

Abstract
Bile secretion is dependent on the coordinated functions of a number of hepatobiliary transport systems. Cholestasis may be caused by an impairment of bile secretion, an obstruction of bile flow or a combination of the two. The common consequence of all forms of cholestasis is retention of bile acids and other potentially toxic compounds in the hepatocytes leading to apoptosis or necrosis of hepatocytes and eventually to chronic cholestatic liver disease. In certain cholestatic disorders there is also leakage of bile acids into the peribiliary space causing portal inflammation and fibrosis. The following pharmacological targets for treatment of intrahepatic cholestasis can be identified: stimulation of orthograde biliary secretion and retrograde secretion of bile acids and other toxic cholephils into the systemic circulation for excretion via the kidneys to reduce their retention in the hepatocytes; stimulation of the metabolism of hydrophobic bile acids and other toxic compounds to more hydrophilic, less toxic metabolites; protection of injured cholangiocytes against toxic effects of bile; inhibition of apoptosis caused by elevated levels of cytotoxic bile acids; inhibition of fibrosis caused by leakage of bile acids into the peribiliary space. The clinical results of ursodeoxcholic acid therapy of primary biliary cirrhosis may be regarded as the first success of this strategy.
Keywords: Bile secretion, Biliary transport, Cholestasis, Nuclear receptors, Cholestatic liver disease, Primary biliary cirrhosis, Ursodeoxycholic acid
INTRODUCTION
Great progress has been made in the last decade in our understanding of the molecular basis of bile formation and the pathobiology of cholestasis[1-3]. Targets for medical therapy of cholestasis have been identified which help to understand the established treatments and facilitate the development of new drugs for cholestatic liver disease. In this short review, present concepts of bile formation and cholestasis are briefly summarized and medical treatment of cholestatic liver diseases is illustrated using primary biliary cirrhosis (PBC), the model disease for chronic cholestatic liver disease, as an example.
MOLECULAR MECHANISMS OF BILE FORMATION
Hepatocellular bile is formed by active transport of solutes into the bile canaliculi. Thereby, a local osmotic gradient is established between canalicular bile and sinusoidal plasma. This causes a flow of water, electrolytes and small solutes into the bile canaliculi, mainly via a paracellular pathway through the tight junctions which exhibit perm selectivity, and are impermeable for large and negatively charged solutes[4].
The most important driving force for hepatocellular bile formation is the secretion of bile acids from the sinusoidal blood into the bile[3]. Conjugated bile acids, which represent the major fraction of bile acids in the blood, are transported across the basolateral membrane of hepatocytes together with sodium by the sodium-taurocholate cotransporter (NTCP, SLC10A1). Un-conjugated bile acids and a large variety of other organic anions including bilirubin are taken up by the hepatocytes via the organic anion-transporting polypeptide 2 (OATP2, SLC21A6). The rate limiting step for bile formation is the active transport of bile acids and other solutes across the canalicular membrane of hepatocytes. This concentrative step is driven by a number of ATP-dependent export pumps (ATP-binding-cassette-transport proteins also known as ABC-transporters). Bile salts are transported by the bile salt export pump (BSEP, ABCB11), whereas bilirubin diglucuronide, glutathione, divalent bile acids conjugates and a large variety of other conjugated organic anions are transported by the multidrug resistance associated protein 2 (MRP2, ABCC2)[2].
A special ABC-transporter, namely the multidrug resistance P-glycoprotein 3 (MDR3, ABCB4), flipps phospholipids from the inner to the outer leaflet of the canalicular membrane. This flippase provides phosphotidylcholine for bile which forms mixed micelles with bile acids and cholesterol[2].
The formation and final composition of bile depends on additional transporters in the canalicular membrane of hepatocytes as well as transporters in cholangiocytes which add cholangiocellular bile to hepatocellular bile. Among those, the chloride-bicarbonate anion exchanger 2 (AE2, SLC10A2) is present in the apical membrane of both hepatocytes and bile duct epithelial cells, whereas the cystic fibrosis transmembrane conductance regulator (CFTR, ABCC7), a chloride channel, is located in the apical membrane of bile duct epithelial cells only[2].
The basolateral membrane of hepatocytes possesses a number of transporters which are expressed during cholestasis and transport solutes in a retrograde fashion back into the blood (see below). These are MRP4 (ABCC4) which transports bile acids together with glutathione[5], MRP3 (ABCC3) which transports conjugated bilirubin and other organic anions[2,6] and OSTα/OSTβ, a heteromeric organic solute transporter which transports bile acids[7]. During cholestasis, MRP3[8] and OSTα/OSTβ are also upregulated in the basolateral membrane of cholangiocytes[9].
MOLECULAR MECHANISMS OF CHOLE-STASIS
Cholestasis can be defined as an impairment of bile flow. The consequences are retention of bile acids, bilirubin and other cholephils in the liver and blood and a deficiency of bile acids in the intestine. Various forms of cholestasis can be caused by an impairment of bile secretion, an obstruction of bile flow or a combination of the two (Figure 1).
Figure 1.
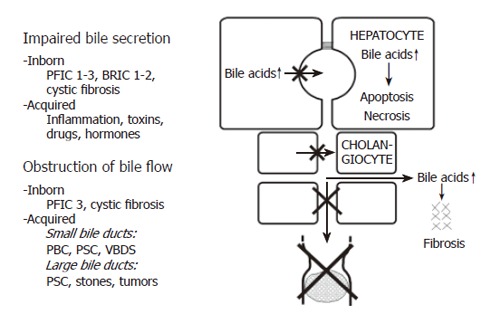
Causes of cholestasis. PBC: Primary biliary cirrhosis; PFIC: Progressive familial intrahepatic cholestasis; PSC: Primary sclerosing cholangitis; VBDS: Vanishing bile duct syndrome. For details see text.
Impairment of bile secretion can be inborn, for instance in different forms of progressive familial intrahepatic cholestasis (PFIC), benign recurrent intrahepatic cholestasis (BRIC), or cystic fibrosis, and it also can be acquired by inflammation, toxins, drugs or hormones[8,10].
Inborn defects of bile secretion: If BSEP is defective because of a gene mutation, PFIC2 or BRIC2 [11] can occur. PFIC2 can be identified by immunostaining of BSEP in liver biopsies[12].
Mutations of MRP2 cause the Dubin Johnson syndrome, which is not a complete cholestasis, but a more selective defect of biliary secretion of organic anions such as bilirubin glucuronide. Mutations of MDR3 cause PFIC3 and mutations of CFTR cause cystic fibrosis[2,8].
Acquired impairment of bile secretion: In inflammatory disorders such as sepsis, bacterial infections, viral hepatitis as well as toxin or drug-induced hepatitis, inflammatory cytokines can impair bile secretion. Thus, TNFα and IL-1β down regulate NTCP and BSEP which are responsible for bile acid transport, as well as OATP2 and MRP2 which are responsible for transport of bilirubin and a variety of other organic ions[13,14].
Drugs can cause cholestasis by inhibiting the function of hepatobiliary transport proteins. Some drugs are known to inhibit BSEP directly from the inside of hepatocytes, which is called cis-inhibition. Examples are cyclosporine A, glibenclamide, troglitazone and bosentan[15,16]. Other drugs, such as estradiol 17β-D-glucuronide, must first be transported into the canalicular lumen by MRP2 and then act on BSEP from the luminal side, which is called trans- inhibition[17].
Obstruction of bile flow can also be caused by inborn disorders, e.g. in cystic fibrosis or in PFIC3, and it can be acquired for instance in PBC, primary sclerosing cholangitis (PSC) or the vanishing bile duct syndrome (VBDS). Much more often obstructive cholestasis is caused by stones or tumours.
In cholestatic disorders caused by an initial injury of cholangiocytes (e.g. an immunological injury in the case of PBC), hydrophobic bile acids in bile (in millimolar concentrations) may aggravate the bile duct lesion and contribute to the destruction and loss of bile ducts resulting in progressive obstructive cholestasis. This may be called extracellular bile acid cytotoxicity in contrast to intracellular bile acid toxicity when bile acids accumulate in hepatocytes (in micromolar concentrations). Extracellular bile acid toxicity also occurs towards normal biliary epithelium when phospholipids in bile are low, as in the inborn defect of PFIC3 or in other “low phospholipid syndromes”, in low phospholipid gallstone disease[18] or in bile acid phospholipid imbalance in bile after liver transplantation[19].
ADAPTIVE RESPONSES TO CHOLESTASIS
In order to compensate for the loss of biliary excretory function in cholestasis and to limit hepatocellular accumulation of potentially toxic biliary constituents, adaptive responses to cholestasis occur in the liver[6,8,14,20,21], the kidney[20-23] and the intestine[22,24]. In the following only the adaptive changes in the liver are discussed.
Down regulation of NTCP and OATP2 reduces the uptake of bile acids and other organic anions in cholestasis and thus protects the hepatocytes against an overload of bile acids and bilirubin[3,25,26]. At the same time there is upregulation of MRP3 and MRP4 in the basolateral membrane[14,22,27-29]. These transporters normally are expressed at a low level only. MRP4 pumps bile salts and bile salt conjugates together with gluthatione from the cells into the blood and thus decreases bile acid retention in cholestatic hepatocytes. MRP3 mainly exports other organic anions. Prior to their extrusion from hepatocytes, hydrophobic bile acids and many xenobiotics are metabolized to more hydrophilic and less toxic compounds by cytochrome P-450(CYP) 3A enzymes. A large fraction of bile acids is sulphated by the enzyme sulfotransferase 2A1 (Figure 2).
Figure 2.
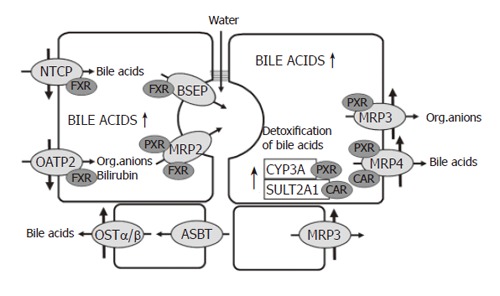
Adaptive responses to cholestasis. BSEP: Bile salt export pump; CAR: Constitutive androstane receptor; CYP3A: Cytochrome P450 enzyme 3A; FXR: Farnesoid X receptor; OATP: Organic anion transporting polypeptide; OST: Organic solute transporter; MRP: Multidrug resistance associated protein; NTCP: Sodium taurocholate co-transporting polypeptide; PXR: Pregnane X receptor; SULT2A1: Sulphotranferase 2A1. For details see tect.
The major players in these adaptive regulations are the nuclear receptors FXR, PXR and CAR[30]. The farnesoid X- receptor (FXR), a bile acid sensor, is mainly involved in the down regulation of NTCP, in the maintenance of BSEP function and in the up-regulation of MRP4 and MDR3. The pregnane X receptor (PXR), to which many xenobiotics bind, is mainly responsible for the up-regulation of MRP3 and various CYP enzymes, especially the family of CYP3A enzymes. There is evidence that more than one of these nuclear receptors can act on the same transporter. Recently, it has been demonstrated that the constitutive androstane receptor (CAR) up-regulates sulfotransferase 2A1 and MRP4 in a coordinated fashion facilitating the conjugation and export of hydrophobic bile acids[31]. In addition to PXR, the perioxisome–proliferator-activated receptor α (PPARα) up-regulates MDR3 (Figure 2).
It is of considerable interest that besides natural bile acids, bile acid derivatives such as ethyl-chenodeoxycholic acid are ligands for FXR[32,33]. Ligands for PXR are many xenobiotics and drugs like rifampicin. Bilirubin and phenobarbital are ligands for CAR and fibrates as well as statins (e.g. pravastatin) bind to PPARα.
These findings open an avenue for the development of drugs which bind to nuclear receptors which enhance normal compensatory mechanisms in cholestasis for the elimination of toxic compounds via alternative excretory routes.
TARGETS FOR PHARMACOLOGICAL THERAPY
The common consequence of all forms of cholestasis is retention of bile acids in hepatocytes. Elevated levels of bile acids then can lead to apoptosis or necrosis of hepatocytes and eventually to chronic cholestatic liver disease[34]. In certain cholestatic disorders there is also leakage of bile acids into the peribiliary space, causing portal inflammation and fibrosis via induction of chemokines and cytokines[35]. Accordingly, the following pharmacological targets for treatment of intrahepatic cholestasis can be identified (Figure 3): stimulation of orthograde biliary secretion and retrograde secretion of bile acids and other toxic cholephils into the systemic circulation for excretion by the kidneys to reduce their retention in the hepatocytes; stimulation of the metabolism of hydrophobic bile acids and other toxic compounds to more hydrophilic but less toxic metabolites; protection of injured cholangiocytes against toxic effects of bile; inhibition of apoptosis caused by elevated levels of cytotoxic bile acids; inhibition of fibrosis caused by leakage of bile acids into the peribiliary space.
Figure 3.
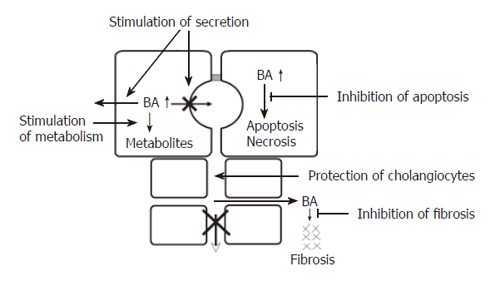
Targets for medical treatment of intrahepatic cholestasis. BA: Bile acids. For details see text.
Stimulation of secretion
Secretion of bile acids and other potentially toxic compounds into the bile and blood may be stimulated by enhancing transporter expression and/or function at different levels, namely the levels of transcription, translation, targeting and protein activation.
In mice both cholic acid (CA) and UDCA stimulate the expression of Bsep and Mrp2 mRNA[36]. One must, however, be aware that these findings may not hold true for men, because considerable species differences exist with regard to binding of bile acids to nuclear receptors and regulation of transporter expression by nuclear receptors. Rifampicin, a ligand of PXR, stimulates the expression of MRP2 at the transcriptional level in man[37].
Ursodeoxycholic acid (UDCA) stimulates targeting of the transporters Bsep and Mrp2 to the canalicular membrane in the rat via at least two different signalling cascades[38-40]. Immunoelectronmicroscopy with gold particles is employed to assess localization of Bsep in the canalicular membrane and in a subapical compartment of rat liver. Bsep and Mrp2 in the canalicular membrane are markedly reduced when taurolithocholic acid (TLCA) is administered in the perfused rat liver, but is maintained when tauroursodeoxycholic acid (TUDCA) is added[38,40]. Enhanced expression of BSEP under UDCA treatment in men may contribute to a better elimination of bile acids from the blood. As shown by Poupon et al[41], in collaboration with our group, UDCA decreases serum levels of the hydrophobic bile acid, chenodeoxycholic acid (CDCA) in PBC. As shown by Zollner et al[14], expression of MRP2 mRNA and protein increases with the enrichment of UDCA in the liver during treatment of patients with PBC and UDCA. Accordingly, as shown by Poupon et al[42], UDCA improves excretory function in PBC. Thus, in a randomised, placebo controlled study over two years, in patients with PBC, serum bilirubin was significantly lower in the UDCA group than in the placebo group.
Activation of transporters in the canalicular membrane by UDCA and phosphorylation may also occur[43], which has not yet been sufficiently studied.
Stimulation of metabolism
Stimulation of the metabolism of hydrophobic bile acids produces more hydrophilic and less toxic compounds. Rifampicin, a drug used for the treatment of cholestatic pruritus, stimulates the expression of CYP3A4 mRNA in patients with gallstones. In line with this, Dilger et al[44] showed that in patients with early stage PBC, rifampicin stimulates CYP3A metabolic activity as assessed by urinary 6β-hydroxy cortisol, whereas UDCA has no effect.
Protection of cholangiocytes
Protection of cholangiocytes by making the bile more hydrophilic and less toxic appears to be an important therapeutic target. UDCA fulfils this requirement because it renders bile acid composition of bile more hydrophilic and increases biliary phospholipid secretion[45].
Inhibition of apoptosis
Inhibition of apoptosis caused by elevated levels of hydrophobic bile acids[46,47] may also be a therapeutic target in cholestasis. As shown by Rodrigues et al[48,49], feeding of the hydrophobic bile acid deoxycholic acid (DCA) to rats increases hepatocyte apoptosis as assessed by the number of tunnel positive hepatocytes. Addition of UDCA inhibits this effect. Toxic bile acids such as CDCA can cause apoptosis of hepatocytes via the CD95 receptor with formation of a death inducing signalling complex (DISC) and activation of caspase 8. Caspase 8 then causes mitochondrial membrane permeability transition (MMPT) which leads to activation of effector caspases and apoptosis. In addition, UDCA stabilizes the mitochondrial membrane and inhibits MMPT and apoptosis[48,49]. The antiapoptotic effect of UDCA has also been demonstrated in human hepatocytes[50].
Inhibition of fibrosis
Inhibition of fibrosis may become an important therapeutic target in the future. In the rat with common bile duct ligation, fibrosis can be inhibited by 6-ethyl CDCA (6-ECDCA). The antifibrotic effect of 6-ECDCA appears to be mediated via FXR and SHP[33]. Recently, an antifibrotic effect of NOR-UDCA has been described in the Mdr2 knock-out mouse[51]. It remains to be shown whether these findings are relevant to human cholestatic liver diseases, but they point towards a promising new way for the development of drugs to inhibit cholestatic fibrosis.
PHARMACOLOGICAL TREATMENT OF CHRONIC CHOLESTATIC LIVER DISEASES
In the following, primary biliary cirrhosis (PBC), the model disease for chronic cholestatic liver disease, is used as an example for the treatment of chronic cholestatic liver diseases by UDCA. PBC is characterized by an inflammatory lesion of interlobular bile ducts, which results in bile duct destruction and may progress to fibrosis and cirrhosis. Since the etiology of the disease is unknown, presently available therapies aim at inhibiting the underlying pathogenetic processes and delaying the progression of the disease.
The pathogenesis of this slowly progressive disease involves a still unknown immunologic injury of small interlobular bile ducts; aggravation of the bile duct lesion by cytotoxic bile acids; obstruction and loss of small bile ducts followed by cholestasis and retention of bile acids; hepatocyte injury, apoptosis, necrosis, fibrosis and eventually cirrhosis with liver failure.
UDCA, at present, is the only approved drug for PBC. It appears to exert its beneficial effects by rendering bile composition less toxic for the injured biliary epithelium, reducing the retention of bile acids in hepatocytes and inhibiting apoptosis[10,34]. Immunosuppressive agents have met with limited success. They have been found useful in combination with UDCA in selected patients[52,53].
In randomized, double-blind placebo-controlled trials UDCA at doses of 13-15 mg/kg body weight per day could improve serum liver tests including serum bilirubin and other serum markers of cholestasis[42,54-56], the Mayo risk score[56] and liver histology[42,55]. As shown by Pares et al[54] and Poupon et al[57], UDCA inhibits histological progression in early stage PBC. As shown by Corpechot et al[58], UDCA inhibits progression to severe liver fibrosis or cirrhosis in early stage PBC. In line with this is the observation that UDCA delays the onset of esophageal varices[59]. A combined analysis of three of the largest trials showed that treatment with UDCA at doses of 13-15 mg/kg per day for up to 4 years can delay the time of liver transplantation or death[60]. Within the first 2 years of treatment, however, a survival benefit was not seen. Doses lower than 10 mg/kg per day of UDCA are of little benefit in PBC[61]. A meta-analysis of 8 randomized trials which showed no difference between UDCA and placebo in the effects on incidence of death, liver transplantation and death or liver transplantation[62] has a number of shortcomings. In 6 of the 8 studies treatment was evaluated up to 24 mo only and the dose of UDCA was 10 mg/kg per day or lower in two of the studies. Therefore, improvement of transplant free survival by UDCA as shown in the combined analysis of the three largest studies with doses of 13-15 mg/kg per day and a follow-up of 4 years may not have been detectable in this meta-analysis.
CONCLUSION
Better insight into the pathobiology of cholestasis has provided new concepts for pharmacological therapies of cholestatic liver diseases. Among those, therapy with UDCA has been studied most extensively. In PBC, the model disease for cholestatic liver diseases which has been highlighted in this review, the beneficial effects of UDCA have been documented by randomized controlled trials. Treatment with UDCA appears to be beneficial also in a number of other cholestatic disorders, such as primary sclerosing cholangitis (PSC)[63-65], intrahepatic cholestasis of pregnancy[66,67], liver disease in cystic fibrosis[68-70], progressive familial intrahepatic cholestasis (PFIC)[71] and some forms of drug-induced cholestasis[10].
Footnotes
S- Editor Wang J L- Editor Wang XL E- Editor Ma WH
References
- 1.Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339:1217–1227. doi: 10.1056/NEJM199810223391707. [DOI] [PubMed] [Google Scholar]
- 2.Kullak-Ublick GA, Beuers U, Paumgartner G. Hepatobiliary transport. J Hepatol. 2000;32:3–18. doi: 10.1016/s0168-8278(00)80411-0. [DOI] [PubMed] [Google Scholar]
- 3.Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126:322–342. doi: 10.1053/j.gastro.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Nathanson MH, Boyer JL. Mechanisms and regulation of bile secretion. Hepatology. 1991;14:551–566. [PubMed] [Google Scholar]
- 5.Rius M, Nies AT, Hummel-Eisenbeiss J, Jedlitschky G, Keppler D. Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology. 2003;38:374–384. doi: 10.1053/jhep.2003.50331. [DOI] [PubMed] [Google Scholar]
- 6.Bohan A, Chen WS, Denson LA, Held MA, Boyer JL. Tumor necrosis factor alpha-dependent up-regulation of Lrh-1 and Mrp3(Abcc3) reduces liver injury in obstructive cholestasis. J Biol Chem. 2003;278:36688–36698. doi: 10.1074/jbc.M304011200. [DOI] [PubMed] [Google Scholar]
- 7.Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, Madejczyk MS, Li N. OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270–1279. doi: 10.1002/hep.20961. [DOI] [PubMed] [Google Scholar]
- 8.Trauner M, Wagner M, Fickert P, Zollner G. Molecular regulation of hepatobiliary transport systems: clinical implications for understanding and treating cholestasis. J Clin Gastroenterol. 2005;39:S111–S124. doi: 10.1097/01.mcg.0000155551.37266.26. [DOI] [PubMed] [Google Scholar]
- 9.Soroka C, Zollner G, Mennone A, Ballatori N, Trauner M, Boyer JL. The heteromeric organic solute transporter, OST alpha-OST beta, is up-regulated in the liver of patients with primary biliary cirrhosis (PBC) and variably induced in rat and mouse cholangiocytes following bile duct ligation. Hepatology. 2005;42:414A. [Google Scholar]
- 10.Paumgartner G, Beuers U. Mechanisms of action and therapeutic efficacy of ursodeoxycholic acid in cholestatic liver disease. Clin Liver Dis. 2004;8:67–81, vi. doi: 10.1016/S1089-3261(03)00135-1. [DOI] [PubMed] [Google Scholar]
- 11.van Mil SW, van der Woerd WL, van der Brugge G, Sturm E, Jansen PL, Bull LN, van den Berg IE, Berger R, Houwen RH, Klomp LW. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379–384. doi: 10.1053/j.gastro.2004.04.065. [DOI] [PubMed] [Google Scholar]
- 12.Jansen PL, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal EM, Hooiveld GJ, Koning JH, De Jager-Krikken A, Kuipers F, Stellaard F, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–1379. doi: 10.1016/s0016-5085(99)70287-8. [DOI] [PubMed] [Google Scholar]
- 13.Trauner M, Fickert P, Stauber RE. Inflammation-induced cholestasis. J Gastroenterol Hepatol. 1999;14:946–959. doi: 10.1046/j.1440-1746.1999.01982.x. [DOI] [PubMed] [Google Scholar]
- 14.Zollner G, Fickert P, Zenz R, Fuchsbichler A, Stumptner C, Kenner L, Ferenci P, Stauber RE, Krejs GJ, Denk H, et al. Hepatobiliary transporter expression in percutaneous liver biopsies of patients with cholestatic liver diseases. Hepatology. 2001;33:633–646. doi: 10.1053/jhep.2001.22646. [DOI] [PubMed] [Google Scholar]
- 15.Fattinger K, Funk C, Pantze M, Weber C, Reichen J, Stieger B, Meier PJ. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69:223–231. doi: 10.1067/mcp.2001.114667. [DOI] [PubMed] [Google Scholar]
- 16.Byrne JA, Strautnieks SS, Mieli-Vergani G, Higgins CF, Linton KJ, Thompson RJ. The human bile salt export pump: characterization of substrate specificity and identification of inhibitors. Gastroenterology. 2002;123:1649–1658. doi: 10.1053/gast.2002.36591. [DOI] [PubMed] [Google Scholar]
- 17.Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, Meier PJ. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000;118:422–430. doi: 10.1016/s0016-5085(00)70224-1. [DOI] [PubMed] [Google Scholar]
- 18.Rosmorduc O, Hermelin B, Poupon R. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology. 2001;120:1459–1467. doi: 10.1053/gast.2001.23947. [DOI] [PubMed] [Google Scholar]
- 19.Geuken E, Visser D, Kuipers F, Blokzijl H, Leuvenink HG, de Jong KP, Peeters PM, Jansen PL, Slooff MJ, Gouw AS, et al. Rapid increase of bile salt secretion is associated with bile duct injury after human liver transplantation. J Hepatol. 2004;41:1017–1025. doi: 10.1016/j.jhep.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Azzaroli F, Wang L, Soroka CJ, Gigliozzi A, Setchell KD, Kramer W, Boyer JL. Adaptive regulation of bile salt transporters in kidney and liver in obstructive cholestasis in the rat. Gastroenterology. 2001;121:1473–1484. doi: 10.1053/gast.2001.29608. [DOI] [PubMed] [Google Scholar]
- 21.Denson LA, Bohan A, Held MA, Boyer JL. Organ-specific alterations in RAR alpha:RXR alpha abundance regulate rat Mrp2 (Abcc2) expression in obstructive cholestasis. Gastroenterology. 2002;123:599–607. doi: 10.1053/gast.2002.34758. [DOI] [PubMed] [Google Scholar]
- 22.Zollner G, Fickert P, Fuchsbichler A, Silbert D, Wagner M, Arbeiter S, Gonzalez FJ, Marschall HU, Zatloukal K, Denk H, et al. Role of nuclear bile acid receptor, FXR, in adaptive ABC transporter regulation by cholic and ursodeoxycholic acid in mouse liver, kidney and intestine. J Hepatol. 2003;39:480–488. doi: 10.1016/s0168-8278(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 23.Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. 2003;83:633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 24.Dietrich CG, Geier A, Salein N, Lammert F, Roeb E, Oude Elferink RP, Matern S, Gartung C. Consequences of bile duct obstruction on intestinal expression and function of multidrug resistance-associated protein 2. Gastroenterology. 2004;126:1044–1053. doi: 10.1053/j.gastro.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 25.Jung D, Kullak-Ublick GA. Hepatocyte nuclear factor 1 alpha: a key mediator of the effect of bile acids on gene expression. Hepatology. 2003;37:622–631. doi: 10.1053/jhep.2003.50100. [DOI] [PubMed] [Google Scholar]
- 26.Gartung C, Ananthanarayanan M, Rahman MA, Schuele S, Nundy S, Soroka CJ, Stolz A, Suchy FJ, Boyer JL. Down-regulation of expression and function of the rat liver Na+/bile acid cotransporter in extrahepatic cholestasis. Gastroenterology. 1996;110:199–209. doi: 10.1053/gast.1996.v110.pm8536857. [DOI] [PubMed] [Google Scholar]
- 27.Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, Zatloukal K, Guo GL, Schuetz JD, Gonzalez FJ, et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology. 2003;125:825–838. doi: 10.1016/s0016-5085(03)01068-0. [DOI] [PubMed] [Google Scholar]
- 28.Keitel V, Burdelski M, Warskulat U, Kühlkamp T, Keppler D, Häussinger D, Kubitz R. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology. 2005;41:1160–1172. doi: 10.1002/hep.20682. [DOI] [PubMed] [Google Scholar]
- 29.Denk GU, Soroka CJ, Takeyama Y, Chen WS, Schuetz JD, Boyer JL. Multidrug resistance-associated protein 4 is up-regulated in liver but down-regulated in kidney in obstructive cholestasis in the rat. J Hepatol. 2004;40:585–591. doi: 10.1016/j.jhep.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Boyer JL. Nuclear receptor ligands: rational and effective therapy for chronic cholestatic liver disease. Gastroenterology. 2005;129:735–740. doi: 10.1016/j.gastro.2005.06.053. [DOI] [PubMed] [Google Scholar]
- 31.Assem M, Schuetz EG, Leggas M, Sun D, Yasuda K, Reid G, Zelcer N, Adachi M, Strom S, Evans RM, et al. Interactions between hepatic Mrp4 and Sult2a as revealed by the constitutive androstane receptor and Mrp4 knockout mice. J Biol Chem. 2004;279:22250–22257. doi: 10.1074/jbc.M314111200. [DOI] [PubMed] [Google Scholar]
- 32.Pellicciari R, Costantino G, Camaioni E, Sadeghpour BM, Entrena A, Willson TM, Fiorucci S, Clerici C, Gioiello A. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J Med Chem. 2004;47:4559–4569. doi: 10.1021/jm049904b. [DOI] [PubMed] [Google Scholar]
- 33.Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, Orlandi S, Pellicciari R, Morelli A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127:1497–1512. doi: 10.1053/j.gastro.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525–531. doi: 10.1053/jhep.2002.36088. [DOI] [PubMed] [Google Scholar]
- 35.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127:261–274. doi: 10.1053/j.gastro.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 36.Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Pojer C, Zenz R, Lammert F, Stieger B, Meier PJ, Zatloukal K, et al. Effects of ursodeoxycholic and cholic acid feeding on hepatocellular transporter expression in mouse liver. Gastroenterology. 2001;121:170–183. doi: 10.1053/gast.2001.25542. [DOI] [PubMed] [Google Scholar]
- 37.Marschall HU, Wagner M, Zollner G, Fickert P, Diczfalusy U, Gumhold J, Silbert D, Fuchsbichler A, Benthin L, Grundström R, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129:476–485. doi: 10.1016/j.gastro.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Beuers U, Bilzer M, Chittattu A, Kullak-Ublick GA, Keppler D, Paumgartner G, Dombrowski F. Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C-dependent mechanisms in cholestatic rat liver. Hepatology. 2001;33:1206–1216. doi: 10.1053/jhep.2001.24034. [DOI] [PubMed] [Google Scholar]
- 39.Kurz AK, Graf D, Schmitt M, Vom Dahl S, Haussinger D. Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology. 2001;121:407–419. doi: 10.1053/gast.2001.26262. [DOI] [PubMed] [Google Scholar]
- 40.Dombrowski F, Stieger B, Beuers U. Tauroursodeoxycholic acid inserts the bile salt export pump into canalicular membranes of cholestatic rat liver. Lab Invest. 2006;86:166–174. doi: 10.1038/labinvest.3700371. [DOI] [PubMed] [Google Scholar]
- 41.Poupon RE, Chretien Y, Poupon R, Paumgartner G. Serum bile acids in primary biliary cirrhosis: effect of ursodeoxycholic acid therapy. Hepatology. 1993;17:599–604. doi: 10.1002/hep.1840170412. [DOI] [PubMed] [Google Scholar]
- 42.Poupon RE, Balkau B, Eschwège E, Poupon R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N Engl J Med. 1991;324:1548–1554. doi: 10.1056/NEJM199105303242204. [DOI] [PubMed] [Google Scholar]
- 43.Noe J, Hagenbuch B, Meier PJ, St-Pierre MV. Characterization of the mouse bile salt export pump overexpressed in the baculovirus system. Hepatology. 2001;33:1223–1231. doi: 10.1053/jhep.2001.24171. [DOI] [PubMed] [Google Scholar]
- 44.Dilger K, Denk A, Heeg MH, Beuers U. No relevant effect of ursodeoxycholic acid on cytochrome P450 3A metabolism in primary biliary cirrhosis. Hepatology. 2005;41:595–602. doi: 10.1002/hep.20568. [DOI] [PubMed] [Google Scholar]
- 45.Stiehl A, Rudolph G, Sauer P, Theilmann L. Biliary secretion of bile acids and lipids in primary sclerosing cholangitis. Influence of cholestasis and effect of ursodeoxycholic acid treatment. J Hepatol. 1995;23:283–289. [PubMed] [Google Scholar]
- 46.Reinehr R, Becker S, Keitel V, Eberle A, Grether-Beck S, Häussinger D. Bile salt-induced apoptosis involves NADPH oxidase isoform activation. Gastroenterology. 2005;129:2009–2031. doi: 10.1053/j.gastro.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 47.Yoon JH, Gores GJ. Death receptor-mediated apoptosis and the liver. J Hepatol. 2002;37:400–410. doi: 10.1016/s0168-8278(02)00209-x. [DOI] [PubMed] [Google Scholar]
- 48.Rodrigues CM, Fan G, Ma X, Kren BT, Steer CJ. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest. 1998;101:2790–2799. doi: 10.1172/JCI1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodrigues CM, Fan G, Wong PY, Kren BT, Steer CJ. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med. 1998;4:165–178. [PMC free article] [PubMed] [Google Scholar]
- 50.Benz C, Angermuller S, Otto G, Sauer P, Stremmel W, Stiehl A. Effect of tauroursodeoxycholic acid on bile acid-induced apoptosis in primary human hepatocytes. Eur J Clin Invest. 2000;30:203–209. doi: 10.1046/j.1365-2362.2000.00615.x. [DOI] [PubMed] [Google Scholar]
- 51.Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, Zatloukal K, Liu J, Waalkes MP, Cover C, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–481. doi: 10.1053/j.gastro.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 52.Rautiainen H, Kärkkäinen P, Karvonen AL, Nurmi H, Pikkarainen P, Nuutinen H, Färkkilä M. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: a three-year randomized trial. Hepatology. 2005;41:747–752. doi: 10.1002/hep.20646. [DOI] [PubMed] [Google Scholar]
- 53.Chazouillères O, Wendum D, Serfaty L, Rosmorduc O, Poupon R. Long term outcome and response to therapy of primary biliary cirrhosis-autoimmune hepatitis overlap syndrome. J Hepatol. 2006;44:400–406. doi: 10.1016/j.jhep.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 54.Pares A, Caballeria L, Rodes J, Bruguera M, Rodrigo L, Garcia-Plaza A, Berenguer J, Rodriguez-Martinez D, Mercader J, Velicia R. Long-term effects of ursodeoxycholic acid in primary biliary cirrhosis: results of a double-blind controlled multicentric trial. UDCA-Cooperative Group from the Spanish Association for the Study of the Liver. J Hepatol. 2000;32:561–566. doi: 10.1016/s0168-8278(00)80216-0. [DOI] [PubMed] [Google Scholar]
- 55.Heathcote EJ, Cauch-Dudek K, Walker V, Bailey RJ, Blendis LM, Ghent CN, Michieletti P, Minuk GY, Pappas SC, Scully LJ. The Canadian Multicenter Double-blind Randomized Controlled Trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology. 1994;19:1149–1156. [PubMed] [Google Scholar]
- 56.Lindor KD, Dickson ER, Baldus WP, Jorgensen RA, Ludwig J, Murtaugh PA, Harrison JM, Wiesner RH, Anderson ML, Lange SM. Ursodeoxycholic acid in the treatment of primary biliary cirrhosis. Gastroenterology. 1994;106:1284–1290. doi: 10.1016/0016-5085(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 57.Poupon RE, Lindor KD, Pares A, Chazouilleres O, Poupon R, Heathcote EJ. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J Hepatol. 2003;39:12–16. doi: 10.1016/s0168-8278(03)00192-2. [DOI] [PubMed] [Google Scholar]
- 58.Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology. 2000;32:1196–1199. doi: 10.1053/jhep.2000.20240. [DOI] [PubMed] [Google Scholar]
- 59.Lindor KD, Jorgensen RA, Therneau TM, Malinchoc M, Dickson ER. Ursodeoxycholic acid delays the onset of esophageal varices in primary biliary cirrhosis. Mayo Clin Proc. 1997;72:1137–1140. doi: 10.4065/72.12.1137. [DOI] [PubMed] [Google Scholar]
- 60.Poupon RE, Lindor KD, Cauch-Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology. 1997;113:884–890. doi: 10.1016/s0016-5085(97)70183-5. [DOI] [PubMed] [Google Scholar]
- 61.Eriksson LS, Olsson R, Glauman H, Prytz H, Befrits R, Ryden BO, Einarsson K, Lindgren S, Wallerstedt S, Weden M. Ursodeoxycholic acid treatment in patients with primary biliary cirrhosis. A Swedish multicentre, double-blind, randomized controlled study. Scand J Gastroenterol. 1997;32:179–186. doi: 10.3109/00365529709000190. [DOI] [PubMed] [Google Scholar]
- 62.Goulis J, Leandro G, Burroughs AK. Randomised controlled trials of ursodeoxycholic-acid therapy for primary biliary cirrhosis: a meta-analysis. Lancet. 1999;354:1053–1060. doi: 10.1016/S0140-6736(98)11293-X. [DOI] [PubMed] [Google Scholar]
- 63.Stiehl A, Rudolph G, Sauer P, Benz C, Stremmel W, Walker S, Theilmann L. Efficacy of ursodeoxycholic acid treatment and endoscopic dilation of major duct stenoses in primary sclerosing cholangitis. An 8-year prospective study. J Hepatol. 1997;26:560–566. doi: 10.1016/s0168-8278(97)80421-7. [DOI] [PubMed] [Google Scholar]
- 64.Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121:900–907. doi: 10.1053/gast.2001.27965. [DOI] [PubMed] [Google Scholar]
- 65.Stiehl A, Rudolph G, Kloters-Plachky P, Sauer P, Walker S. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. J Hepatol. 2002;36:151–156. doi: 10.1016/s0168-8278(01)00251-3. [DOI] [PubMed] [Google Scholar]
- 66.Palma J, Reyes H, Ribalta J, Hernandez I, Sandoval L, Almuna R, Liepins J, Lira F, Sedano M, Silva O, et al. Ursodeoxycholic acid in the treatment of cholestasis of pregnancy: a randomized, double-blind study controlled with placebo. J Hepatol. 1997;27:1022–1028. doi: 10.1016/s0168-8278(97)80146-8. [DOI] [PubMed] [Google Scholar]
- 67.Mazzella G, Rizzo N, Azzaroli F, Simoni P, Bovicelli L, Miracolo A, Simonazzi G, Colecchia A, Nigro G, Mwangemi C, et al. Ursodeoxycholic acid administration in patients with cholestasis of pregnancy: effects on primary bile acids in babies and mothers. Hepatology. 2001;33:504–508. doi: 10.1053/jhep.2001.22647. [DOI] [PubMed] [Google Scholar]
- 68.Colombo C, Battezzati PM, Podda M, Bettinardi N, Giunta A. Ursodeoxycholic acid for liver disease associated with cystic fibrosis: a double-blind multicenter trial. The Italian Group for the Study of Ursodeoxycholic Acid in Cystic Fibrosis. Hepatology. 1996;23:1484–1490. doi: 10.1002/hep.510230627. [DOI] [PubMed] [Google Scholar]
- 69.Lindblad A, Glaumann H, Strandvik B. A two-year prospective study of the effect of ursodeoxycholic acid on urinary bile acid excretion and liver morphology in cystic fibrosis-associated liver disease. Hepatology. 1998;27:166–174. doi: 10.1002/hep.510270126. [DOI] [PubMed] [Google Scholar]
- 70.van de Meeberg PC, Houwen RH, Sinaasappel M, Heijerman HG, Bijleveld CM, Vanberge-Henegouwen GP. Low-dose versus high-dose ursodeoxycholic acid in cystic fibrosis-related cholestatic liver disease. Results of a randomized study with 1-year follow-up. Scand J Gastroenterol. 1997;32:369–373. doi: 10.3109/00365529709007686. [DOI] [PubMed] [Google Scholar]
- 71.Jacquemin E, Hermans D, Myara A, Habes D, Debray D, Hadchouel M, Sokal EM, Bernard O. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology. 1997;25:519–523. doi: 10.1002/hep.510250303. [DOI] [PubMed] [Google Scholar]