCR products were separated by electrophoresis on a 6% polyacrylamide gel. The gel was then either dried and exposed to film, or stained with SybrGold and imaged on a Storm 840 fluorescent gel imager.
2.12. rAAV-mediated gene targeting
pAAV-HPRT-Puro, pAAV-helper, and pAAV-RC vectors were transfected into 70% confluent AAV-293 cells in 10 cm cell culture dishes and infectious rAAV-HPRT-Puro virus was harvested 3 days later by freeze/thawing as described [33, 40]. The virus was subsequently purified using a rAAV virus purification kit and the viral titer was quantitated by qPCR. A day before infection, cells were subcultured into 2 × 105 cells/well in 6-well cell culture dishes in duplicate. Before adding virus, the exact number of cells was determined by counting one of the duplicate wells, and then virus at a MOI of 1 × 104 was added to the other well. Two days after infection, 1% of the cells were plated into a 10 cm culture dish without any drug selection and used to determine the plating efficiency. The remaining cells were plated into 10 cm culture dishes with 2 μg/ml puromycin. This medium was replaced 5 days later with puromycin-containing medium supplemented with 5 μg/ml of 6TG (6-thioguanine) except for one plate, which was used to quantitate the random integration frequency (i.e., those clones that were just G418 positive). When the cells had formed visible colonies at approximately 10 to 14 days later, the plates were fixed and stained with crystal violet.
3. Results
3.1. Generation of a conditionally-null LIGIII HCT116 cell line
To generate a HCT116 cell line that was conditionally-null for LIGIII expression, a rAAV gene-targeting methodology was adopted [33, 40], similar to the targeting strategy that was used to generate a Ku86 conditionally-null HCT116 cell line [41]. The conditional targeting vector contained three LoxP sites flanking a NEO (neomycin-resistance) gene and exon 5 of LIGIII, respectively (Fig. S1A). Eight correctly targeted first round clones (LIGIIINEO/+; Fig. S1B) from 210 G418-resistant clones were identified (relative gene targeting frequency: 3.8%). One of these clones was transiently treated with pGK-Cre to remove the NEO selection cassette (Fig. S1C). The resulting LIGIIIflox/+ cell line was subjected to a second round of gene targeting using a non-conditional knockout vector, in which exon 5 was designed to be simply replaced by a floxed NEO gene (Fig. S1C). Ten correctly targeted second round clones were identified from 711 G418-resistant clones (relative gene targeting frequency: 1.4%). Additional analysis (data not shown) demonstrated that 9 of the clones were re-targeted (and therefore biologically uninteresting) whereas one clone was correctly targeted to the second allele (i.e., LIGIIIflox/NEO; Fig. S1D), which was subsequently infected with an AdCre virus to remove the NEO gene. The resulting LIGIIIflox/− cell line (Fig. S1E) was viable and when needed, Cre recombinase could be re-introduced to generate LIGIII−/− cells (Fig. S1F).
PCR analyses were used to molecularly confirm the genetic designation of the cell lines. A 662 bp PCR product is diagnostic for the presence of the wild-type LIGIII exon 5 whereas a 748 bp product should be generated when the floxed exon 5 DNA is used as a substrate (Fig. 1A). As expected, PCR of the parental wild-type LIGIII+/+ cell line generated only the 662 bp product, whereas PCR of the LIGIIIflox/+ cell line produced both the wild type (662 bp) and the floxed allele (748 bp) fragments (Fig. 1A). In contrast, LIGIII conditionally null cell lines — with or without the NEO selection cassette — generated only the 748 bp PCR product corresponding to the floxed allele (Fig. 1A).
Fig. 1.

Generation of a LIGIII conditionally-null cell line. (A) PCR confirmation of LIGIIIflox/− cells. A primer set, LIG3_LArm_F3 (F3) and LIG3_RArm_R2 (R2), which yield differently sized PCR products from the floxed allele (748 bp) and wild-type allele (662 bp) were used. With the same primer set, an allele containing the NEO gene generated a band over 3 kb and the loxP-only allele yielded a 97 bp product, but they are not shown in this figure. (B) The floxed allele disappears after AdCre infection. With increasing amounts of AdCre virus, the 748 bp PCR product derived from the floxed allele disappeared in the LIGIIIflox/− clone. In contrast, the expression of Cre did not affect the LIGIII+/+ control cells. (C) Western blot analysis confirms the loss of LIGIII protein. Protein samples were produced in parallel to the DNA samples in (B). Increasing amount of AdCre virus correlated with decreasing amount of LIGIII protein only in the experimental cell line.
To assess the conditionality of the cell line, LIGIII+/+ and LIGIIIflox/− cells were infected with an increasing amount of AdCre virus, and 5 days later genomic DNA was purified and analyzed by PCR and whole cell extracts were prepared and analyzed by immunoblotting. As the amount of Cre recombinase increased, the PCR signal from the genomic floxed allele (748 bp) decreased until the signal was undetectable (Fig. 1B). Similarly, at the protein level, the increasing presence of Cre completely ablated detectable LIGIII protein expression (Fig. 1C). In contrast, Cre expression in the parental wild-type LIGIII+/+ cells had no effect on formation of the exon 5-derived PCR product (662 bp) nor little effect on LIGIII protein expression (Fig. 1B and C). From these experiments, we concluded that we had successfully constructed a human LIGIIIflox/− (i.e., conditionally null) cell line.
3.2. The mitochondrial form of LIGIII is essential for human somatic cell survival
To assess the essential nature of LIGIII, we initially infected the LIGIIIflox/NEO cell line (Fig. S1D) with AdCre and then isolated 22 individual G418-sensitive colonies by limiting dilution. Theoretically, two possible cell lines could have been recovered: LIGIIIflox/− and LIGIII−/− (Fig. S1E and F, respectively), however all 22 recovered clones had a LIGIIIflox/− genotype and none were LIGIII−/− (data not shown). This extreme asymmetry in the recovery of Cre-treated survivors strongly suggested that the LIGIII−/− cell line was not viable.
Because the mitochondrial form of LIGIII is essential in mice [30, 32, 45], we tested whether this activity was conserved in human LIGIII. To this end, we complemented the LIGIIIflox/− cells with a modified LIGIII cDNA that could be expressed only in the mitochondria (mL3), and generated a stable LIGIIIflox/-:mL3 cell line. The mL3 expression construct was made by mutating the second and third LIGIII ATGs to ATCs (Fig. S2A). The nuclear form of LIGIII is normally translated from the second ATG, and without the second ATG only the longer mitochondrial-specific version of the protein should be made [36]. We mutated the third ATG simply as a precaution to ensure that no N-terminally truncated nuclear protein could be expressed. Importantly, after infecting LIGIIIflox/-:mL3 cells with AdCre virus and isolating single cell clones by limiting dilution, we observed that 25 out of 43 were genotypically LIGIII-null (LIGIII−/−:mL3; data not shown) — a result that contrasted sharply with 0 out of 22 obtained when we tried to establish LIGIII-null cells in the absence of mL3 expression.
We next investigated whether the mitochondrial form of LIGIII could directly rescue the lethality of LIGIII-deficient cells. When AdCre virus was used for this experiment, it was often difficult to differentiate the viral toxicity caused by the adenovirus from the apoptosis induced by the absence of LIGIII. To improve upon this experimental set-up, a tamoxifen-inducible Cre recombinase was stably introduced into LIGIIIflox/+, LIGIIIflox/−, LIGIIIflox/-:mL3 and LIGIIIflox/-:nL3 (see below) cell lines. 4-OHT (4-hydroxyltamoxifen) treatment completely killed LIGIIIflox/− cells (clone #5) but had little effect on LIGIIIflox/+ cells (clones #101-1 and #101-21), which still had a functional LIGIII allele even after Cre treatment. Significantly, the lethality of LIGIII-null cells was significantly rescued by expression of the mitochondrial form of LIGIII (Fig. S2B; clones #73-5 and #73-6). From these experiments we concluded that the mitochondrial form of LIGIII is essential for human somatic cell viability.
Finally, a mitochondrial-exclusive expression pattern of mL3 was verified by immunohistochemistry. In the parental wild-type HCT116 LIGIII+/+ cells, LIGIII protein was expressed ubiquitously throughout the cell, whereas in LIGIII−/−:mL3 cells fluorescent signal was detected virtually exclusively in the cytoplasm (Fig. S2C).
3.3. Complementation of LIGIII−/−:mL3 with a nuclear LIGIII cDNA
With a viable LIGIII-null cell line in hand (i.e., LIGIII−/−:mL3 cells), we were equipped to investigate the phenotypes resulting from the loss of nuclear LIGIII expression. Before beginning these analyses, however, we augmented our reagents with a derivative cell line that re-expressed a nuclear-specific LIGIII cDNA. For this approach, a nuclear-only LIGIII cDNA, nL3, was generated by deleting the N-terminal MLS from the wild-type cDNA and by adding a C-terminal FLAG epitope-tag (Fig. S2A). We isolated stable LIGIII−/−:nL3 and LIGIII−/−:mL3:nL3 cell lines, the later of which exhibited a strong LIGIII ICC signal from the nucleus in addition to pan-cytoplasmic staining (Fig. S2C). Importantly, the nuclear-exclusive form of LIGIII — in contrast to the mitochondrial-exclusive form — was incapable of rescuing the lethality of LIGIII-nulls (Fig. S2B; clone #12-12).
3.4. A LIGIII deficiency causes a growth defect
While the mitochondrial form of LIGIII rescued the lethality of LIGIII-deficient cells, the rescued phenotype was not as robust as even LIGIII heterozygous cells (Fig. S2B, compare clones #73-5 and #73-6 with #101-1 and #101-21). Thus, we investigated whether there was a growth defect associated with the absence of LIGIII. Three thousand cells were seeded on day 0 into each well of a 6-well tissue culture dish and the number of cells in each well was counted on days 4 to 8 (Fig. 2A). LIGIIIflox/− cells showed a slight haploinsufficiency for growth, which was significantly exacerbated in the LIGIII−/−:mL3 cell line. From these experiments we concluded either that the absence of nuclear LIGIII — or inadequate mitochondrial LIGIII — expression results in a proliferation defect.
Fig. 2.
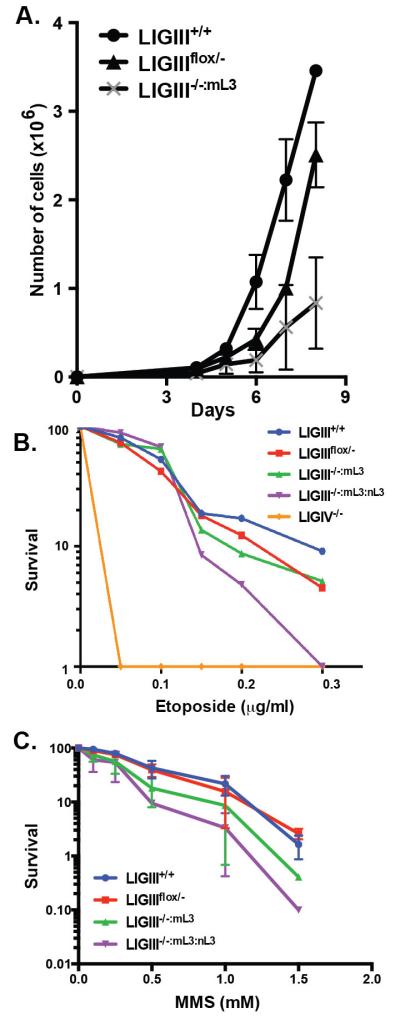
A LIGIII deficiency causes growth retardation, but not hypersensitivity to DNA damaging agents. (A) Three thousand cells were plated on day 0 and their growth was subsequently assessed by counting trypan blue-excluding cells at the indicated days. The average of two independent experiments, each done in triplicate, is shown. (B) Etoposide sensitivity. Three hundred cells were plated a day before etoposide treatment. Survival was normalized by setting the value obtained from no etoposide treatment as 100 percent. As a positive control, a LIGIV−/− cell line, which is known to be extremely sensitive to etoposide, was used. The plotted values are the average of three independent experiments. (C) MMS sensitivity was performed in a similar way to the etoposide sensitivity test, except for the drug-treatment time: cells were incubated in MMS-containing media for 1 hr.
3.5. LIGIII-null cells are not sensitive to DNA damaging agents
Given that LIGIII has been implicated in both SSB (single-strand break) and DSB repair pathways [16, 46], colony-forming assays were used to examine the sensitivity of LIGIII-null cells to a variety of DNA damaging agents. In these survival experiments, the wild-type cells were grown for 7 days and the LIGIII mutant lines were grown for 10 days to accommodate the latter’s slower growth phenotype. Etoposide is a topoisomerase II inhibitor and a powerful radiomimetic drug that induces DNA DSBs [47]. As a positive control, a LIGIV−/− cell line [35] was, as expected, exquisitely sensitive to etoposide, even at the lowest concentration (Fig. 2B). In contrast, LIGIIIflox/−, LIGIII−/−:mL3 and LIGIII−/−:mL3:nL3 did not exhibit any increased sensitivity to etoposide compared to the wild-type parental cells (Fig. 2B). Similarly, the LIGIII-deficient cell lines were slightly, but not significantly, sensitive to MMS (methyl methanesulfonate), an alkylating agent that induces SSBs at low doses and DSBs at high doses (Fig. 2C). In conclusion, the absence of LIGIII (and presumably the A-NHEJ DSB repair pathway) did result in a slower growth phenotype, but this was not reflected in a hypersensitivity to DNA damaging agents.
3.6. The absence of LIGIII does not affect the overall DNA EJing activity of human cells
The total DNA EJing activity of LIGIII-null cells was measured using pEGFP-Pem1-Ad2, an extra-chromosomal reporter assay vector [13, 43, 48]. The reporter plasmid consists of the GFP gene, which is interrupted by a 2.4 kb intron derived from the rat Pem1 gene. An exon (Ad2) derived from adenovirus serotype 2 has been introduced into the middle of the intron and is flanked by HindIII and I-SceI restriction enzyme recognition sites (Fig. 3A). Without modification, the GFP gene is not expressed because the Ad2 exon is incorporated into GFP mRNA (Fig. 3C). Pre-digestion of the plasmid with HindIII or I-SceI removes the Ad2 exon and generates a linear plasmid with compatible (i.e., ends that can be joined by simple ligation) or incompatible (i.e., ends that require some sort of processing before they are rejoined) ends, respectively (Fig. 3B). Productive end joining of the linear plasmid after it is transfected into the experimental cell line can be quantitated using FACS analysis of GFP expression (Fig. 3C).
Fig. 3.
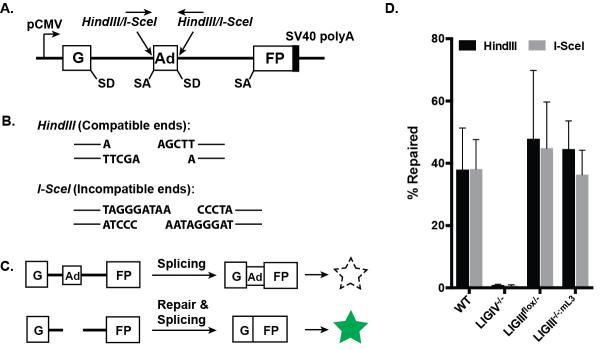
A LIGIII deficiency does not affect total end-joining activity. (A) A cartoon of the pEGFP-Pem1-Ad2 repair substrate used for analysis of DNA end-joining activity. Expression of the GFP cassette is driven by the CMV promoter and terminated by the SV40 polyA sequence. The GFP coding sequence is interrupted by a 2.4 kb intron containing an adenovirus exon (Ad), which is flanked by HindIII and I-SceI restriction enzyme recognition sites. Splice donor (SD) and splice acceptor (SA) sites are also shown. (B) HindIII digestion generates compatible ends with 4 nt overhangs, whereas I-SceI digestion produces incompatible ends, that require some processing before they can be rejoined. (C) The starting substrate is GFP negative because the Ad exon is efficiently spliced into the middle of the GFP ORF, inactivating the GFP activity. Cleavage with either HindIII or I-SceI removes the Ad exon and, upon successful intracellular plasmid circularization, GFP expression is restored and can be quantitated by flow cytometry. (D) The impact of LIGIII deficiency on end joining. A LIGIV-null cell line was used as a negative control. The plotted values are the average of three independent experiments.
When the pEGFP-Pem1-Ad2 plasmid was transfected into the parental wild-type cell line, approximately 35% of the plasmid was productively end joined regardless of whether it had been digested by either HindIII or I-SceI (Fig. 3D). As expected, LIGIV-null cells were profoundly impaired in this DNA EJing activity and showed only a few percent of activity. In contrast, LIGIIIflox/− and LIGIII−/−:mL3 cell lines had DNA EJing activities indistinguishable from wild-type cells (Fig. 3D). Thus, partial or complete deficiencies in nuclear LIGIII did not appear to affect the overall DNA EJing activity of human somatic cells.
3.7. Microhomology-mediated EJing is still detectable in human somatic cells lacking nuclear LIGIII expression
Even though the overall DNA EJing activity was unchanged in LIGIII-null cells, it was still possible that distinct DNA repair pathways were being used. To examine this possibility, we next quantitated microhomology-mediated EJing. To this end, a reporter substrate, pDVG94 that is biased towards detecting microhomology-mediated EJing events [35, 43, 49, 50] was used in vivo to measure this activity of LIGIII-deficient cells. EcoRV and AfeI digestion of pDVG94 generates a blunt-ended, linear, double-stranded substrate with 6 bp direct repeats at both ends (Fig. 4A). Repair of this substrate by C-NHEJ generally generates a product that retains at least some of either repeat, whereas microhomology-mediated EJing (i.e., A-NHEJ) produces a unique product that contains only a single repeat, and which now forms the recognition sequence for the BstXI restriction enzyme. The linearized pDVG94 plasmid was introduced into the relevant cell lines and 24 hr later the DNA was recovered from the cells and repaired junctions were amplified by PCR with radiolabeled primers (Fig. 4A). The resulting ~180 bp PCR products were then digested with BstXI and subjected to agarose gel electrophoresis. BstXI-resistant DNA corresponds to C-NHEJ-mediated repair events whereas a 120 bp product is diagnostic of A-NHEJ/microhomology-mediated EJing. Wild-type cells generated only ~1% of the 120 bp product (Fig. 4B), consistent with most of the EJing in human somatic cells resulting from C-NHEJ [35, 43]. Similarly, and again as expected [35, 43], a LIGIV-null cell line showed highly elevated levels of the 120 bp product indicative of a virtually exclusive reliance on A-NHEJ (Fig. 4B). In contrast, in either the LIGIIIflox/− or two independent LIGIII−/−:mL3 cell lines (clones #29 and #35) the amount of the 120 bp product was either slightly elevated or unchanged in comparison to wild-type cells (Fig. 4B), indicating that LIGIII-deficient cells were still able to carry out microhomology-mediated EJing.
Fig. 4.
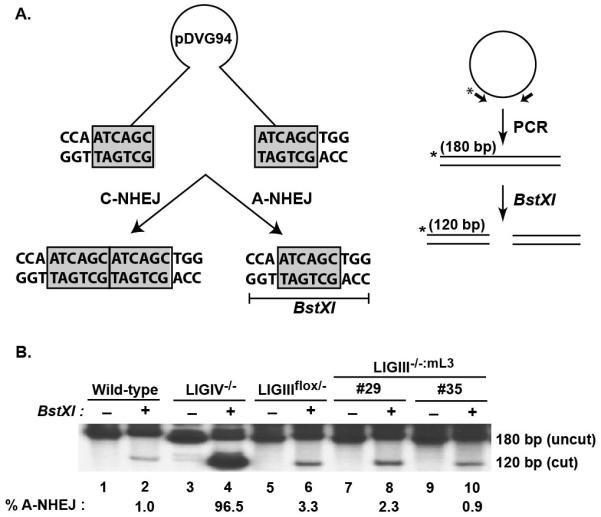
LIGIII-null cells have normal microhomology-mediated end-joining activity. (A) After EcoRV and AfeI restriction enzyme digestion, the reporter substrate, pDVG94, becomes a blunt-ended linear plasmid with 6 bp direct repeats at both ends. Repair via C-NHEJ generally retains at least part of either repeat, whereas A-NHEJ generates only a single repeat and a novel restriction enzyme site that can be cleaved by BstXI. Repaired junctions were amplified by PCR using radiolabeled primers and the 180 bp PCR product was subjected to BstXI restriction enzyme digestion. The 180 bp uncut product represents repair via C-NHEJ whereas the 120 bp digested product represents A-NHEJ-mediated repair. (B) Both LIGIII heterozygous (LIGIIIflox/−) and LIGIII-null (LIGIII−/−:mL3) cells have similar microhomology end-joining activity to the wild-type control. A LIGIV−/− cell line was used as a positive control.
3.8. A LIGIII deficiency does not affect the overall rAAV-mediated gene-targeting rate
AAV infections in humans are apathogenic and this feature has made rAAV-mediated gene targeting technology one of the more promising candidates for therapeutic use [33]. The utility of rAAV, however, is offset by random viral integration events, which are not desired and potentially mutagenic. Thus, increasing the overall correct gene targeting frequency is one of the most sought-after advances for rAAV-mediated gene targeting technology and for gene therapy in general. Previous studies from our laboratory have indicated that neither HR nor C-NHEJ was responsible for rAAV random integration events [34, 35]. In addition, sequencing results from other laboratories had indicated that rAAV random integration events could be mediated by microhomology usage [51, 52]. Together, these observations led to the hypothesis that rAAV random integrations are mediated by A-NHEJ, and to the prediction that disruption of A-NHEJ (e.g., by functionally inactivating LIGIII) would ablate random integrations and thus improve the correct rAAV-mediated gene-targeting rate.
To test this hypothesis, we performed gene targeting in a LIGIII-null cell line at the HPRT (hypoxanthine phosphoribosyl transferase) locus using a rAAV gene targeting vector designed to disrupt exon 3 of HPRT. HPRT is an X-chromosome linked gene and the enzyme encoded by HPRT is needed to generate nucleotides through the purine salvage pathway [53]. Cells expressing a wild-type HPRT gene are lethally poisoned by the toxic nucleoside analog 6-TG (6-thioguanine), whereas cells with a defective HPRT gene can survive in the presence of 6-TG. Because HCT116 was derived from a male patient, it contains only a single X-chromosome and therefore after a single round of gene targeting, 6-TG selection could be used to isolate correctly targeted clones. Interestingly, LIGIII-null cells showed a 2-fold increase in the frequency of correct targeting (Fig. 5B). Unexpectedly, however, the frequency of random integration events was not reduced in LIGIII-null cells, but was also actually enhanced (Fig. 5A). Consequently, there was no statistical difference in the relative gene-targeting rate between the wild type and LIGIII-null cell lines (Fig. 5C). These data demonstrated (quite unexpectedly) that while LIGIII is a general suppressor of rAAV integrations, it does not preferentially affect random versus correct targeting events.
Fig. 5.
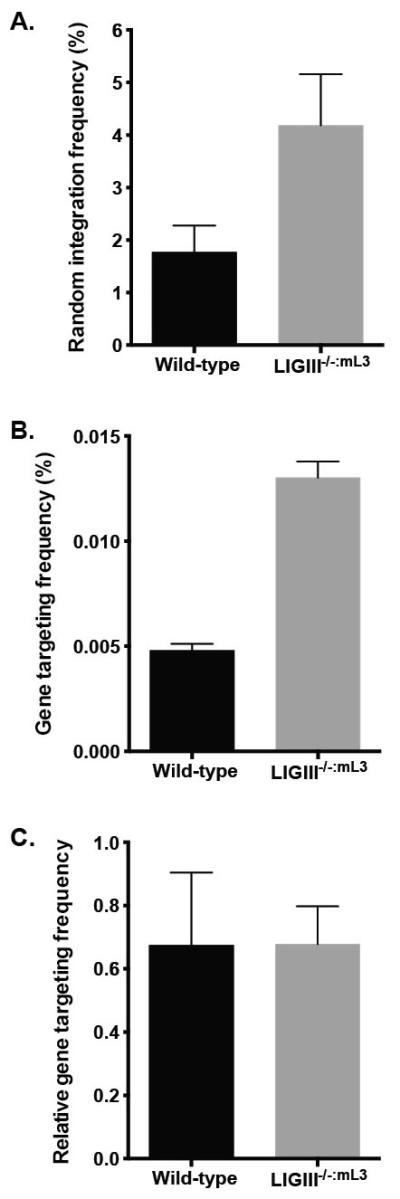
A LIGIII deficiency does not affect the relative frequency of rAAV-mediated gene targeting at the HPRT locus. (A) rAAV random integration frequency. The random integration frequency was determined as the number of puromycin-resistant clones normalized by plating efficiency. (B) rAAV gene targeting frequency. The gene targeting frequency was determined as the number of both puromycin and 6-thioguanine-resistant clones normalized by plating efficiency. (C) The relative frequency of rAAV gene targeting represents the ratio of the correct targeting events versus the total viral integration events. LIGIII-null cells showed both higher random integration and gene targeting frequencies and hence overall there was no difference in relative gene targeting frequency.
3.9. The absence of Ku reveals a requirement for LIGIII
The above experiments demonstrated that while the absence of LIGIII resulted in a slower growth phenotype, it did not i) make the cells hypersensitive to DNA damaging agents, ii) manifest itself in any detectable DNA DSB repair deficiency nor iii) deleteriously impact on the process of gene targeting. Our laboratory [34] and others [44, 54-56] have shown that cells reduced or deficient for the Ku heterodimer show a marked increase in the activity of A-NHEJ. We therefore reasoned that if LIGIII is mediating A-NHEJ repair reactions that are too rare to detect in an otherwise wild-type cell, then there might be an observable effect of the absence of nuclear LIGIII in a Ku-null cell. To test this hypothesis, we constructed a Ku86flox/−:LIGIIImito/− doubly mutant human cell line. This was achieved by “knocking-in” ATG > ATC mutations into one endogenous LIGIII allele, and knocking out the second LIGIII allele by rAAV-mediated gene targeting in a conditionally-null Ku86 cell line (Fig. S3A). After also engineering in the above-mentioned tamoxifen-inducible Cre recombination system, we confirmed by immunofluorescence (Fig. S3B) and PCR (Fig. S3C) that the Ku86flox/−:LIGIIImito/− cells only expressed mitochondrial LIGIII and were conditionally-null for Ku86, respectively. Furthermore, in order to directly compare LIGIII to another DNA ligase in this context, we also created a Ku86flox/−:LIGIV−/− Cre-inducible cell line (Fig. S3C). Although both doubly mutant cell lines eventually die due to Ku-regulated telomere defects [41], they do survive 3 to 7 days post Cre treatment, which was a sufficient time window to assay the levels of DNA repair in the cells.
Consequently, we performed a microhomology assay using the aforementioned pDVG94 reporter plasmid (Fig. 6A). We confirmed our previously published finding [43] that in the absence of the Ku, there is a large (~20-fold) increase in the relative amount of A-NHEJ repair (Fig. 6B). Importantly, the absence of nuclear LIGIII in the Ku86-deficient cells completely abrogated this increased A-NHEJ (compare Ku86−/− with Ku86−/−:LIGIIImito/−; Fig. 6B). Notably, although the absence of LIGIV by itself resulted in exceedingly few total repair events (Fig. 3D), those that did occur were almost exclusively biased towards microhomology and the presence or absence of Ku in the cells had no discernible effect on this bias (compare LIGIV−/− with Ku86−/−:LIGIV−/−; Fig. 6B). Interestingly, the converse was not true. Thus, the absence of LIGIV in the cells always shifted the repair bias towards A-NHEJ regardless of the status of Ku expression (compare Ku86−/− with Ku86−/−:LIGIV−/−; Fig. 6B).
Fig. 6.

LIGIII is required for microhomology-mediated A-NHEJ events normally repressed by Ku. (A) The reporter substrate, pDVG94, described in Figure 5 was used with the indicated cell lines. The 180 bp uncut product represents repair via C-NHEJ whereas the 120 bp digested product represents A-NHEJ-mediated repair. Note that the enhanced A-NHEJ repair observed in a Ku86−/− cell line was completely abrogated in a Ku86−/−:LIGIIImito/− cell line. (B) Four independent assays comparable to that shown in panel (A) were averaged and the error bars represent the standard error of the mean.
The pDVG94 microhomology assay measures relative A-NHEJ activity. To determine the impact of ligation on absolute levels of A-NHEJ, we performed an additional analysis with the aforementioned cell lines using the pEJ2 reporter plasmid. This plasmid contains an I-SceI restriction enzyme site flanked by 8 bp of microhomology situated between a promoter and GFP coding sequences {Fig. 7A; [44]}. In addition, stop codons have been engineered in all three reading frames to disrupt the translation that starts at an N-terminal epitope tag. Thus, this plasmid can be linearized by I-SceI restriction enzyme digestion and transfected into cells. Microhomology-mediated repair removes the stop codons and allows GFP expression, whereas repair by C-NHEJ does not. Consequently, cells were transfected with I-SceI-digested pEJ2 plasmid and mCherry (as a transfection control) and assayed for A-NHEJ activity. First, it was obvious that in all the cell lines that A-NHEJ is a rare event (Fig. 7B), consistent with our other analyses. Nonetheless, the absence of Ku caused a 4-fold increase in GFP-positive cells and almost all of the increase could be abrogated by the absence of LIGIII, but not LIGIV (Fig. 7C).
Fig. 7.

LIGIII, and not LIGIV, is required for microhomology-mediated EJing events normally repressed by Ku. (A) A diagram of the reporter substrate, pEJ2. The gray rectangle with the arrow represents a transcriptional promoter. The brown rectangle (tag) represents and in-frame epitope tag. The orange rectangle is expanded below to show an I-SceI restriction enzyme recognition site and the red rectangle represents translational stops in all 3 reading frames (stop). These elements are flanked by 8 bp of a direct repeat (CAAGCCCG) that permits microhomology-mediated repair. The green rectangle (GFP) represents the coding sequences for green fluorescent protein. Repair of the I-SceI-linearized pEJ2 by C-NHEJ retains the stop cassette and does not permit GFP expression whereas repair by A-NHEJ deletes out the stop element and allows GFP expression. (B) The indicated cell lines were transfected with the I-SceI-linearized pEJ2 reporter and then either left untreated (-Cre) or treated with 4-OHT to induce Cre (+Cre) expression. (C) Four independent experiments similar to the one shown in panel (B) were averaged and presented as the fold change relative to wild type.
In toto, these data clearly demonstrated a separation of function between the two ligases and strongly suggested that nuclear LIGIII does play a significant role in A-NHEJ, but only in the absence of Ku.
3.10. In the absence of Ku, most EJing remains LIGIV-dependent
All of the above data were consistent with an old model in which the vast majority of EJing events in human somatic cells are Ku- and LIGIV-dependent and that only a minority of repair events are normally repaired through a Ku-suppressible, LIGIII-dependent pathway (Fig. 8A). This model predicts that in a Ku-deficient human cell, the total EJing events should be sensitive to the status of LIGIII and unaffected by the presence in LIGIV. To test this prediction, we quantitated the total amount of EJing in our cell lines using the pEGFP-Pem1-Ad2 reporter plasmid (Fig. 9A). The Ku86−/−, LIGIII−/−:mL3 and Ku86−/−:LIGIIImito/− cell lines had virtually indistinguishable robust total EJing activity whereas a Ku86−/−:LIGIV−/− cell line was extremely deficient (Fig. 9B). Thus, very unexpectedly, in a Ku-deficient human cell line, the vast majority of EJing events remain LIGIV-dependent.
Fig. 8.
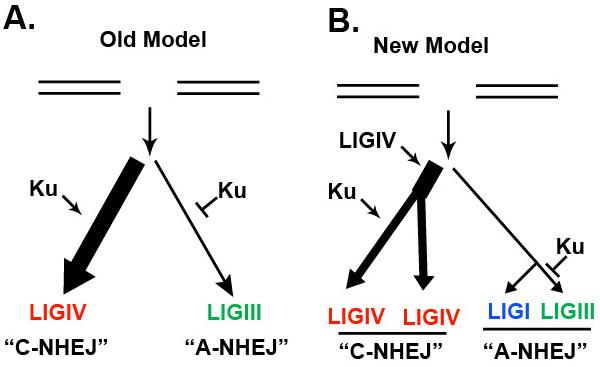
A new model for EJing pathways in human somatic cells (A) The old model for EJing. A DSB could be repaired by one of two pathways. The first and most active was the Ku-dependent, LIGIV-dependent C-NHEJ pathway and the second was the minor Ku-suppressible, LIGIII-dependent A-NHEJ pathway. (B) A proposed new model posits that all DSB substrates are first subjected to a LIGIV-mediated rejoining event, which is relatively successful. Only when this rejoining event fails is Ku recruited for facilitating additional processing events. LIGIV is, however, subsequently re-recruited to finish these repair events. Thus, C-NHEJ is actually composed of 2 separate pathways, one of which is Ku dependent and one of which is not. Both pathways are, however, dependent upon LIGIV. Moreover, the A-NHEJ pathway also bifurcates and can either be mediated by LIGI or LIGIII and only the later of which is Ku-suppressible.
Fig. 9.

Ku-independent EJing is LIGIV-dependent. (A) Profiles of EJing assays using the reporter pEGFP-Pem1-Ad2 described in Figure 4 transfected into the indicated cell lines. (B) Four independent assays comparable to that shown in panel (A) were averaged and the error bars represent the standard error of the mean. (C) Repaired plasmids were recovered from the indicated cell lines, propagated thorough bacteria and then either left untreated (-HindIII) or subjected to restriction enzyme digestion (+HindIII) and then analyzed by agarose gel electrophoresis. The appearance of a diagnostic doublet following restriction enzyme digestion indicates that perfect rejoining occurred. All of the gels show a marker lane on the left followed by 4 lanes containing independently isolated plasmids. (D) 18 to 40 independent plasmids similar to the four shown in panel (C) were quantitated for each of the indicated cell lines and scored for the occurrence of perfect joins.
To extend these findings, we recovered the repaired pEGFP-Pem1-Ad2 plasmids out of cells 24 hr post transfection. These plasmids were analyzed by quantifying the plasmids that had simply religated their HindIII-linearized ends {“perfect rejoins”; [43]}. The repaired plasmids recovered from cells were amplified by colony PCR, and the products were then re-digested with HindIII and analyzed by gel electrophoresis (Fig. 9C). As we had previously observed [43], the absence of Ku increased the frequency of perfect rejoins (Fig. 9D), however the absence of LIGIII had no impact on this process (compare Ku86−/− with Ku86−/−:LIGIIImito/−; Fig. 9D). Interestingly, Ku86−/−:LIGIV−/− (and LIGIV−/−) cells carried out almost exclusively perfect rejoining, implicating therefore, by default, LIGI in this process [57].
Altogether, these data demonstrated that LIGIV is still required for most of the Ku-independent EJing events in human cells. Moreover, they demonstrate that LIGIII is required for much of the Ku-suppressible A-NHEJ activity. And finally, the data suggest that at least some of the perfect rejoining and A-NHEJ observed in either wild type or Ku-deficient cells can be mediated by an additional pathway, which we infer perforce to be LIGI-mediated [57].
4. Discussion
4.1. LIGIII has an essential mitochondrial function
We have generated a viable human somatic cell line that lacks the expression of nuclear LIGIII. Somewhat paradoxically, we nonetheless demonstrate that LIGIII is an essential gene. Thus, no living null cells could be isolated after an AdCre infection of the LIGIIIflox/− cell line. In contrast, after complementing the LIGIIIflox/− cell line with a mitochondrial-specific cDNA isoform of LIGIII, we readily isolated viable LIGIII-null (LIGIII−/−:mL3) cells. Therefore, LIGIII is dispensable in the nucleus of human cells but essential in mitochondria.
A similar conclusion that LIGIII is essential for mitochondrial function was reached using the chicken cell line, DT40 [38], and in the mouse [30, 32, 39]. What the essential activity of mitochondrial LIGIII is, however, is unclear. One obvious function would be a requirement for LIGIII in Okazaki fragment maturation during mitochondrial DNA replication. LIGI mediates Okazaki fragment maturation in the nucleus [58, 59], but since LIGI lacks a MLS and it is not detected in mitochondria, it is clear that some other ligase must perform this function in mitochondria [38, 60]. Similarly, since LIGIV also lacks a MLS, is non-essential, and appears to be involved exclusively in C-NHEJ, it is also a poor candidate [35]. In contrast, the mitochondrial-specific isoform of LIGIII should be able to mediate the ligation of Okazkai fragments, which are very similar to the repair intermediates that LIGIII is known to ligate together during BER [19, 61].
4.2. rAAV random integrations are not mediated solely by LIGIII/A-NHEJ
Correct gene targeting events require HR, and the (much more frequent) random integrations of the targeting vector are presumably facilitated by DNA EJing pathways (i.e., C-NHEJ, A-NHEJ or both). Indeed, large-scale whole-genome sequencing results demonstrated that rAAV random integration events were mediated by some form of NHEJ [51, 52]. Reductions, however, in the expression of Ku did not significantly impact the frequency of random integrations of rAAV vectors during gene targeting [34]. Similar observations have been made for LIGIV-deficient cells [35] suggesting strongly that C-NHEJ is not required for this process. Together, these observations led to the hypothesis that rAAV random integrations are instead mediated by A-NHEJ, and to the prediction that disruption of A-NHEJ (e.g., by functionally inactivating LIGIII) would significantly improve the correct rAAV-mediated gene-targeting rate.
Surprisingly, no such effect was observed (Fig. 5) and the random rAAV integration frequency was actually (~3-fold) elevated, demonstrating that LIGIII could even be viewed as a suppressor of these events. A parsimonious explanation for these results is that if random rAAV integrations are neither mediated by LIGIV nor LIGIII then they may be mediated by LIGI (see also Fig. 8B). Although LIGI is predominately thought of as a DNA replication-linked ligase, it is enzymatically capable of resealing the staggered ends of the DSBs generated during random rAAV integration [58]. Unfortunately, LIGI is an essential gene in the HCT116 cell line (unpublished data), which precludes gene targeting experiments and a direct test of the hypothesis.
Alternatively, we propose that in the absence of any one particular ligase that rAAV integration is performed by the remaining ligases. There is precedent from other cellular activities for the mammalian ligases being redundant. This is especially true of LIGI and LIGIII. Thus, viable murine LIGI-null MEFs actually proliferate rather normally, suggesting that some other ligase must be compensating for LIGI during DNA replication [60, 62, 63]. Similarly, the co-depletion of LIGI in murine LIGIII-knockout cells sensitized the cells to MMS exposure, an activity previously thought to be solely the purview of LIGIII [30]. Perhaps the most compelling case for genetic redundancy comes from work in the chicken DT40 cell line, where single, double and or triple mutants were generated to establish that LIGI and LIGIII are functionally redundant for viability [38]. A similar conclusion that LIGI and LIGIII are functionally redundant for in vitro end joining in human extracts was obtained by reducing the levels of each, or both, protein(s) using RNA interference or neutralizing antibodies [64]. If this redundancy between LIGI and LIGIII extends to rAAV random integration it provides a plausible explanation for the lack of a compelling phenotype for this process in the LIGIII-null human cells.
4.3. LIGIII is required for A-NHEJ events normally suppressed by Ku
In order to generate a 120 bp BstXI-dependent restriction product from the pDVG94 plasmid a unique repair event is required: both ends of the blunt-ended plasmid must be resected and the 6 nt long complementary strands must be precisely annealed and ligated. This repair product can be detected only at low levels (a few percent of the total) in wild-type cells and has been widely interpreted as being produced by A-NHEJ [43, 49]. Moreover, when cells are mutated for any of the canonical C-NHEJ genes, the 120 bp fragment becomes virtually the sole repair product {Fig. 4 and 6; [43]}. All of these observations led to the prediction that the ablation of A-NHEJ should block the formation of this repair product. However, the appearance of the 120 bp product was completely unaffected by the absence of nuclear LIGIII (Fig. 4 and 6). A trivial explanation is that the cell line is leaky and expresses some LIGIII in the nucleus. While we cannot unequivocally rule out this possibility, we do not believe that this is a likely explanation. First, we mutated not only the normally-used ATG for the nuclear form of LIGIII, but a downstream ATG as well to ensure that not even truncated forms of the protein would be expressed. In addition, a mitochondrial-exclusive expression pattern of the mL3 construct was confirmed by immunocytochemistry (Fig. S2C and S3B). Still if mL3 protein did leak into nucleus, even at undetectable levels, this would likely have functional relevance since even low levels of LIGIII are sufficient for effective A-NHEJ [65]. Finally, we also note that all of our repair data was generated using episomal reporter vectors that may not always faithfully represent endogenous DNA repair events, which are, perforce, chromosomal in nature [66].
An alternative attractive possibility is that LIGIII is redundant with LIGI in the A-NHEJ pathway [67]. This hypothesis, which we elaborated above as a possible explanation for the absence of an anticipated effect on gene targeting, would explain the continued production of the 120 bp product even in the absence of LIGIII. In almost every experimental situation where the expression of both LIGI and LIGIII has been reduced, the resultant repair activity is significantly less than when either ligase is individually reduced [30, 38, 64]. Thus, our data suggests that there are redundant LIGI- and LIGIII-dependent forms of A-NHEJ, although only the LIGIII-branch appears to be suppressible by Ku (see also Fig. 8B).
It is unclear what biological function this Ku-suppressible, LIGIII-dependent A-NHEJ activity serves. To our knowledge, the only nuclear DSB “activity” that can currently be ascribed to LIGIII in C-NHEJ-proficient cells is the ability to facilitate chromosomal translocations [31]; an activity that seems untenable as an evolutionary explanation for the presence of LIGIII. We note, however, that although nuclear LIGIII-null cells exhibit no obvious DSB repair phenotypes per se, they do exhibit a growth defect, implying that the absence of LIGIII is deleterious for the normal progression of cell division. However, this requirement for LIGIII could simply be related to its role in BER, an activity likely needed for efficacious cell cycle progression [68]. In addition, we propose that there may be transient periods in the cell cycle when Ku’s activity is suppressed, and where LIGIII-dependent A-NHEJ can be engaged in a beneficial manner. An obvious time for this would be during S-phase. Thus, cells normally require HR to accurately re-start/repair stalled/broken replication forks [69], and the repression of C-NHEJ via the suppression of Ku is likely instrumental in this process. The DNA ligase required for these HR transactions is unknown but our data is consistent with the recent suggestion that LIGIII may be required for at least Fanconi anemia pathway-mediated HR processes [70]. Whether LIGIII-dependent A-NHEJ activities could also be utilized in this situation is clearly a question that deserves additional investigation.
4.4. A new model for EJing in human somatic cells
Previous studies of EJing in human cells had postulated a model of DSB repair thati consisted of a predominant Ku-dependent, LIGIV-dependent C-NHEJ pathway and a minor Ku-suppressible, LIGIII-dependent pathway {Fig. 8A; [4, 43, 44]}. Here we demonstrate that this model is, at best, over-simplified. Thus, our data show that there is a constant low level of A-NHEJ in human cells that is unaffected by the presence or absence of LIGIII (Fig. 4 and 7). We ascribe this activity to LIGI and propose that the LIGI-dependent branch of A-NHEJ is separable from the LIGIII-dependent branch (Fig. 8B). Moreover, we demonstrate that the LIGIII-dependent branch of A-NHEJ is uniquely Ku-suppressible (Fig. 6 and 8B). Most interestingly, however, this work has demonstrated that there is a large fraction of EJing events that are Ku-independent, but nonetheless LIGIV-dependent (Fig. 9). In a previous study we had demonstrated a robust EJing activity in Ku-deficient human somatic cells and had inferred that this activity was likely due to LIGIII-dependent A-NHEJ [43]. However, the experiments presented here show that little of this Ku-independent activity is actually LIGIII-dependent, but is instead LIGIV-dependent. Interestingly, but in retrospect not unexpectedly, this Ku-independent, LIGIV-dependent activity is capable of performing sticky-ended ligations at very high frequency {Fig. 9C; [43]}. Thus, we propose that there may a hierarchical order to C-NHEJ in which direct ligation of repair events is first attempted and only if that fails is Ku recruited to enable more complicated downstream processes such as end resection and polymerization before ligation ensues (Fig. 8B). Our postulated model is consistent with the recent demonstration that XRCC4 and XLF can facilitate the bridging of DNA ends for subsequent LIGIV-dependent ligation [71].
In summary, these studies have revealed that human LIGIII is an essential gene due to its requirement in mitochondrial function. Moreover, we demonstrate that while nuclear LIGIII is not required for random gene targeting integrations, it is required for mediating the enhanced A-NHEJ repair events observed in the absence of Ku. Finally, we have identified a novel EJing activity observed in the absence of Ku that is LIGIV-dependent. Altogether, these observations enhance our understanding of EJing in human somatic cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights of this study.
-
➢
Construction of the first human nuclear LIGIII-deficient cell line
-
➢
Demonstration that the absence of LIGIII confers no significant DNA repair or gene targeting defect to otherwise normal cells
-
➢
Demonstration that LIGIII plays a role in A-NHEJ only in the absence of Ku
-
➢
Demonstration that the majority of DSB repair remains LIGIV-dependent in a Ku-deficient cell line
-
➢
A new model for the mechanism of NHEJ pathways in human cells
ACKNOWLEDGMENTS
The authors thank Dr. Anja-Katrin Bielinsky and members of the Hendrickson laboratory for their critical comments on the manuscript.
FUNDING
This work was supported by grants from the National Institutes of Health {GM088351}, the National Cancer Institute {CA154461} and by a research contract provided by Horizon Discovery, Ltd.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement. E.A.H. declares that he is a member of the scientific advisory board of Horizon Discovery, Ltd., a company that specializes in rAAV-mediated gene targeting technology and that his laboratory was funded, in part, through a research contract from the same company.
References
- [1].Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584:3703–3708. doi: 10.1016/j.febslet.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- [5].Yano K, Morotomi-Yano K, Lee KJ, Chen DJ. Functional significance of the interaction with Ku in DNA double-strand break recognition of XLF. FEBS Lett. 2011;585:841–846. doi: 10.1016/j.febslet.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Andres SN, Vergnes A, Ristic D, Wyman C, Modesti M, Junop M. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012;40:1868–1878. doi: 10.1093/nar/gks022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Roy S, Andres SN, Vergnes A, Neal JA, Xu Y, Yu Y, Lees-Miller SP, Junop M, Modesti M, Meek K. XRCC4’s interaction with XLF is required for coding (but not signal) end joining. Nucleic Acids Res. 2012;40:1684–1694. doi: 10.1093/nar/gkr1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cottarel J, Frit P, Bombarde O, Salles B, Negrel A, Bernard S, Jeggo PA, Lieber MR, Modesti M, Calsou P. A noncatalytic function of the ligation complex during nonhomologous end joining. J Cell Biol. 2013;200:173–186. doi: 10.1083/jcb.201203128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res. 2011;711:61–72. doi: 10.1016/j.mrfmmm.2011.02.005. [DOI] [PubMed] [Google Scholar]
- [11].Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131:223–225. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- [12].Simsek D, Jasin M. DNA ligase III: a spotty presence in eukaryotes, but an essential function where tested. Cell Cycle. 2011;10:3636–3644. doi: 10.4161/cc.10.21.18094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zha S, Boboila C, Alt FW. Mre11: roles in DNA repair beyond homologous recombination. Nat Struct Mol Biol. 2009;16:798–800. doi: 10.1038/nsmb0809-798. [DOI] [PubMed] [Google Scholar]
- [15].Gottlich B, Reichenberger S, Feldmann E, Pfeiffer P. Rejoining of DNA double-strand breaks in vitro by single-strand annealing. Eur J Biochem. 1998;258:387–395. doi: 10.1046/j.1432-1327.1998.2580387.x. [DOI] [PubMed] [Google Scholar]
- [16].Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, Iliakis G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–4030. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- [17].Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- [18].Cheng Q, Barboule N, Frit P, Gomez D, Bombarde O, Couderc B, Ren GS, Salles B, Calsou P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011;39:9605–9619. doi: 10.1093/nar/gkr656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- [21].Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhuang J, Jiang G, Willers H, Xia F. Exonuclease function of human Mre11 promotes deletional nonhomologous end joining. J Biol Chem. 2009;284:30565–30573. doi: 10.1074/jbc.M109.059444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].You Z, Bailis JM. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010;20:402–409. doi: 10.1016/j.tcb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. 2011;18:80–84. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bentley J, Diggle CP, Harnden P, Knowles MA, Kiltie AE. DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res. 2004;32:5249–5259. doi: 10.1093/nar/gkh842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mattarucchi E, Guerini V, Rambaldi A, Campiotti L, Venco A, Pasquali F, Lo Curto F, Porta G. Microhomologies and interspersed repeat elements at genomic breakpoints in chronic myeloid leukemia. Genes Chromosomes Cancer. 2008;47:625–632. doi: 10.1002/gcc.20568. [DOI] [PubMed] [Google Scholar]
- [27].Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013;13:443–454. doi: 10.1038/nrc3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–416. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, McKinnon PJ. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature. 2011;471:240–244. doi: 10.1038/nature09773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ, Gregory PD, Holmes MC, Jasin M. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011;7:e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Simsek D, Furda A, Gao Y, Artus J, Brunet E, Hadjantonakis AK, Van Houten B, Shuman S, McKinnon PJ, Jasin M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature. 2011;471:245–248. doi: 10.1038/nature09794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Khan IF, Hirata RK, Russell DW. AAV-mediated gene targeting methods for human cells. Nat Protoc. 2011;6:482–501. doi: 10.1038/nprot.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fattah FJ, Lichter NF, Fattah KR, Oh S, Hendrickson EA. Ku70, an essential gene, modulates the frequency of rAAV-mediated gene targeting in human somatic cells. Proc Natl Acad Sci U S A. 2008;105:8703–8708. doi: 10.1073/pnas.0712060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Oh S, Wang Y, Zimbric J, Hendrickson EA. Human LIGIV is synthetically lethal with the loss of Rad54B-dependent recombination and is required for certain chromosome fusion events induced by telomere dysfunction. Nucleic Acids Res. 2013;41:1734–1749. doi: 10.1093/nar/gks1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lakshmipathy U, Campbell C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol Cell Biol. 1999;19:3869–3876. doi: 10.1128/mcb.19.5.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Perez-Jannotti RM, Klein SM, Bogenhagen DF. Two forms of mitochondrial DNA ligase III are produced in Xenopus laevis oocytes. J Biol Chem. 2001;276:48978–48987. doi: 10.1074/jbc.M107177200. [DOI] [PubMed] [Google Scholar]
- [38].Arakawa H, Bednar T, Wang M, Paul K, Mladenov E, Bencsik-Theilen AA, Iliakis G. Functional redundancy between DNA ligases I and III in DNA replication in vertebrate cells. Nucleic Acids Res. 2012;40:2599–2610. doi: 10.1093/nar/gkr1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Puebla-Osorio N, Lacey DB, Alt FW, Zhu C. Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol Cell Biol. 2006;26:3935–3941. doi: 10.1128/MCB.26.10.3935-3941.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang Y, Ghosh G, Hendrickson EA. Ku86 represses lethal telomere deletion events in human somatic cells. Proc Natl Acad Sci U S A. 2009;106:12430–12435. doi: 10.1073/pnas.0903362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Doherty JE, Huye LE, Yusa K, Zhou L, Craig NL, Wilson MH. Hyperactive piggyBac gene transfer in human cells and in vivo. Hum Gene Ther. 2012;23:311–320. doi: 10.1089/hum.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fattah F, Lee EH, Weisensel N, Wang Y, Lichter N, Hendrickson EA. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ruhanen H, Ushakov K, Yasukawa T. Involvement of DNA ligase III and ribonuclease H1 in mitochondrial DNA replication in cultured human cells. Biochim Biophys Acta. 2011;1813:2000–2007. doi: 10.1016/j.bbamcr.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Caldecott KW. Mammalian single-strand break repair: mechanisms and links with chromatin. DNA Repair (Amst) 2007;6:443–453. doi: 10.1016/j.dnarep.2006.10.006. [DOI] [PubMed] [Google Scholar]
- [47].Pacchierotti F, Ranaldi R. Mechanisms and risk of chemically induced aneuploidy in mammalian germ cells. Curr Pharm Des. 2006;12:1489–1504. doi: 10.2174/138161206776389859. [DOI] [PubMed] [Google Scholar]
- [48].Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc Natl Acad Sci U S A. 2004;101:7624–7629. doi: 10.1073/pnas.0400726101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Verkaik NS, Esveldt-van Lange RE, van Heemst D, Bruggenwirth HT, Hoeijmakers JH, Zdzienicka MZ, van Gent DC. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur J Immunol. 2002;32:701–709. doi: 10.1002/1521-4141(200203)32:3<701::AID-IMMU701>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- [50].Weterings E, Verkaik NS, Keijzers G, Florea BI, Wang SY, Ortega LG, Uematsu N, Chen DJ, van Gent DC. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol Cell Biol. 2009;29:1134–1142. doi: 10.1128/MCB.00971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Miller DG, Trobridge GD, Petek LM, Jacobs MA, Kaul R, Russell DW. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J Virol. 2005;79:11434–11442. doi: 10.1128/JVI.79.17.11434-11442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nakai H, Wu X, Fuess S, Storm TA, Munroe D, Montini E, Burgess SM, Grompe M, Kay MA. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J Virol. 2005;79:3606–3614. doi: 10.1128/JVI.79.6.3606-3614.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hladnik U, Nyhan WL, Bertelli M. Variable expression of HPRT deficiency in 5 members of a family with the same mutation. Arch Neurol. 2008;65:1240–1243. doi: 10.1001/archneur.65.9.1240. [DOI] [PubMed] [Google Scholar]
- [54].Guirouilh-Barbat J, Rass E, Plo I, Bertrand P, Lopez BS. Defects in XRCC4 and KU80 differentially affect the joining of distal nonhomologous ends. Proc Natl Acad Sci U S A. 2007;104:20902–20907. doi: 10.1073/pnas.0708541104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mansour WY, Schumacher S, Rosskopf R, Rhein T, Schmidt-Petersen F, Gatzemeier F, Haag F, Borgmann K, Willers H, Dahm-Daphi J. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res. 2008;36:4088–4098. doi: 10.1093/nar/gkn347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schulte-Uentrop L, El-Awady RA, Schliecker L, Willers H, Dahm-Daphi J. Distinct roles of XRCC4 and Ku80 in non-homologous end-joining of endonuclease- and ionizing radiation-induced DNA double-strand breaks. Nucleic Acids Res. 2008;36:2561–2569. doi: 10.1093/nar/gkn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Paul K, Wang M, Mladenov E, Bencsik-Theilen A, Bednar T, Wu W, Arakawa H, Iliakis G. DNA ligases I and III cooperate in alternative non-homologous end-joining in vertebrates. PloS one. 2013;8:e59505. doi: 10.1371/journal.pone.0059505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zheng L, Shen B. Okazaki fragment maturation: nucleases take centre stage. J Mol Cell Biol. 2011;3:23–30. doi: 10.1093/jmcb/mjq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Le Chalony C, Hoffschir F, Gauthier LR, Gross J, Biard DS, Boussin FD, Pennaneach V. Partial complementation of a DNA ligase I deficiency by DNA ligase III and its impact on cell survival and telomere stability in mammalian cells. Cell Mol Life Sci. 2012;69:2933–2949. doi: 10.1007/s00018-012-0975-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- [62].Bentley D, Selfridge J, Millar JK, Samuel K, Hole N, Ansell JD, Melton DW. DNA ligase I is required for fetal liver erythropoiesis but is not essential for mammalian cell viability. Nat Genet. 1996;13:489–491. doi: 10.1038/ng0896-489. [DOI] [PubMed] [Google Scholar]
- [63].Bentley DJ, Harrison C, Ketchen AM, Redhead NJ, Samuel K, Waterfall M, Ansell JD, Melton DW. DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability. J Cell Sci. 2002;115:1551–1561. doi: 10.1242/jcs.115.7.1551. [DOI] [PubMed] [Google Scholar]
- [64].Liang L, Deng L, Nguyen SC, Zhao X, Maulion CD, Shao C, Tischfield JA. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008;36:3297–3310. doi: 10.1093/nar/gkn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Windhofer F, Wu W, Iliakis G. Low levels of DNA ligases III and IV sufficient for effective NHEJ. J Cell Physiol. 2007;213:475–483. doi: 10.1002/jcp.21120. [DOI] [PubMed] [Google Scholar]
- [66].Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7:2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Katyal S, McKinnon PJ. Disconnecting XRCC1 and DNA ligase III. Cell Cycle. 2011;10:2269–2275. doi: 10.4161/cc.10.14.16495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res. 2013;2:107–129. doi: 10.3978/j.issn.2218-676X.2013.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Huang Y, Li L. DNA crosslinking damage and cancer - a tale of friend and foe. Transl Cancer Res. 2013;2:144–154. doi: 10.3978/j.issn.2218-676X.2013.03.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mahaney BL, Hammel M, Meek K, Tainer JA, Lees-Miller SP. XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochem Cell Biol. 2013;91:31–41. doi: 10.1139/bcb-2012-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.