

Abstract
A scarlet fever outbreak occurred in Hong Kong in 2011. The majority of cases resulted in the isolation of Streptococcus pyogenes emm12 with multiple antibiotic resistances. Phylogenetic analysis of 22 emm12 scarlet fever outbreak isolates, 7 temporally and geographically matched emm12 non–scarlet fever isolates, and 18 emm12 strains isolated during 2005–2010 indicated the outbreak was multiclonal. Genome sequencing of 2 nonclonal scarlet fever isolates (HKU16 and HKU30), coupled with diagnostic polymerase chain reaction assays, identified 2 mobile genetic elements distributed across the major lineages: a 64.9-kb integrative and conjugative element encoding tetracycline and macrolide resistance and a 46.4-kb prophage encoding superantigens SSA and SpeC and the DNase Spd1. Phenotypic comparison of HKU16 and HKU30 with the S. pyogenes M1T1 strain 5448 revealed that HKU16 displays increased adherence to HEp-2 human epithelial cells, whereas HKU16, HKU30, and 5448 exhibit equivalent resistance to neutrophils and virulence in a humanized plasminogen murine model. However, in contrast to M1T1, the virulence of HKU16 and HKU30 was not associated with covRS mutation. The multiclonal nature of the emm12 scarlet fever isolates suggests that factors such as mobile genetic elements, environmental factors, and host immune status may have contributed to the 2011 scarlet fever outbreak.
Scarlet fever, also known as scarlatina, is a toxin-mediated disease caused by Streptococcus pyogenes (group A Streptococcus [GAS]), characterized by a scarlet-colored rash, fever, and exudative pharyngitis. Once viewed as a significant cause of pandemic childhood morbidity and mortality in the 19th and early 20th centuries, scarlet fever is now considered a rare disease [1, 2]. Streptococcus pyogenes remains a major cause of human infection, morbidity, and mortality resulting from benign diseases such as pharyngitis and impetigo; severe diseases including puerperal sepsis, bacteremia, streptococcal toxic shock–like syndrome, and necrotizing fasciitis; and the postinfectious immune complications rheumatic fever, rheumatic heart disease, and acute poststreptococcal glomerulonephritis [3]. The production of pyrogenic exotoxins such as SpeA, SpeC, SSA, and other superantigens is important in the pathogenesis of toxin-mediated diseases such as scarlet fever and toxic shock syndrome [4].
Newly emergent clonal GAS strains may arise and cause significant outbreaks and pandemic disease. Acquisition of large regions of foreign DNA by transduction or recombination events has produced a number of GAS strains with improved fitness or altered tissue tropisms. In the last 30 years, a single clone of serotype M1T1 GAS has disseminated globally and now accounts for almost 20% of all clinical GAS isolates in developed countries [5]. Emergence of the M1T1 clone has been linked to its acquisition, through horizontal gene transfer events, of a 36-kb chromosomal DNA region and 2 bacteriophages encoding the DNase Sda1 and the SpeA superantigen, respectively [6–8]. Outbreak clones of M3 GAS have also been linked to its acquisition of speA [9]. The propensity of serotype M28 GAS to cause puerperal sepsis has been attributed to acquisition of an approximately 37-kb region of DNA from group B Streptococcus, encoding surface proteins that enable colonization of the human urogenital tract [10]. Localized outbreaks also occur as a result of GAS clones with more modest genomic changes, presumably as a result of host selective pressure. An outbreak of M3 GAS in Canada was attributed to a 4-amino-acid duplication in the N-terminus of M protein, resulting in a strain with increased resistance to phagocytosis by human polymorphonuclear leukocytes [9]. Thus, clonal GAS strains that can cause globally disseminated disease or localized outbreaks may arise through the acquisition of large segments of foreign DNA through horizontal gene transfer events or relatively minor genetic changes, each providing a selective advantage for the newly generated clone. Environmental factors such as overcrowding and poor hygiene also contribute to GAS epidemics, as has been recently reported in outbreaks of ecthyma (M81 GAS) in active military populations [11] and cellulitis (M59 GAS) in socially disadvantaged populations [12].
Scarlet fever has been a notifiable disease to the Hong Kong Department of Health since 1946, usually with <200 cases per year and critical cases or deaths rarely encountered [13]. In 2011, an alarming increase in the number of scarlet fever notifications was reported. In this work, we have undertaken a genomic and phenotypic analysis of outbreak strains in order to investigate the molecular basis of disease emergence.
MATERIALS AND METHODS
GAS Strains
GAS strains collected in this study were typed as emm12 using standard procedures as recommended by the Centers for Disease Control and Prevention (http://www.cdc.gov/ncidod/biotech/strep/doc.htm) [14]. A total of 22 emm12 GAS isolates from medically diagnosed scarlet fever cases were collected from Queen Mary Hospital and the Hong Kong Department of Health laboratories over the course of the outbreak. An additional 7 emm12 GAS isolates not associated with scarlet fever patients were collected over the same timeframe. In addition, 18 emm12 strains isolated in the 6 years prior to the current outbreak were included. Additional clinical data include date of isolation, patient sex and age, clinical manifestation, site of specimen collection, and antibiotic resistance profile (Table 1).
Table 1.
Group A Streptococcus emm12 isolates Examined in This Study
| HKU Nos. | Hospital | Received Date | Sex | Age (y) | Specimen | Pen | Ery | Cld | Van | Tet | Clinical Information | ICE-emm12 | ΦHKU.vir |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 16 | QMH | 06/11 | Female | 6 | Blood culture | S | R | R | S | R | Scarlet fever | + | + |
| 19 | DH | 03/11 | Male | 13 | Throat swab | S | R | R | S | S | Tonsillitis | − | − |
| 27 | DH | 01/11 | Female | 39 | Midstream urine | S | S | S | S | S | Urinary tract infection | − | − |
| 29 | DH | 01/11 | Female | 7 | Throat swab | S | R | R | S | R | Tonsillitis | + | + |
| 30 | DH | 02/11 | Male | 4 | Throat swab | S | R | R | S | R | Scarlet fever | − | + |
| 32 | DH | 02/11 | Female | 33 | High vaginal swab | S | R | R | S | R | Vaginal discharge | + | + |
| 34 | DH | 02/11 | Female | 61 | Sputum | S | R | R | S | R | Cough | + | − |
| 35 | DH | 02/11 | Female | 1 | Wound swab | S | R | R | S | R | Eczema | + | + |
| 57 | DH | 06/11 | Female | 38 | Throat swab | S | S | S | S | S | Tonsillitis | − | − |
| 84 | KWH | 2005 | Male | 3 | Throat swab | S | R | R | S | R | Acute tonsillitis | + | + |
| 86 | KWH | 2005 | Male | 4 | Throat swab | S | R | R | S | R | Pharyngitis | + | + |
| 98 | KWH | 2006 | Male | 4 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 111 | KWH | 2007 | Female | 5 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 116 | KWH | 2007 | Female | 8 | Throat swab | S | R | R | S | R | Acute tonsillitis | + | + |
| 153 | KWH | 2010 | Male | 5 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 157 | KWH | 2010 | Female | 5 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 161 | KWH | 2010 | Male | 5 | Throat swab | S | R | R | S | R | Henoch-Schönlein purpura | + | − |
| 189 | KWH | 2005 | Male | <1 | Throat swab | S | R | R | S | R | Pharyngitis | + | + |
| 283 | KWH | 2008 | Female | 4 | Throat swab | S | R | R | S | R | Pharyngitis | + | + |
| 288 | KWH | 2008 | Female | 6 | Throat swab | S | R | R | S | R | Acute tonsillitis | + | − |
| 294 | KWH | 2008 | Male | 7 | Throat swab | S | R | R | S | R | URTI & sore throat | + | − |
| 362 | DH | 2008 | Female | 50 | Throat swab | S | S | S | S | S | NA | − | − |
| 365 | DH | 2009 | Female | 59 | Low vaginal swab | S | S | S | S | S | Vaginitis | − | + |
| 366 | DH | 2009 | Female | 35 | Throat swab | S | S | S | S | S | Acute tonsillitis | − | − |
| 367 | DH | 2010 | Female | 29 | High vaginal swab | S | S | S | S | S | Vaginal discharge | − | + |
| 368 | DH | 2010 | Female | 15 | Vulval swab | S | S | S | S | S | Vaginal discharge | − | − |
| 371 | DH | 2010 | Female | 29 | Throat swab | S | S | S | S | S | NA | − | + |
| 373 | DH | 06/11 | Male | 6 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 374 | DH | 06/11 | Female | 3 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 375 | DH | 06/11 | Male | 6 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 378 | DH | 06/11 | Female | 8 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 379 | DH | 06/11 | Female | 8 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 382 | DH | 06/11 | Female | 4 | Throat swab | S | S | S | S | S | Scarlet fever | − | − |
| 383 | DH | 06/11 | Female | 3 | Throat swab | S | R | R | S | S | Scarlet fever | + | + |
| 385 | DH | 06/11 | Male | 6 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 386 | DH | 06/11 | Male | 4 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 387 | DH | 06/11 | Male | 3 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 390 | DH | 06/11 | Female | 3 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 392 | DH | 06/11 | Male | 3 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 395 | DH | 06/11 | Female | 6 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 396 | DH | 06/11 | Male | 10 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 397 | DH | 06/11 | Female | 14 | Throat swab | S | R | R | S | R | Scarlet fever | + | − |
| 398 | DH | 06/11 | Male | 4 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 399 | DH | 06/11 | Male | 6 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 400 | DH | 06/11 | Male | 6 | Throat swab | S | R | R | S | R | Scarlet fever | + | + |
| 401 | DH | 06/11 | Male | 8 | Throat swab | S | S | S | S | S | Scarlet fever | − | − |
| 402 | DH | 06/11 | Male | 6 | Throat swab | S | R | R | S | S | Scarlet fever | + | + |
Abbreviations: Cld, clindamycin; DH, Hong Kong Department of Health; Ery, erythromycin; ICE, integrative and conjugative element; KWH, Kwong Wah Hospital; NA, not available; Pen, penicillin; QMH, Queen Mary Hospital; R, resistant; S, sensitive; Tet, tetracycline; Van, vancomycin; +, PCR positive; −, PCR negative.
Pulsed-Field Gel Electrophoresis and Phylogenetic Analyses
Pulsed-field gel electrophoresis (PFGE) was performed as previously described [15] using the CHEF Mapper XA system (Bio-Rad) and restriction endonuclease SmaI. The PFGE image was analyzed with Bionumerics (Applied Maths), and a dendrogram was constructed by the unweighted pair-group method with arithmetic mean method using the Dice similarity coefficient. PFGE clusters were defined as isolates with ≥80% similarity [15].
Genome Sequence Assembly of GAS HKU16 and HKU30
Streptococcus pyogenes strain HKU16 genomic DNA was sequenced and assembled by a combination of 454-pyrosequencing (Roche 454 Life Science) and Illumina 76-base paired-end sequencing (Illumina). The longer reads from 454-pyrosequencing were used to generate a high-quality draft assembly followed by the use of Illumina paired-end reads to resolve repeats and homopolymeric sequences, correct errors, and cross-validate the assembly. Assembly verification was performed by concordance measurements using Illumina paired-end reads, and by comparison with PFGE patterns obtained using restriction enzymes AscI and SmaI, as previously described [16, 17]. Protein coding sequences were predicted using Glimmer3, and automated genome annotation was performed on the RAST server [18].
Streptococcus pyogenes strain HKU30 genomic DNA was sequenced by multiplex 75-bp Illumina Hi-seq paired-end reads with a mean library size of 300 bp. Mean sequence coverage was 219-fold. Illumina sequence reads were submitted to the European Nucleotide Archive (accession number ERS046934). De novo assembly of HKU30 was performed using Velvet [19], which generated 84 assembled contigs. Contigs were then ordered against the HKU16 complete genome sequence using ABACAS [20] and subsequently improved by iteratively mapping the Illumina data to the draft assembly using IMAGE [21]. Contigs were manually corrected by BLAST analyses against the emm12 MGAS9429 (accession number CP000259). The final HKU30 draft genome comprised 23 contigs. Examination of the large chromosomal inversion identified in HKU16 relative to HKU30 was determined by polymerase chain reaction (PCR) of both HKU16 and HKU30 using the primers oMD44 5′-GTCAAAGTCCCTTTAATCCAGC-3′ and oMD43 5′-TGGTAACTTCCAATATGAGTAGC-3′ (3090 bp, 5′ inversion); oMD47 5′-TGCATCTTGATAGGGATAGGC-3′ and oMD46 5′-TCTTTGGATTAGTAGCTCATATC-3′ (1658 bp, 3′ inversion). Lack of inversion in HKU30 was confirmed by switching the primer combinations to oMV44 and oMV47 (5′ region) and oMV43 and oMV46 (3′ inversion).
A genome map of HKU16 and BLAST comparisons against the HKU30 draft genome sequence and MGAS9429 and MGAS2096 complete genome sequences was created using BRIG [22]. BLASTn comparisons were run using BLAST+ with an E cutoff of 10.0. Overall genome architecture of the whole genome sequences of HKU16, MGAS9429, MGAS2096, and the 23 contig draft genome sequence of HKU30 was determined using the Artemis Comparison Tool [23].
Development of Diagnostic PCR for the Detection of Integrative and Conjugative Element–emm12 and ΦHKU.vir
Diagnostic PCR assays for the presence or absence of the 64.9-kb composite transposon-like element containing tetM and ermB and the ΦHKU.vir prophage encoding the superantigens SSA and SpeC and the DNase Spd1 were performed using a HotStarTaq MasterMix Kit (Qiagen) with an annealing temperature of 50°C using standard procedures. PCR of 3 regions of the 64.9-kb integrative and conjugative element (ICE)–emm12 was conducted using primers LPW17791 (5′-CAAGGTATATCCCAACATGAGTA-3′) and LPW17793 (5′-GAGGGCTGGCGATACGTT-3′) (232 bp, CT-PCR1); LPW17795 (5′-GGCATTACACTAAGCATCTT-3′) and LPW17797 (5′-TTGCTGGTATTATTGCTGAA-3′) (202 bp, CT-PCR2); and LPW17799 (5′-CCAGTATGAAATCTATTCCAT-3′) and LPW17801 (5′-AGATATTACAGAAAATGCAGAA-3′) (258 bp, CT-PCR3). PCR of 2 regions of prophage ΦHKU.vir was undertaken using primers LPW17901 (5′-ATTACTACGTTATTTACTACGTT-3′) and LPW17903 (5′-AAACCCACAGACAGGACAGGAA-3′) (949 bp, VIR-PCR1); and LPW17931 (5′-AATGATGCCAGTTGAATGCTA-3′) and LPW17882 (5′-CAAGTGGATTGATGAAGAGAA-3′) (1,602 bp, VIR-PCR2). Amplicon identity was checked by Sanger sequence analysis.
Phenotypic Comparison of GAS Strains
SpeB assays were performed according to standard procedures [24]. The capacity of GAS to adhere to the HEp-2 human epithelial cell line, resist human neutrophil killing, and switch to the SpeB-negative (covRS mutant) form was undertaken using standard procedures [25, 26]. A humanized plasminogen mouse model of invasive infection, approved by the University of Queensland Animal Ethics Committee, was used to assess GAS virulence [25].
RESULTS
Description of the Hong Kong Scarlet Fever Outbreak
Commencing in March 2011, an outbreak of scarlet fever comprising >1000 reported cases occurred in Hong Kong (Figure 1). Moreover, several children with invasive GAS infection and septic shock were admitted to intensive care. The majority of GAS isolates recovered from scarlet fever cases were genotype emm12 [13] and were resistant to tetracycline and macrolide antibiotics (Table 1).
Figure 1.

Monthly notifications of scarlet fever cases to the Hong Kong Department of Health from 2005 to 2011.
Phylogenetic Comparison of GAS emm12 isolates
A total of 22 emm12 GAS isolates from medically diagnosed scarlet fever cases were collected from Queen Mary Hospital and the Hong Kong Department of Health laboratories over the course of the outbreak and subjected to PFGE. An additional 7 emm12 GAS isolates from patients without scarlet fever collected over the same timeframe and 18 emm12 strains isolated in the 6 years prior to the current outbreak were also included for comparison (Table 1). A phylogenetic tree constructed based on these PFGE data suggests the outbreak was multiclonal in nature, with scarlet fever isolates occurring across multiple lineages (Figure 2).
Figure 2.

Pulsed-field gel electrophoresis dendrogram of Streptococcus pyogenes isolates from Hong Kong, showing multiple clones organized in at least 6 clusters based on a Dice similarity coefficient cutoff of 80% (dashed line). Isolate names highlighted in boldface/italics signify isolation from a scarlet fever case.
Genomic Architecture of Hong Kong emm12 Scarlet Fever Isolates HKU16 and HKU30
To identify potential GAS genomic elements that contributed to the Hong Kong outbreak, the complete genome sequence of the nonclonal scarlet fever isolates HKU16 and a draft genome sequence of HKU30 were determined. The genome sequence of S. pyogenes HKU16 (GenBank accession number AFRY00000000) comprised a circular 1 908 100-bp chromosome with G + C content of 38.5%. The genome contains 6 ribosomal RNA operons, 67 transfer RNA genes for all 20 amino acids, and 1861 predicted protein-coding genes. The draft genome sequence of S. pyogenes HKU30 (sequence reads submitted to the European Nucleotide Archive with the accession number ERS046934) was de novo assembled from 75 base paired-end reads. A final draft HKU30 genome sequence representing 23 contigs was determined by ordering and gap closure to the complete HKU16 genome sequence. The resulting HKU30 draft genome is comprised of 1 890 596 bp with a G + C content of 38.35% (Figure 3).
Figure 3.

Circular genome map of Streptococcus pyogenes emm12 genome HKU16 with BLAST comparisons to the S. pyogenes emm12 genomes HKU30, MGAS9429, and MGAS2096. The map was created using BRIG [22]. The innermost rings show G+C content (black) and G+C skew (purple/green) of HKU16. The 3 outer rings show BLAST comparisons (using BLASTn and an E-value cutoff of 10.0) to the draft genome sequence of HKU30 (red) and complete genome sequences of MGAS9429 (blue) and MGAS2096 (orange). Legend shows percentage of identity of BLASTn hits to the HKU16 reference. Labels around the outer ring refer to the insertion sites for the 46.4-kb ΦHKU.vir prophage and 64.9-kb composite transposon-like element ICE-emm12. The insertion site of the Φ9429.1-like prophage that is present in MGAS9429, MGAS2096, and HKU30 but absent in HKU16, is indicated in red. Prophage Φ9429.2 is present in MGAS9429, HKU30, and HKU16, but absent in MGAS2096. Prophage Φ9429.3 is present in all 4 genomes. Abbreviation: kbp = kilobase pairs.
The genomes of both HKU16 and HKU30 share the same exotoxin profile, which differs from that of the non–scarlet fever–associated strains MGAS9429 and MGAS2096 [27]. In the GAS emm12 reference strain MGAS9429, the speC and spd1 genes are located within the Φ9429.1 prophage. However, HKU16 does not contain the Φ9429.1 prophage, yet carries speC and spd1 within a novel prophage, designated ΦHKU.vir, highlighting the chimeric nature of GAS phage. ΦHKU.vir also encodes the superantigen SSA, a toxin not present in either MGAS9429 or MGAS2096. HKU.vir was also identified in HKU30 (see below). Similar to MGAS9429, the genomes of HKU16 and HKU30 contain the speH- and speI-positive Φ9429.2 prophage and the sdaD2-positive Φ9429.3 prophage (Figure 3). We also determined that HKU16 differs from HKU30, MGAS9429, and MGAS2096 by a large genomic inversion encompassing 81% of the genome. The HKU16 inversion is likely to have occurred between 2 transposase-like elements, an observation supported by the presence of a characteristic 885-kb PFGE fragment following digestion of HKU16 DNA with AscI (Figure 4).
Figure 4.
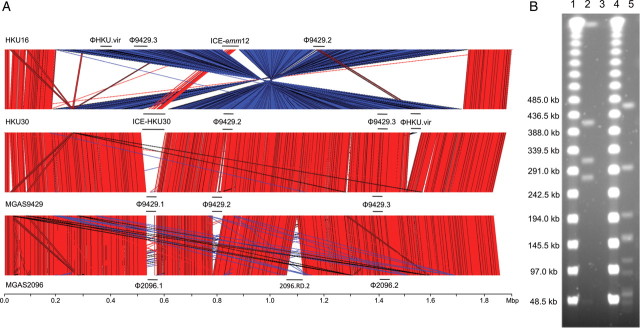
A, Whole genome comparison of Streptococcus pyogenes emm12 strains HKU16, HKU30, MGAS9429, and MGAS2096. Regions of genetic similarity were determined using BLASTn with graphical representation of syntenic gene content designated using the Artemis Comparison Tool (http://www.sanger.ac.uk/Software/ACT/). Red bars between the stacked genome sequences reflect conserved gene content; blue regions indicate inverted, yet conserved matches. These analyses indicate overall chromosome synteny between emm12 isolates. Large chromosomal features depicting prophage and the composite transposon-like element are indicated by a horizontal bar labeled with the feature identity. A large chromosomal inversion identified in HKU16 is depicted (blue region) relative to HKU30, MGAS9429, and MGAS2096. 2096-RD.2 is an ICE element encoding for tetracycline (tetO) resistance in MGAS2096 that shares limited overall nucleotide identity with either ICE-emm12 or ICE-HKU30. HKU30 is a draft genome sequence, de novo assembled using Velvet with contigs ordered and gaps closed using HKU16 as the reference genome. B, Pulsed-field gel electrophoresis analysis of AscI- and SmaI-digested S. pyogenes HKU16 genomic DNA. Lanes 1 and 4: 48.5-kb lambda ladder size markers; lane 2: AscI-digested HKU16 genomic DNA; lane 3: undigested HKU16 genomic DNA; lane 5: SmaI-digested HKU16 genomic DNA. These restriction fragmentation patterns are concordant with the predicted in silico digestion of the assembled HKU16 genome sequence in the 30- to 900-kb range (AscI: 885 052 bp, 405 111 bp; 304 985 bp, 272 520 bp, and 40 432 bp; SmaI: 458 928 bp, 292 656 bp, 196 544 bp, 154 764 bp, 152 373 bp, 112 012 bp, 86 707 bp, 59 305 bp, 56 267 bp, 54 778 bp, 51 979 bp, 51 005 bp, 42 501 bp, 42 154 bp, 41 347 bp, and 34 375 bp).
Novel Mobile Genetic Elements Distributed Across emm12 Lineages
Two previously unreported genomic insertions of 64.9 kb and 46.4 kb were identified in the HKU16 genome (Figure 5). The larger insert is a 64.9-kb ICE comprising a composite transposon-like element containing a Tn916-type transposon embedded within another conjugative transposon (Figure 5A). This ICE, designated ICE-emm12, contains 54 open reading frames including the ermB and tetM genes encoding macrolide-lincomycin-streptogramin resistance and tetracycline resistance, respectively (Figure 5A). Also present is a MATE-type efflux pump and multidrug ABC-type transporter that may confer additional drug resistance of unknown specificity. An ICE-emm12–like element was identified in the draft genome of HKU30, termed ICE-HKU30. The element shared the same overall genetic architecture to ICE-emm12 including the ermB and tetM genes. The overall nucleotide homology between the 2 ICE elements is 67%, suggesting a common yet distant evolutionary relationship. In the draft HKU30 genome sequence, ICE-HKU30 is located within a 2096.1-like prophage and as such localizes to a genomic location different from that of ICE-emm12 (Figure 4). Both ICE-emm12 and ICE-HKU30 share <50% nucleotide homology to the tetracycline (tetO) and erythromycin (ermA) harboring ICE elements of 2096-RD.2 (MGAS2096, emm12) and 10750-RD.2 (MGAS10750, emm4), respectively [28], suggesting that the multidrug-resistant ICE elements in HKU16 and HKU30 are genetically distinct. In HKU16, the 46.4-kb insertion is a prophage designated ΦHKU.vir encoding 66 open reading frames, including genes encoding streptococcal superantigens SSA and SpeC, and the DNase Spd1 virulence factor (Figure 5B). The ssa gene is arranged in the opposite orientation to the speC and spd1 genes. ΦHKU.vir was also identified in the draft HKU30 genome sequence.
Figure 5.
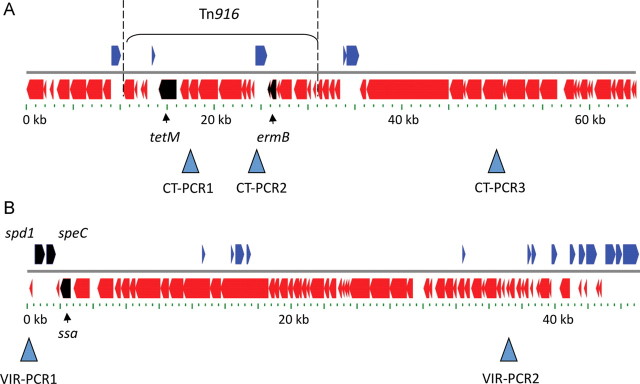
Diagrammatic representation of (A) the 64.9-kb composite transposon-like element ICE-emm12 and (B) 46.4-kb ΦHKU.vir prophage. A, The position of Tn916 in the composite transposon-like element is indicated by a bracket. The tetM and ermB genes are given as black arrows. The positions of 3 diagnostic polymerase chain reaction (PCR) assays for CT-PCR1, CT-PCR2, and CT-PCR3 are indicated by arrowheads. Scale bar is given in kb. B, The spd1, speC, and ssa genes are given as black arrows. The positions of 2 diagnostic PCR assays for VIR-PCR1 and VIR-PCR2 are indicated by arrowheads. Scale bar is given in kb.
PCR assays were developed for clinical diagnostic purposes and to detect ICE-emm12–containing tetM and ermB (CT-PCR1, CT-PCR2, and CT-PCR3) and phage ΦHKU.vir (VIR-PCR1 and VIR-PCR2) (Figure 5) in the GAS strain set. In total, 86% of the 2011 emm12 scarlet fever isolates contained ICE-emm12 (19 of 22 isolates), 86% harbored prophage ΦHKU.vir (19 of 22 isolates), and 81% contained both ICE-emm12 and prophage ΦHKU.vir (18 of 22 isolates). Of the temporally and geographically matched non–scarlet fever GAS, 4 of 7 carried ICE-emm12, 3 of 7 harbored ΦHKU.vir, and 3 of 7 contained both elements. Of the 18 Hong Kong GAS strains isolated between 2005 and 2010, 66% carried ICE-emm12 (12 of 18), 72% harbored ΦHKU.vir (13 of 18), and 66% contained both elements (12 of 18) (Table 1 and Figure 2).
Comparison of HKU16 and HKU30 With the Globally Disseminated M1T1 Clone Strain 5448
The GAS M1T1 clone and serotype M12 strains share a number of genomic features including a 36-kb chromosomal region encoding the extracellular toxins NAD+-glycohydrolase and streptolysin, and the homologous prophage Φ9429.3/Φ5005.3 encoding the streptodornase SdaD2/Sda1 [8, 27]. Several recent studies have documented an association between mutations in the covRS 2-component gene regulatory system of S. pyogenes with enhanced virulence in mouse models of infection and invasive disease in humans. Such mutations result in the loss of expression of the cysteine protease SpeB and upregulation of multiple virulence factors including SdaD2/Sda1 [7, 25, 29]. HKU16, HKU30, and 5448 were found to express SpeB, as determined by Western immunoblot with SpeB-specific antisera (Figure 6A). A phenotypic comparison of the ability of HKU16, HKU30, and the M1T1 strain 5448 to adhere to the HEp-2 human epithelial cell line, resist human neutrophil killing, and exhibit virulence in mouse models of infection was undertaken. HKU16 was found to display significantly higher levels of adherence to the HEp-2 human epithelial cell line in comparison with 5448; the adherence of HKU30 did not differ significantly from either HKU16 or 5448 (Figure 6B). HKU16, HKU30, and 5448 exhibited equivalent levels of resistance to killing by human neutrophils (Figure 6C). To compare the invasive potential of HKU16 and HKU30 with 5448, virulence was examined in a humanized plasminogen mouse model. While the SpeB-positive S. pyogenes strain 5448 is virulent in this model of invasive infection [25], several nonserotype M1 SpeB-positive strains are avirulent owing to an inability to switch to an SpeB-negative invasive phenotype [24]. Similar to the M1T1 strain 5448, HKU16 and HKU30 are capable of causing lethal infection in mice (Figure 6D–6F). To further explore the mechanism of virulence in the mouse model, we examined the capacity of 5448, HKU16, and HKU30 to switch to the SpeB-negative form 3 days after infection. In contrast to strain 5448, lethal infection in a murine model by HKU16 and HKU30 was not linked to mutations in the covRS 2-component regulatory system that led to a SpeB-negative phenotype (Figure 6G–I) [24].
Figure 6.

Phenotypic comparison of Streptococcus pyogenes strains HKU16, HKU30, and 5448. A, Western blot revealed production of the 28-kDa mature cysteine protease SpeB (arrowhead). B, Bacterial adherence to the human epithelial HEp-2 cell line in vitro (mean ± SEM). Compared with the M1T1 strain 5448, the outbreak strain HKU16 displayed significantly increased adherence (P < .01, unpaired 2-tailed Student t test). C, No significant difference between the percentage of survival of HKU16, HKU30, and 5448 was observed after 30 min coincubation with human neutrophils in vitro (1-way analysis of variance). D and E, The percentage of survival of humanized plasminogen AlbPLG1 (solid line; n = 10) and wild-type control C57BL/J6 (dashed line; n = 10) mice, over 10 days, following subcutaneous challenge with (D) HKU16 (5.2 × 107 colony-forming units [CFUs]/dose), (E) HKU30 (5.2 × 107 CFUs/dose), and (F) 5448 (3.9 × 107 CFUs/dose). Data in (F) taken from previous work [24]. No significant difference between the virulence of these strains in humanized plasminogen mice was detected (log-rank test). G–I, The capacity of group A Streptococcus (GAS) to SpeB switch to an invasive covRS mutant form in vivo. G, Total CFUs subcutaneously administered to C57BL/J6 mice (n = 10). H, Total CFUs recovered per gram of lesion (infection site). Each data point represents a single mouse. I, Percentage of SpeB-negative CFUs retrieved following subcutaneous passage (n = 50 CFUs/mouse). Only the M1T1 strain 5448 was observed to readily SpeB-switch during local infection.
DISCUSSION
Given the rapid surge in scarlet fever cases documented in 2011, it is perhaps a surprising observation that the Hong Kong outbreak was not caused by a single, newly emergent clone. PFGE analysis indicates this epidemic is multiclonal in nature, suggesting a lack of evolutionary pressure for a single clone. The unifying characteristic of the majority of emm12 scarlet fever isolates is the presence of ICE-emm12 and ΦHKU.vir. PCR analysis of 18 emm12 GAS isolated in the 6 years preceding the outbreak shows that 66% of these strains carried ICE-emm12, 72% contained ΦHKU.vir, and 66% harbored both elements. Given the isolation of strains containing these horizontally acquired elements in the Hong Kong GAS population prior to the outbreak, it seems likely that the GAS emm12 strains causing the sharp rise in scarlet fever in 2011 may have been circulating in the Hong Kong population for a number of years. Increased resistance to antibiotics may have led to a gradual increase in their relative abundance in the Hong Kong population over time. Furthermore, scarlet fever is a disease whose symptoms occur primarily as a result of the host's immune response to infection, and thus disease outcome will depend on both the presence of a GAS strain with the ability to cause disease and a host immune state predisposed to disease [30]. Although acquisition of ΦHKU.vir may have provided an increased likelihood of scarlet fever owing to the presence of superantigens SSA and SpeC, it is unlikely that the dramatic increase in cases in 2011 is due solely to bacterial factors. However, it is possible to speculate that the outbreak was triggered by a combination of either population immune status and/or other unknown environmental factor(s) that resulted in a population that is particularly susceptible to disease. Thus, we hypothesize that a complex of factors underpins the emergence of this multiclonal emm12 scarlet fever infection cluster.
Scarlet fever is a toxin-mediated disease. As such, the ability to produce pyrogenic exotoxins such as SpeA, SpeC, SSA, and other superantigens is important in the pathogenesis of scarlet fever and toxic shock syndrome [4]. The acquisition and carriage of the superantigens SpeC and SSA on the ΦHKU.vir prophage in the majority of emm12 scarlet fever isolates results in a combination of superantigens which, we suggest, is one factor that has contributed to disease severity and upsurge of scarlet fever in Hong Kong. The acquisition of superantigen SpeA and streptodornase Sda1/SdaD2 is thought to have played a major role in global dissemination and invasive disease severity of the M1T1 clone of S. pyogenes [6–8, 25]. Similarly, acquisition of a Shiga toxin 2–encoding prophage is also predicted to have driven the Escherichia coli O104:H4 outbreak that occurred in May and June 2011 in Germany [31, 32]. Such outbreaks exemplify the important role of bacteriophage acquisition by bacterial pathogens in the development of epidemic disease.
The presence of ICE-emm12 or other genetic elements encoding tetracycline and macrolide resistance in emm12 strains such as HKU16 may confer bacterial survival advantage as these antimicrobials are commonly used for treatment of upper respiratory tract infections of childhood. This has important implications for clinical management, as most guidelines recommend clindamycin to decrease toxin production in addition to bactericidal penicillin G in patients with streptococcal toxic shock [33]. However, in vitro studies suggest that toxin production might paradoxically increase in clindamycin resistant S. pyogenes incubated with clindamycin [34]. ICE-emm12 also encodes a MATE efflux pump that may confer environmental survival advantage by possible resistance against chemical disinfectants such as cetrimide, chlorhexidine, or dequalinium [35]. ICE-emm12 shares most homology with genes found in other gram-positive species. Finally, comparative genomic analysis with emm12 strains MGAS9426 and MGAS2096 found that HKU16, and not HKU30, contains a major genomic inversion occurring between 2 transposase-like elements. A similar change has been previously reported to be associated with an invasive M3 strain of S. pyogenes [36].
The Hong Kong scarlet fever epidemic emm12 strains HKU16 and HKU30 share a number of genetic and phenotypic traits with the globally disseminated M1T1 strain 5448. Common genetic features include carriage of a 36-kb chromosomal region encoding the extracellular toxins NAD+-glycohydrolase and streptolysin O, and the prophage Φ9429.3/Φ5005.3 encoding streptodornase SdaD2/Sda1 [8, 27]. Phenotypically, these strains share similar resistance profiles against human neutrophils and virulence potential in a humanized plasminogen mouse model of invasive disease. However, in contrast to M1T1 strain 5448, HKU16 exhibits significantly higher adherence to HEp-2 cells while HKU16 and HKU30 display virulence potential unlinked to mutations in the covRS 2-component regulatory [7]. Each of these phenotypes may play a role in GAS niche adaption, colonization, immune resistance, and/or virulence.
In this study, we have described the use of rapid whole genome sequencing and complementary phylogenetic analysis to demonstrate the multiclonal nature of the 2011 Hong Kong scarlet fever outbreak, suggesting that a complex of factors underpin this epidemic. The unifying characteristic of the majority of emm12 scarlet fever isolates is the presence of 2 novel horizontally acquired elements, ICE-emm12 and ΦHKU.vir. Diagnostic PCR assays for ICE-emm12 and ΦHKU.vir have been validated to monitor the distribution of these mobile genetic elements in the GAS population.
Notes
Acknowledgments. The authors thank Dr Matthew Holden, Wellcome Sanger Institute, for bioinformatics assistance; Dr Samson Wong; and staff of the Public Health Laboratory Centre, Centre for Health Protection, Department of Microbiology at Queen Mary Hospital, Kwong Wah Hospital, and the University of Hong Kong for their kind support.
Financial support. The work was supported by donations from Ms Eunice Yin-Nei Lam, Mr Sai-Hong Yeung, and Mr Tony Yeung; a Research Fund for the Control of Infectious Disease–commissioned grant and government consultancy project from the Department of Health of Hong Kong; the National Health and Medical Research Council of Australia; and the Wellcome Trust, United Kingdom.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Katz AR, Morens DM. Severe streptococcal infections in historical perspective. Clin Infect Dis. 1992;14:298–307. doi: 10.1093/clinids/14.1.298. [DOI] [PubMed] [Google Scholar]
- 2.Morens DM, Folkers GK, Fauci AS. The challenge of emerging and re-emerging infectious diseases. Nature. 2004;430:242–9. doi: 10.1038/nature02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–94. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis. 2009;9:611–6. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 6.Cleary PP, Kaplan EL, Handley JP, et al. Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet. 1992;339:518–21. doi: 10.1016/0140-6736(92)90339-5. [DOI] [PubMed] [Google Scholar]
- 7.Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–36. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 8.Sumby P, Porcella SF, Madrigal AG, et al. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis. 2005;192:771–82. doi: 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 9.Beres SB, Sylva GL, Studevant DE, et al. Genome-wide molecular dissection of serotype M3 group A Streptococcus strains causing two epidemics of invasive infections. Proc Natl Acad Sci U S A. 2004;101:11833–8. doi: 10.1073/pnas.0404163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green NM, Zhang S, Porcella SF, et al. Genome sequence of a serotype M28 strain of group A Streptococcus: potential new insights into puerperal sepsis and bacterial disease specificity. J Infect Dis. 2005;192:760–70. doi: 10.1086/430618. [DOI] [PubMed] [Google Scholar]
- 11.Wasserzug O, Valinski L, Klement E, et al. A cluster of ecthyma outbreaks caused by a single clone of invasive and highly infective Streptococcus pyogenes. Clin Infect Dis. 2009;48:1213–19. doi: 10.1086/597770. [DOI] [PubMed] [Google Scholar]
- 12.Tyrrell GJ, et al. Epidemic of group A Streptococcus M/emm59 causing invasive disease in Canada. Clin Infect Dis. 2010;51:1290–7. doi: 10.1086/657068. [DOI] [PubMed] [Google Scholar]
- 13.Lau MCK. Increase in scarlet fever cases in 2011. Communicable Diseases Watch. 2011;8:48–9. [Google Scholar]
- 14.Beall B, Facklam R, Thompson T. Sequencing emm-specific PCR products for routine and accurate typing of group A streptococci. J Clin Microbiol. 1996;34:953–8. doi: 10.1128/jcm.34.4.953-958.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carrico JA, Silva-Costa C, Melo-Cristina J, et al. Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J Clin Microbiol. 2006;44:2524–32. doi: 10.1128/JCM.02536-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tse H, Tsoi HW, Leung SP, et al. Complete genome sequence of Staphylococcus lugdunensis strain HKU09–01. J Bacteriol. 2010;192:1471–2. doi: 10.1128/JB.01627-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tse H, Tsoi HW, Leung SP, et al. Complete genome sequence of the veterinary pathogen Staphylococcus pseudintermedius strain HKU10-03, isolated in a case of canine pyoderma. J Bacteriol. 2011;193:1783–4. doi: 10.1128/JB.00023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 2009;25:1968–9. doi: 10.1093/bioinformatics/btp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11:R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carver T, Berriman M, Tivey A, et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–76. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maamary PG, Sanderson-Smith ML, Aziz RK, et al. Parameters governing invasive disease propensity of non-M1 serotype group A streptococci. J Innate Immun. 2010;2:596–606. doi: 10.1159/000317640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker MJ, Hollands A, Sanderson-Smith ML, et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med. 2007;13:981–5. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 26.Hollands A, Pence MA, Timmer AM, et al. Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group A Streptococcus M1T1 clone. J Infect Dis. 2010;202:11–9. doi: 10.1086/653124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beres SB, Richter EW, Nagiec MJ, et al. Molecular genetic anatomy of inter- and intraserotype variation in the human bacterial pathogen group A Streptococcus. Proc Natl Acad Sci U S A. 2006;103:7059–64. doi: 10.1073/pnas.0510279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beres SB, Musser JM. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One. 2007;2:e800. doi: 10.1371/journal.pone.0000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2006;2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casadevall A, Pirofski L. Host-pathogen interactions: the attributes of virulence. J Infect Dis. 2001;184:337–44. doi: 10.1086/322044. [DOI] [PubMed] [Google Scholar]
- 31.Rasko DA, Webster DR, Sahl JW, et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med. 2011;365:709–17. doi: 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohde H, Qin J, Cui Y, et al. Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N Engl J Med. 2011;365:718–24. doi: 10.1056/NEJMoa1107643. [DOI] [PubMed] [Google Scholar]
- 33.Lappin E, Ferguson AJ. Gram-positive toxic shock syndromes. Lancet Infect Dis. 2009;9:281–90. doi: 10.1016/S1473-3099(09)70066-0. [DOI] [PubMed] [Google Scholar]
- 34.Minami M, Kamimura T, Isaka M, et al. Clindamycin-induced CovS-mediated regulation of the production of virulent exoproteins streptolysin O, NAD glycohydrolase, and streptokinase in Streptococcus pyogenes. Antimicrob Agents Chemother. 2010;54:98–102. doi: 10.1128/AAC.00804-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuroda T, Tsuchiya T. Multidrug efflux transporters in the MATE family. Biochim Biophys Acta. 2009;1794:763–8. doi: 10.1016/j.bbapap.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa I, Kurokawa K, Yamashita A, et al. Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res. 2003;13:1042–55. doi: 10.1101/gr.1096703. [DOI] [PMC free article] [PubMed] [Google Scholar]