

Abstract
OBJECTIVE
Our goal was to evaluate the effects of simvastatin on endometrial cancer cell lines and primary cultures of endometrial cancer cells.
METHODS
Cell proliferation in the ECC-1 and Ishikawa endometrial cancer cell lines and primary cultures of endometrial cancer cells was assessed by MTT assay. Apoptosis and cell cycle were detected by Annexin V assay and propidium iodide staining, respectively. Reactive oxygen species and cell adhesion were assessed using ELISA assays. Invasion was analyzed using a transwell invasion assay. Mitochondrial DNA damage was confirmed using qPCR. The effects of simvastatin on the AKT/mTOR and MAPK pathways were determined by Western blotting.
RESULTS
Simvastatin inhibited cell proliferation in a dose-dependent manner in both endometrial cancer cell lines and 5/8 primary cultures of endometrial cancer cells. Simvastatin treatment resulted in G1 cell cycle arrest, a reduction in the enzymatic activity of HMG-CoA, induction of apoptosis as well as DNA damage and cellular stress. Treatment with simvastatin resulted in inhibition of the MAPK pathway and exhibited differential effects on the AKT/mTOR pathway in the ECC-1 and Ishikawa cells. Minimal change in AKT phosphorylation was seen in both cell lines. An increase in phosphorylated S6 was seen in ECC-1 and a decrease was seen in Ishikawa. Treatment with simvastatin reduced cell adhesion and invasion (p<0.01) in both cell lines.
CONCLUSION
Simvastatin had significant anti-proliferative and anti-metastatic effects in endometrial cancer cells, possibly through modulation of the MAPK and AKT/mTOR pathways, suggesting that statins may be a promising treatment strategy for endometrial cancer.
Keywords: endometrial cancer, statins, simvastatin, MAPK pathway, AKT/mTOR pathway, invasion
INTRODUCTION
Endometrial cancer is the fourth most common cancer among women with an estimated 49,560 new cases diagnosed in the United States in 2013 (1). The incidence of this disease has increased over the past few decades, largely as a result of the growing obesity epidemic (1). Obesity, diabetes and insulin resistance are well known risk factors for endometrial cancer and may be associated with worse outcomes for this disease (2). Women with early stage disease and low-grade endometrioid histology have a relatively good prognosis with surgery alone or surgery plus radiation. However, those patients with advanced stage III or IV endometrial cancer are unlikely to be cured by surgery, chemotherapy, radiation or a combination of these treatment modalities. Given the rising incidence of this disease and the paucity of effective treatments for advanced endometrial cancer, it is imperative to search for novel agents for the effective management of this obesity-driven cancer.
Simvastatin, one of the 3-hydoxy-3methylglutaryl coenzyme A reductase (HMGCR) competitive inhibitors, is a well-tolerated cholesterol reducing agent used to treat hypercholesterolemia. HMGCR catalyzes the rate-limiting step in mevalonate synthesis, which is essential for cellular synthesis of cholesterol and a variety of nonsteriod isoprenoid derivatives involved in cell proliferation, differentiation and survival (3, 4). In addition to its cholesterol-reducing effects, in vitro and in vivo studies suggest that simvastatin inhibits cancer cell growth by inducing apoptosis and inhibiting cell cycle progression through multiple cell signaling pathways (4–8). An association between long-term statin use and a relative reduction in the risk of cancer has been illustrated in several studies (9–11). A recent epidemiological study found that the use of statins was protective against the development of endometrial cancer and was associated with improvements in endometrial cancer survival (12). Phase II clinical trials have shown some cancer patients may benefit from simvastatin combined with other chemotherapeutic agents (13, 14).
Little is known of whether statins impact endometrial cancer cell growth. Given that endometrial cancer incidence and obesity are on the rise and simvastatin has demonstrated anti-proliferative effects in other types of cancers, the aim of this study was to investigate the effect of simvastatin on cell proliferation, apoptosis, and adhesion/invasion in endometrial cancer cell lines and primary cultures of endometrial cancer cells.
MATERIALS AND METHODS
Cell culture and reagents
The ECC-1 and Ishikawa cell lines were provided as a gift from Dr Bruce Lessey (Department of OB/GYN Greenville Memorial Hospital) (15). Both cell lines are estrogen receptor-alpha positive and progesterone receptor weakly positive, which was recently confirmed in our laboratory by chloramphenicol acetyltransferase (CAT) activity. The ECC-1 cells were maintained in RPMI 1640 containing 5% fetal bovine serum, 300 mM l-glutamine, 5 μg/ml bovine insulin, 10,000 U/ml penicillin and 10,000 μg/ml streptomycin under 5% CO2. The Ishikawa cells were grown in MEM supplemented with 5% fetal bovine serum, 300 mM l-glutamine, 10,000 U/ml penicillin and 10,000 μg/ml streptomycin under 5% CO2. Simvastatin, MTT (3-5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and RNase A were purchased from Sigma (St. Louis, MO). The anti-phosphorylated-AKT, anti-pan-AKT, anti-phosphorylated-p42/44, anti-pan-p42/44, anti-phosphorylated-S6, anti-pan-S6, anti-cleaved caspase 3, anti-BCL-2, and anti-MCL-1 antibodies were purchased from Cell Signaling (Beverly, MA). The anti-HMGCoA antibody was from Santa Cruz (Dallas, Texas). Enhanced chemiluminescence Western blotting detection reagents were purchased from Amersham (Arlington Heights, IL). All other chemicals were purchased from Sigma.
Cell proliferation assays
The ECC-1 and Ishikawa cells were plated and grown in 96-well plates at a concentration of 4000 cells/well for 24 h. Cells were subsequently treated with varying doses of simvastatin for 72 h. MTT (5 mg/ml) was added to the 96-well plates at 10 μl/well, followed by an additional hour of incubation. The MTT reaction was terminated through the addition of 100 μl of DMSO. The results were read by measuring absorption at 570 nm with a Microplate Reader (Tecan, Morrisville, NC). The effect of simvastatin was calculated as a percentage of control cell growth obtained from DMSO treated cells grown in the same 96-well plates. Each experiment was performed in triplicate to assess for consistency of results.
Apoptosis assay
Apoptosis was detected with the Annexin V FITC kit (Biolegend, San Diego, CA) on the Cellometer (Nexelom, Lawrence, MA). Briefly, 2×105 cells/well were seeded into 6-well plates, incubated overnight and then treated with simvastatin at different doses for 24 h. The cells were then collected, washed with PBS and resuspended in 100 ul binding buffer. Subsequently, 1 ul of annexin V-FITC (100 ug/ml) and 0.5 uL of propidium iodide (2 mg/ml) were added in the binding buffer and placed in the dark for 15 minutes. The samples were immediately measured by Cellometer. The results were analyzed by FCS4 express software (Molecular Devices, Sunnyvale, CA). All experiments were performed in triplicate to assess for consistency of response.
Cell cycle assay
The effects of simvastatin on cell cycle progression were measured by Cellometer. Briefly, 2.5×105 cells/well were seeded into 6-well plates, incubated overnight and then treated with simvastatin at different concentrations for 24 h. The cells were harvested and washed with phosphate buffered saline (PBS). The pellet was resuspended and fixed in 90% pre-chilled methanol and stocked overnight at −20°C. The cells were then washed with PBS again and resuspended in 50 μl RNase solution (Thermo Fisher 250 ug/mL) and 10 mM EDTA for 30 minutes. Finally, 50 μl staining solution [containing 2 mg/ml PI (Biotium, Hayward, MA), 0.1 mg/ml Azide (Sigma-Aldrich), and 0.05% Triton X-100 (Sigma-Aldrich)] was added, and the final mixture was incubated for 15 minutes in the dark before being analyzed on Cellometer. The measured results were analyzed using the FCS4 express software. Cell cycle analysis assay was performed in duplicate.
Adhesion assay
Each well in a 96-well plate was coated with 100 ul laminin-1 (10 ug/ml) and incubated at 37°C or 1 h. The fluid was then aspirated, and 200 ul blocking buffer was added to each well for 45–60 min at 37°C. The wells were washed with PBS, and the plate was allowed to chill on ice. To each well, 2.5 × 103 cells were added with PBS and varying concentrations of simvastatin directly. The plate was then allowed to incubate at 37°C for 2 h. The medium was then aspirated, and the cells were fixed by directly adding 100 ul of 5% glutaraldehyde and incubating for 30 min at room temperature. Adhered cells were then washed with PBS and stained with 100 ul of 0.1% crystal violet for 30 min. The cells were washed repeatedly with water, and 100 ul of 10% acetic acid was added to each well to solubilize the dye. After 5 minutes of shaking, the absorbance was measured at 570 nm using a microplate reader from Tecan (Mannedorf, Switzerland). All experiments were performed in duplicate to assess for consistency of response.
Invasion assay
Cell invasion assays were performed using 96-well HTS transwells (Corning Life Sciences, Wilmington, NC) coated with 0.5-1X BME (Trevigen, Gaithersburg, Maryland). Starved (serum-free media for 12 h) ECC-1 and Ishikawa cells (50,000 cells/well) were seeded for 12 h in the upper chambers of the wells in 50 μl FBS-free medium, and the lower chambers were filled with 150 μl regular medium with different concentrations of simvastatin. The plate was incubated for 24 h at 37°C to allow invasion into the lower chamber. After washing the upper and lower chambers with PBS once, 100 ul Calcein AM solution was added into the lower chamber and incubated at 37°C for 30–60 min. The lower chamber plate was measured by the plate reader for reading fluorescence at EX/EM 485/520 nM. All experiments were performed in duplicate to assess for consistency of response.
Reactive oxygen species (ROS) assay
ROS generation was assessed using the ROS-sensitive fluorescence indicator, DCFH-DA. To determine intracellular ROS scavenging activity, Ishikawa and ECC-1 cells (1.0 × 104 cells/well) were seeded in black 96-well plates. After 24 h, the cells were treated with simvastatin for 8 h to induce ROS generation. After the cells were incubated with DCFH-DA (20 μM) for 30 minutes, the fluorescence intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a fluorescence microplate reader. All experiments were performed in duplicate to assess for consistency of response.
Mitochondrial DNA damage assay
Mitochondrial DNA damage was detected using a qPCR assay previously described by Furda et. al. (16). This assay is based on the principle that many types of DNA lesions can slow or block the progression of DNA polymerase (16). Thus, if equal quantities of DNA from different samples are qPCR amplified under the same conditions, DNA with less damage will amplify to a greater extent than DNA with more damage (16). This assay is particularly sensitive in identifying ROS-mediated mitochondrial DNA damage (16). High-molecular-weight DNA was isolated using a QIAamp DNA mini kit (QIAGEN, Venlo, Limberg) following the recommended protocol. The concentration of total cellular DNA was determined by using the Tecan Nanodrop. Quantitative PCR (qPCR) assays were performed as previously described with minor modifications (16). The primers for large fragments of the mitochondrial (mt) DNA (8.9 kb) are forward 5′ - TCA AAG CCT CCT TAT TCG AGC CGA -3′, reverse 5′ – TTT CATCAT GCG GAG ATG TTG GAT GG - 3′, and primers for small mtDNA (221 bp) fragment are forward 5′ – CCC CAC AAA CCC CAT TAC TAA ACC CA -3′, reverse 5′ – TTT CATCAT GCG GAG ATG TTG GAT GG - 3′. A total volume of 50 μl was used in PCRs containing: 15 ng of template DNA, 5 pmol of each primer, 10X mix buffer and 2.5 units of recombinant Taq DNA polymerase High Fidelity (Invitrogen, Carlsbad, CA). A quantitative control using half the concentration of control template DNA was included in each set of PCR reactions. Small fragments (211 bp) of the mtDNA were also amplified for internal controls, respectively. The internal controls were used to normalize the results obtained from the large fragments and to monitor the mitochondrial copy number. The thermal cycling conditions were as follows: 95°C for 3 min, followed by 19 cycles of 94°C for 1 min, 64°C for 1 min and 68°C for 9 min, primer extension at 72°C for 3 min at the end of these cycles. Every sample was tested in triplicate. qPCR products were quantitated using the Quant-iT dsDNA High Sensitivity Assay Kit (Life Technologies, Grand Island, NY). The average lesion frequency per each fragment was calculated by using the Poisson equation. This experiment was done in duplicate to assess for consistency of response.
Western immunoblotting
The Ishikawa and ECC-1 cells were plated at 2 × 105 cells/well in 6-well plates in their corresponding media and then treated with simvastatin for 20 h. Cell lysates were prepared in RIPA buffer (1% NP40, 0.5 sodium deoxycholate and 0.1% SDS). Equal amounts of protein were separated by gel electrophoresis and transferred onto a PVDF membrane. The membrane was blocked with 5% nonfat dry milk and then incubated with a 1:1000 dilution of primary antibody overnight at 4°C. The membrane was then washed and incubated with a secondary peroxidase-conjugated antibody for 1 h after washing. Antibody binding was detected using an enhanced chemiluminescence detection system on the Alpha Innotech Imaging System (Protein Simple, Santa Clara, CA). After developing, the membrane was stripped and re-probed using antibodies against β-actin or α-tubulin to confirm equal loading. Intensity for each band was measured and normalized to β-actin or α-tubulin as an internal control. Each experiment was repeated two times to assess for consistency of results.
Endometrial cancer tissue sample collection and primary cell culture
Eight tumor specimens were sampled from patients undergoing surgery for endometrial carcinoma at the University of North Carolina at Chapel Hill (UNC-CH). The protocol was reviewed and exemption granted by the Institutional Review Board at UNC-CH. For the culture of primary endometrial cancer cells, the freshly obtained tissues were washed three times with Hank’s Buffered Salt Solution (HBSS), and then minced by scissors in DMEM/F12 medium containing 10% fetal bovine serum (FBS). These tissues were then digested in 0.2% collagenase IA, 100 U/ml penicillin and anti-anti for 30–60 min at 37°C water bath with shaking. After two centrifugations with PBS solution, the cells were re-suspended and diluted to 1×105 cells/ml with DMEM/F12 medium. 2×104 cells/well were seeded into 96-well plates and incubated for 24 h before treatment with simvastatin. Cells were then treated with varying doses of simvastatin. Cell proliferation was measured by MTT assay 72 h after treatment.
HMGCR Assay
HMGCR activity was measured using the HMG-CoA reductase (HMGR) assay kit from Sigma-Aldrich (Saint Louis, MO), following the manufacturer’s instructions. Both cell lines were treated with simvastatin for 24 h, and proteins were collected using mammalian protein extraction buffer. The spectrometer was operated with the Tecon I control software in time driver mode. The absorbance at 340 nm was monitored at a time interval of 1.00 s for a total of 15 min. This experiment was performed in duplicate.
Statistical Analysis
Results for experiments were normalized to the mean of the control and analyzed using the Student t-test. Differences were considered significant if the p value was less than 0.05 (p < 0.05) with a confidence interval of 95%.
RESULTS
Simvastatin inhibits cell growth and decreases HMGCR activity
The effect of simvastatin on cell proliferation was examined in the endometrial cancer cell lines, ECC-1 and Ishikawa. Both cells were exposed to varying doses of simvastatin for 72 h. As shown in Figure 1A, simvastatin effectively inhibited growth in a dose-dependent manner in both endometrial cancer cells. The mean IC50 value for each of these cell lines was approximately 15 uM and 17 uM for ECC-1 and Ishikawa cells, respectively. In order to insure that simvastatin had an inhibitory effect on its molecular target, we examined HMGCR activity in both cell lines after exposure to varying doses of simvastatin (1 and 10uM) for 24 h. A significant decrease in HMGCR activity was seen in the ECC-1 (Figure 1B) and Ishikawa cell lines (Figure 1C), co-incident with inhibition of proliferation.
Figure 1. Simvastatin inhibits the growth of endometrial cancer cells and HMG-CR activity and induces cell cycle G0/G1 arrest.

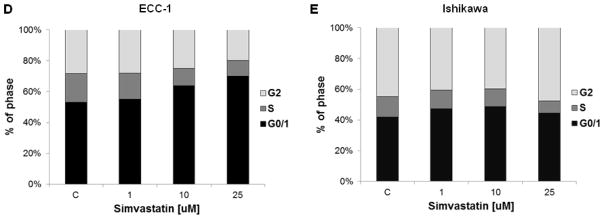
ECC-1 and Ishikawa cells were cultured for 24 h and then treated with varying concentrations of simvastatin in 96 well plates for 72 h. Cell proliferation was assessed by MTT assay (A). The effect of simvastatin on HMGCR activity levels was determined by HMG-CoA Reductase Assay. Treatment with simvastatin for 24 h resulted in a dose-dependent decrease in HMGCR activity in both ECC-1 (B) and Ishikawa (C). Cell cycle analysis was performed by Cellometer after treatment with the indicated doses of simvastatin for 24 h. Simvastatin markedly induced cell cycle G0/G1 arrest in ECC-1 (D) in a dose dependent manner (p<0.05 for doses 10 and 25 μM), while a lesser effect was seen in Ishikawa (E). * p<0.05
Simvastatin induces cell cycle arrest in G0/G1 and apoptosis
To evaluate the underlying mechanism of growth inhibition by simvastatin, the cell cycle profile was analyzed after treating the ECC-1 and Ishikawa cells with varying doses (1–25 uM) of simvastatin for 24 h. As illustrated in Figure 1D, simvastatin treatment resulted in G0/G1 cell cycle arrest and reduced S phase in a dose-dependent manner in ECC-1. Minimal effects on G0/G1 arrest were seen in the Ishikawa cells (Figure 1E). To further confirm whether the growth inhibition of endometrial cancer cells was related to apoptosis, we evaluated the apoptotic effect of simvastatin on ECC-1 and Ishikawa cells by Annexin-V FITC stain analysis, which detects the phospholipid phosphatidylserine (PS) translocation from the inner (cytoplasmic) leaflet of the cell membrane to the external surface in very early apoptotic cells. As shown in Figure 2A–2D, after treatment of the cells with simvastatin at the indicated concentrations for 24 h, the percentage of early apoptotic cells increased in a dose-dependent manner in both cell lines.
Figure 2. Simvastatin induced apoptosis in the ECC-1 (A and C) and Ishikawa (B and D) cell lines, as evidenced by increased cleaved caspase-3 and decreased BCL-2 expression.


ECC-1 and Ishikawa cells were cultured for 24 h and the treated with simvastatin at varying doses for 24 h. Apoptosis was examined by Annexin V assay via Cellometer. Caspase-3, BCL-2 and MCL-1 were determined by Western immunoblotting in ECC-1 (E) and Ishikawa cells (F) after exposure to simvastatin for 16 h. * p<0.05
To further analyze the effects of simvastatin on apoptosis pathways, Western blot analysis was used to detect a change in caspase-3 and other anti-apoptotic proteins. Caspase-3 is a member of the caspase family, which are cysteine proteases that act in a cascade manner to trigger apoptosis and are considered to be effector caspases involved in cell disassembly. BCL-2 and MCL-1 are anti-apoptotic proteins that belong to the BCL-2 family of cell death regulator proteins. Western blotting indicated that simvastatin increased cleaved caspase-3 and decreased BCL-2 protein expression in both cell lines while MCL-1 levels remained constant (Figure 2E and 2F). These results suggest that simvastatin reduces cell proliferation through inducing cell cycle G1 arrest and induction of apoptosis in endometrial cancer cells. Furthermore, simvastatin may be involved in different cell death mechanisms to inhibit cell proliferation in different endometrial cancer cells.
Simvastatin increased levels of intracellular ROS and resulted in DNA damage
To investigate the involvement of oxidative stress in the anti-proliferative effect of simvastatin, intracellular ROS levels were examined by using the ROS fluorescence indicator DCFH-DA. As seen in Figure 3A and 3B, treatment with simvastatin (1–25 uM) for 24 h significantly increased ROS production in a dose-dependent manner in both the ECC-1 and Ishikawa cells. Given that the production of peroxides and free radicals induced in oxidative stress can damage several cell components including nuclear and mitochondrial DNA, we evaluated DNA damage by qPCR and found that simvastatin induced DNA damage in a dose-dependent manner in the ECC-1 (Figure 3C) and Ishikawa (Figure 3D) cell lines after 24 h of treatment. These results indicate that an increase in ROS and DNA damage might also be involved in the anti-tumorigenic effects of simvastatin in endometrial cancer cells.
Figure 3. The effect of simvastatin on reactive oxygen species (ROS) generation and mitochondrial DNA damage in endometrial cancer cell lines.

ECC1 and Ishikawa cells were treated with simvastain at different concentrations for 8 h, and the ROS level was determined using DCFH-DA dye on a plate reader (A and B, respectively). The ECC-1 and Ishikawa cell lines were treated with the indicated concentrations of simvastatin for 24 h, and DNA damage was analyzed by qPCR assay (C and D, respectively). * p<0.05
Effect of simvastatin on the AKT/mTOR and MAPK pathways
To investigate the mechanisms underlying the inhibition of cell growth by simvastatin, we characterized the effect of simvastatin on the extracellular signal regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) and AKT/mammalian target of rapamycin (AKT/mTOR) pathways. Simvastatin reduced phosphorylation of p42/44 (ERK1/2) in a dose-dependent manner in both endometrial cancer cells lines, within 18 h of exposure (Figure 4A and 4B). We then evaluated the effect of simvastatin on downstream targets of the AKT/mTOR pathway, including phosphorylated AKT and phosphorylated ribosomal protein S6. Simvastatin was found to decrease phosphorylation of S6 in the Ishikawa cells, whereas simvastatin increased phosphorylation of S6 in the ECC-1 cells. There was little effect on phosphorylated AKT expression in either cell line after 18 h of treatment. This data suggests that simvastatin may exert its anti-tumor activity via cell signaling pathways other than those downstream of the mTOR pathway.
Figure 4. The effect of simvastatin on the AKT/mTOR and MAPK pathways in endometrial cancer cell lines.

ECC-1 (A) and Ishikawa (B) cells were treated with simvastatin at different concentrations as indicated for 18 h. Expression of phosphorylated-AKT, -S6 and -p42/44 was determined by Western immunoblotting.
Simvastatin inhibits cell adhesion and invasion
In order to determine the effect of simvastatin on adhesion and invasion of endometrial cancer cells, an in vitro laminin adhesion assay and a transwell invasion system were employed. Incubation of ECC-1 and Ishikawa cells with simvastatin (1, 10 and 25 uM) for 2 h showed significant inhibition of cell adhesion (Figure 5A and 5B). Simvastatin significantly blocked endometrial cancer cell invasion after 24 h of treatment as determined by transwell invasion assay (Figure 5C and 5D). Inhibition of cell adhesion and invasion was dose-dependent in both cells. These results suggest that simvastatin may function to inhibit adhesion and invasion in endometrial cancer cells as well as inducing apoptosis, cell cycle arrest and cellular stress.
Figure 5. The effect of simvastatin on adhesion and invasion in endometrial cancer cell lines.

ECC-1 (A) and Ishikawa (B) cells were cultured for 24 h and then treated with simvastain as indicated for 2 h in a laminin coated 96 well plate. Adhesion was assessed using a plate reader. ECC-1 (C) and Ishikawa (D) cells were cultured for 24 h and then treated with simvastain as indicated for 2 h in a BME coated 96 transwell plate. Invasion was determined using a plate reader. Treatment with simvastatin resulted in relative inhibition of adhesion and invasion. * p<0.05
Simvastatin inhibited proliferation of endometrial cancer cells derived from patients
To expand on our work in established cell lines, we further investigated the effects of simvastatin on tumor cell growth in primary cultures of endometrial cancer using the MTT assay. These results demonstrated that the majority of the primary cell cultures responded to simvastatin, with five of eight achieving an IC50 (range: 8 to 15 uM) after 72 h of treatment with simvastatin (Figure 6A and D). To further investigate if HMGCR protein expression was associated with sensitivity to simvastatin, we detected HMGCR protein by Western blot in all of the primary cultures of endometrial cancer cells (Figure 6B and 6C). We found no correlation between level of HMGCR protein expression and response to treatment with simvastatin. For example, primary culture samples 4 and 6 had the least HMGCR expression, yet sample 6 had a strong response to treatment and sample 4 had a non-significant response. However, these findings support the possibility that simvastatin exhibits its anti-proliferative effects through multiple pathways, such as MAPK and AKT/mTOR pathways, instead of exclusively targeting the mevalonate pathway.
Figure 6. The effect of simvastatin on eight primary culture endometrial cancer samples.


Six out of eight primary cultures endometrial cancer tumors had a decrease in cell proliferation with an IC50 ranging from 8–15 uM after treatment with indicated doses of simvastatin for 72 h (A and D). HMGCR expression for each endometrial cancer tumor was quantified via western blot (B and C). There was no correlation found between HMGCR expression and response to simvastatin treatment. *NR= no response
DISCUSSION
The sterols biosynthesis and protein prenylation involved in the mevalonate pathway has been associated with tumor development and progression. The end products of the mevalonate pathway are involved in post-translational modifications, which are responsible for activation of the Ras protein family, regulation of signal transduction and induction of cell growth and apoptosis. The mevalonate pathway is often overactive in cancer cells and is thought to be a potential oncogenic pathway (17, 18). In this study, we investigated the anti-neoplastic activity of simvastatin in endometrial cancer cell lines and primary endometrial cancer cells. Simvastatin was found to inhibit cellular proliferation, suppress HMGCR enzymatic activity, cause mitochondrial DNA damage, induce apoptosis and cellular stress, and block cellular adhesion and invasion. These observations are comparable to recent studies in breast, liver, melanoma and lung cancer, showing promising anti-tumorigenic effects of statins on the growth of cancer in vitro and in vivo (19–23). Studies in other cancer cell types have also shown statins to have anti-metastatic effects such as inhibition of adhesion and invasion (24–26). Lastly, statin treatment has also been found to decrease metastatic burden in cancer mouse models (27, 28).
Simvastatin inhibited the MAPK pathway and exhibited differential effects on the AKT/mTOR pathways in the ECC-1 and Ishikawa cell lines. Treatment with simvastatin has been shown to decrease phosphorylation of p42/44 (ERK1/2) and AKT in other cancer cell types (29, 30). In our study, a minimal change in AKT phosphorylation was seen. Little is known about the effects of statins on phosphorylated S6 expression in cancer cells. In one study, treatment with lovastatin in immortalized rat brain neuroblasts resulted in a decrease in phosphorylation of AKT and its downstream targets including S6 (31). Similar results were seen in the Ishikawa cell line; however, phosphorylation of S6 was increased in the ECC-1 cell line. The discrepancies between the two cell lines may be due to negative feedback or alternative signaling pathways taken advantage of by simvastatin to decrease cell proliferation.
In times of cellular stress, an increase in production of reactive oxygen species (ROS) can result in DNA damage and subsequently cell death. Studies in cancer cell lines have found that statin treatment results in the induction of intracellular ROS (8, 32, 33). In addition, our study demonstrated an associated increase in mitochondrial DNA damage following treatment with simvastatin. These results indicate that the apoptotic effect induced by simvastatin is potentially mediated by oxidative stress and consequently mitochondrial DNA damage.
To date, only one other study has looked at statin’s anti-proliferative effects in endometrial cancer cells (34). This study analyzed the effects of lipophilic versus hydrophobic statins on cell proliferation in the Ishikawa endometrial cancer cell line. This same study also used primary cells from a recurrent endometrial cancer patient, in order to analyze the synergistic effects of statins and chemotherapy on cell viability. The lipophillic (simvastatin and lovastatin) but not hydrophilic (pravastatin) statins were found to inhibit proliferation in endometrial, ovarian and cervical cell lines. Our study demonstrates a more comprehensive look at the anti-proliferative and anti-metastatic effects of simvastatin in established endometrial cancer cell lines and further analyzes simvastatin’s anti-proliferative effect using eight primary culture samples. Our data supports that simvastatin is a potent inhibitor of cell proliferation in human endometrial cancer cells predominantly through apoptosis and cell cycle arrest and that using simvastatin to target the mevalonate pathway may be an effective chemotherapeutic strategy for the treatment of endometrial cancer.
We acknowledge that the IC50 doses of simvastatin used in this in vitro study (14–17 uM for ECC-1 and Ishikawa, 5–9 uM for primary culture cells) are supra-theraputic compared to the doses used in hypercholesterolemia patients. However, the range of doses are very similar to those used in other in vitro studies of simvastatin (1–30 uM) (3–6). The maximum recommended clinical simvastatin dose is 80 mg/day. The therapeutic dose of simvastatin, 1mg/kg/day, correlates to a serum level of 0.1 uM (35). It is important to consider that cells in culture are grown in an environment of excess nutrients, which may explain why concentrations above the therapeutic dose are needed to detect the effects of simvastatin. Studies using statins (ranging from 0.5–1.5 uM) in mice models have used more physiological doses and have shown a decrease in local tumor growth as well as metastasis (20–22). It is possible that using these supra-therapeutic doses may also cause simvastatin to have off-target effects which could be reflected in the lack of correlation between HMGCR expression and response to simvastatin in the primary cultures as well as the variable effects of simvastatin on downstream signaling pathways in the Ishikawa and ECC-1 cell lines. Thus, we plan to assess the anti-tumorigenic effects of simvastatin in endometrial cancer mouse models and correlate these in vivo findings to our work in endometrial cancer cell lines and primary cultures.
Evidence that simvastatin inhibits HMGCR activity, a critical enzyme in the mevalonate pathway, makes it a logical option for development of an anti-cancer agent. Currently, statins are being studied clinically for use in the prevention and treatment of cancer and as an adjuvant therapy in combination with chemotherapeutic agents. Many epidemiological studies have shown a correlation between statin use and a relative reduction in the risk of endometrial, colorectal, gastric, and hepatocellular cancer (9–12, 36, 37). Two colorectal cancer studies reported a 17–51% reduction in colorectal cancer risk with simvastatin (36, 37). Improved overall survival was seen in patients who used statins one year prior to the diagnosis of endometrial and ovarian cancers (12). Reduced rates of advanced disease have been found among prostate cancer patients taking statins at the time of prostatectomy (38). A clinical trial study comparing advanced liver cancer patients taking pravastatin versus a placebo showed increased survival amongst those patients who had been given the statin (39). Breast cancer patients who were given atorvastatin two weeks prior to surgery showed anti-proliferative effects, with a decrease in Ki-67 staining in those tumors that were HMGCR positive (40). Although statins used as a single agent have shown some promise, statins may also be useful in combination with other cytotoxic agents. A phase II study using simvastatin plus irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) as first-line chemotherapy in metastatic colorectal cancer showed that time to progression was prolonged, and no additional adverse effects were seen with the addition of simvastatin (13).
Obese patients often suffer from co-morbidities such as hypercholesterolemia and diabetes and are at increased risk for the development of endometrial cancer with conceivable worse outcomes. Thus, it is important to find new strategies to treat these “high risk” patients. With obesity and endometrial cancer rates on the rise, simvastatin offers a new treatment and possibly prevention strategy for this disease. Simvastatin is a safe drug that is currently used by millions worldwide to treat hypercholesterolemia. Simvastatin is effective at inhibiting cell proliferation through multiple mechanisms in endometrial cancer cell lines and primary endometrial cancer culture cells, suggesting that using statins to target HMGCR may be promising for clinical trials in endometrial cancer.
RESEARCH HIGHLIGHTS.
Simvastatin has anti-proliferative and anti-metastatic effects in endometrial cancer cells, suggesting that statins may have promise for endometrial cancer treatment.
Simvastatin’s anti-tumorigenic effects may be partially mediated through regulation of the MAPK pathway in endometrial cancer cells.
Acknowledgments
VBJ is supported by NIH/NCI 1K23CA143154-01A1 and the Steelman Fund.
Footnotes
Presented as a poster presentation at the 2014 Annual Meeting of the Society of Gynecologic Oncology in Tampa, FL.
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Society AC. Cancer facts and figures. Atlanta (GA): American Cancer Society; 2011. [Google Scholar]
- 2.Zhang Y, Liu H, Yang S, Zhang J, Qian L, Chen X. Overweight, obesity and endometrial cancer risk: results from a systematic review and meta-analysis. The International journal of biological markers. 2014;29(1):e21–9. doi: 10.5301/jbm.5000047. Epub 2013/10/31. [DOI] [PubMed] [Google Scholar]
- 3.Matsuura M, Suzuki T, Suzuki M, Tanaka R, Ito E, Saito T. Statin-mediated reduction of osteopontin expression induces apoptosis and cell growth arrest in ovarian clear cell carcinoma. Oncol Rep. 2011;25(1):41–7. Epub 2010/11/27. [PubMed] [Google Scholar]
- 4.Gopalan A, Yu W, Sanders BG, Kline K. Simvastatin inhibition of mevalonate pathway induces apoptosis in human breast cancer cells via activation of JNK/CHOP/DR5 signaling pathway. Cancer letters. 2013;329(1):9–16. doi: 10.1016/j.canlet.2012.08.031. Epub 2012/09/11. [DOI] [PubMed] [Google Scholar]
- 5.Cho SJ, Kim JS, Kim JM, Lee JY, Jung HC, Song IS. Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis-associated colon cancer in mice. Int J Cancer. 2008;123(4):951–7. doi: 10.1002/ijc.23593. Epub 2008/06/04. [DOI] [PubMed] [Google Scholar]
- 6.Liang YW, Chang CC, Hung CM, Chen TY, Huang TY, Hsu YC. Preclinical Activity of Simvastatin Induces Cell Cycle Arrest in G1 via Blockade of Cyclin D-Cdk4 Expression in Non-Small Cell Lung Cancer (NSCLC) International journal of molecular sciences. 2013;14(3):5806–16. doi: 10.3390/ijms14035806. Epub 2013/03/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Relja B, Meder F, Wilhelm K, Henrich D, Marzi I, Lehnert M. Simvastatin inhibits cell growth and induces apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. International journal of molecular medicine. 2010;26(5):735–41. doi: 10.3892/ijmm_00000520. Epub 2010/09/30. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez CA, Rodriguez E, Varela E, Zapata E, Paez A, Masso FA, et al. Statin-induced inhibition of MCF-7 breast cancer cell proliferation is related to cell cycle arrest and apoptotic and necrotic cell death mediated by an enhanced oxidative stress. Cancer investigation. 2008;26(7):698–707. doi: 10.1080/07357900701874658. Epub 2008/07/09. [DOI] [PubMed] [Google Scholar]
- 9.Singh PP, Singh S. Statins are associated with reduced risk of gastric cancer: a systematic review and meta-analysis. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2013;24(7):1721–30. doi: 10.1093/annonc/mdt150. Epub 2013/04/20. [DOI] [PubMed] [Google Scholar]
- 10.Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144(2):323–32. doi: 10.1053/j.gastro.2012.10.005. Epub 2012/10/16. [DOI] [PubMed] [Google Scholar]
- 11.Jacobs EJ, Newton CC, Thun MJ, Gapstur SM. Long-term use of cholesterol-lowering drugs and cancer incidence in a large United States cohort. Cancer Res. 2011;71(5):1763–71. doi: 10.1158/0008-5472.CAN-10-2953. Epub 2011/02/24. [DOI] [PubMed] [Google Scholar]
- 12.Lavie O, Pinchev M, Rennert HS, Segev Y, Rennert G. The effect of statins on risk and survival of gynecological malignancies. Gynecol Oncol. 2013;130(3):615–9. doi: 10.1016/j.ygyno.2013.05.025. Epub 2013/05/31. [DOI] [PubMed] [Google Scholar]
- 13.Lee J, Jung KH, Park YS, Ahn JB, Shin SJ, Im SA, et al. Simvastatin plus irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) as first-line chemotherapy in metastatic colorectal patients: a multicenter phase II study. Cancer chemotherapy and pharmacology. 2009;64(4):657–63. doi: 10.1007/s00280-008-0913-5. Epub 2009/01/27. [DOI] [PubMed] [Google Scholar]
- 14.Han JY, Lee SH, Yoo NJ, Hyung LS, Moon YJ, Yun T, et al. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2011;17(6):1553–60. doi: 10.1158/1078-0432.CCR-10-2525. Epub 2011/03/18. [DOI] [PubMed] [Google Scholar]
- 15.Lessey BA, Vendrov AE, Yuan L. Endometrial cancer cells as models to study uterine receptivity. In: Kuramoto H, Nishida M, editors. Cell and Molcular Biology of Endometrial Carcinoma. Tokyo: Springer-Verlag; 2003. pp. 267–79. [Google Scholar]
- 16.Furda A, Santos JH, Meyer JN, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2014;1105:419–37. doi: 10.1007/978-1-62703-739-6_31. Epub 2014/03/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Notarnicola M, Messa C, Pricci M, Guerra V, Altomare DF, Montemurro S, et al. Up-regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in left-sided human colon cancer. Anticancer Res. 2004;24(6):3837–42. Epub 2005/03/02. [PubMed] [Google Scholar]
- 18.Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A. 2010;107(34):15051–6. doi: 10.1073/pnas.0910258107. Epub 2010/08/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pich C, Teiti I, Rochaix P, Mariame B, Couderc B, Favre G, et al. Statins Reduce Melanoma Development and Metastasis through MICA Overexpression. Frontiers in immunology. 2013;4:62. doi: 10.3389/fimmu.2013.00062. Epub 2013/03/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kidera Y, Tsubaki M, Yamazoe Y, Shoji K, Nakamura H, Ogaki M, et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. Journal of experimental & clinical cancer research: CR. 2010;29:127. doi: 10.1186/1756-9966-29-127. Epub 2010/09/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H, Wang Z, Li Y, Li W, Chen Y. Simvastatin prevents proliferation and bone metastases of lung adenocarcinoma in vitro and in vivo. Neoplasma. 2013;60(3):240–6. doi: 10.4149/neo_2013_032. Epub 2013/02/05. [DOI] [PubMed] [Google Scholar]
- 22.Yu X, Luo Y, Zhou Y, Zhang Q, Wang J, Wei N, et al. BRCA1 overexpression sensitizes cancer cells to lovastatin via regulation of cyclin D1-CDK4-p21WAF1/CIP1 pathway: analyses using a breast cancer cell line and tumoral xenograft model. International journal of oncology. 2008;33(3):555–63. Epub 2008/08/13. [PubMed] [Google Scholar]
- 23.Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 2011;71(6):2286–97. doi: 10.1158/0008-5472.CAN-10-3367. Epub 2011/01/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denoyelle C, Vasse M, Korner M, Mishal Z, Ganne F, Vannier JP, et al. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis. 2001;22(8):1139–48. doi: 10.1093/carcin/22.8.1139. Epub 2001/07/27. [DOI] [PubMed] [Google Scholar]
- 25.Kusama T, Mukai M, Iwasaki T, Tatsuta M, Matsumoto Y, Akedo H, et al. 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors reduce human pancreatic cancer cell invasion and metastasis. Gastroenterology. 2002;122(2):308–17. doi: 10.1053/gast.2002.31093. Epub 2002/02/08. [DOI] [PubMed] [Google Scholar]
- 26.Yongjun Y, Shuyun H, Lei C, Xiangrong C, Zhilin Y, Yiquan K. Atorvastatin suppresses glioma invasion and migration by reducing microglial MT1-MMP expression. Journal of neuroimmunology. 2013;260(1–2):1–8. doi: 10.1016/j.jneuroim.2013.04.020. Epub 2013/05/28. [DOI] [PubMed] [Google Scholar]
- 27.Matar P, Rozados VR, Roggero EA, Scharovsky OG. Lovastatin inhibits tumor growth and metastasis development of a rat fibrosarcoma. Cancer biotherapy & radiopharmaceuticals. 1998;13(5):387–93. doi: 10.1089/cbr.1998.13.387. Epub 2000/06/14. [DOI] [PubMed] [Google Scholar]
- 28.Jani JP, Specht S, Stemmler N, Blanock K, Singh SV, Gupta V, et al. Metastasis of B16F10 mouse melanoma inhibited by lovastatin, an inhibitor of cholesterol biosynthesis. Invasion & metastasis. 1993;13(6):314–24. Epub 1993/01/01. [PubMed] [Google Scholar]
- 29.Fang Z, Tang Y, Fang J, Zhou Z, Xing Z, Guo Z, et al. Simvastatin inhibits renal cancer cell growth and metastasis via AKT/mTOR, ERK and JAK2/STAT3 pathway. PloS one. 2013;8(5):e62823. doi: 10.1371/journal.pone.0062823. Epub 2013/05/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. The Journal of pharmacology and experimental therapeutics. 2011;336(2):496–505. doi: 10.1124/jpet.110.174870. Epub 2010/11/10. [DOI] [PubMed] [Google Scholar]
- 31.Cerezo-Guisado MI, Garcia-Marin LJ, Lorenzo MJ, Bragado MJ. Lovastatin inhibits the growth and survival pathway of phosphoinositide 3-kinase/protein kinase B in immortalized rat brain neuroblasts. Journal of neurochemistry. 2005;94(5):1277–87. doi: 10.1111/j.1471-4159.2005.03345.x. Epub 2005/08/23. [DOI] [PubMed] [Google Scholar]
- 32.Qi XF, Zheng L, Lee KJ, Kim DH, Kim CS, Cai DQ, et al. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell death & disease. 2013;4:e518. doi: 10.1038/cddis.2013.44. Epub 2013/03/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crescencio ME, Rodriguez E, Paez A, Masso FA, Montano LF, Lopez-Marure R. Statins inhibit the proliferation and induce cell death of human papilloma virus positive and negative cervical cancer cells. International journal of biomedical science: IJBS. 2009;5(4):411–20. Epub 2009/12/01. [PMC free article] [PubMed] [Google Scholar]
- 34.Kato S, Smalley S, Sadarangani A, Chen-Lin K, Oliva B, Branes J, et al. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. Journal of cellular and molecular medicine. 2010;14(5):1180–93. doi: 10.1111/j.1582-4934.2009.00771.x. Epub 2009/05/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan HY, DeVault AR, Wang-Iverson D, Ivashkiv E, Swanson BN, Sugerman AA. Comparative pharmacokinetics and pharmacodynamics of pravastatin and lovastatin. Journal of clinical pharmacology. 1990;30(12):1128–35. doi: 10.1002/j.1552-4604.1990.tb01856.x. Epub 1990/12/01. [DOI] [PubMed] [Google Scholar]
- 36.Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, et al. Statins and the risk of colorectal cancer. The New England journal of medicine. 2005;352(21):2184–92. doi: 10.1056/NEJMoa043792. Epub 2005/05/27. [DOI] [PubMed] [Google Scholar]
- 37.Vinogradova Y, Hippisley-Cox J, Coupland C, Logan RF. Risk of colorectal cancer in patients prescribed statins, nonsteroidal anti-inflammatory drugs, and cyclooxygenase-2 inhibitors: nested case-control study. Gastroenterology. 2007;133(2):393–402. doi: 10.1053/j.gastro.2007.05.023. Epub 2007/08/08. [DOI] [PubMed] [Google Scholar]
- 38.Mondul AM, Han M, Humphreys EB, Meinhold CL, Walsh PC, Platz EA. Association of statin use with pathological tumor characteristics and prostate cancer recurrence after surgery. The Journal of urology. 2011;185(4):1268–73. doi: 10.1016/j.juro.2010.11.089. Epub 2011/02/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawata S, Yamasaki E, Nagase T, Inui Y, Ito N, Matsuda Y, et al. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br J Cancer. 2001;84(7):886–91. doi: 10.1054/bjoc.2000.1716. Epub 2001/04/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bjarnadottir O, Romero Q, Bendahl PO, Jirstrom K, Ryden L, Loman N, et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat. 2013;138(2):499–508. doi: 10.1007/s10549-013-2473-6. Epub 2013/03/09. [DOI] [PubMed] [Google Scholar]