

Abstract
Unambiguous identification of tandem mass spectra is a cornerstone in mass-spectrometry-based proteomics. As the study of post-translational modifications (PTMs) by means of shotgun proteomics progresses in depth and coverage, the ability to correctly identify PTM-bearing peptides is essential, increasing the demand for advanced data interpretation. Several PTMs are known to generate unique fragment ions during tandem mass spectrometry, the so-called diagnostic ions, which unequivocally identify a given mass spectrum as related to a specific PTM. Although such ions offer tremendous analytical advantages, algorithms to decipher MS/MS spectra for the presence of diagnostic ions in an unbiased manner are currently lacking. Here, we present a systematic spectral-pattern-based approach for the discovery of diagnostic ions and new fragmentation mechanisms in shotgun proteomics datasets. The developed software tool is designed to analyze large sets of high-resolution peptide fragmentation spectra independent of the fragmentation method, instrument type, or protease employed. To benchmark the software tool, we analyzed large higher-energy collisional activation dissociation datasets of samples containing phosphorylation, ubiquitylation, SUMOylation, formylation, and lysine acetylation. Using the developed software tool, we were able to identify known diagnostic ions by comparing histograms of modified and unmodified peptide spectra. Because the investigated tandem mass spectra data were acquired with high mass accuracy, unambiguous interpretation and determination of the chemical composition for the majority of detected fragment ions was feasible. Collectively we present a freely available software tool that allows for comprehensive and automatic analysis of analogous product ions in tandem mass spectra and systematic mapping of fragmentation mechanisms related to common amino acids.
In mass spectrometry (MS)-based proteomics, protein mixtures are digested into peptides using standard proteases such as trypsin or Lys-C (1). The complex peptide mixture is separated via liquid chromatography (LC) directly coupled to MS, and the eluting peptide ions are electrosprayed into the vacuum of the mass spectrometer, where a peptide mass spectrum is recorded (2). In the mass spectrometer, selected peptide ions are fragmented, most commonly through the collision of peptide molecular ions with inert gas molecules in a technique referred to as either collision-induced dissociation (CID)1 or collisionally activated dissociation (3, 4). During this energetic collision, some of the deposited kinetic energy is converted into internal energy, which results in peptide bond breakage and fragmentation of the molecular peptide ion into sequence-specific ions (5). Identification of the analyzed peptide is then performed by scanning the measured peptide mass and list of fragment masses against a protein sequence database (6). Overall this approach provides a rapid and sensitive means of determining the primary sequence of peptides.
During the fragmentation step, various types of fragment ions can be observed in the MS/MS spectrum. Their occurrence depends on the primary sequence of the investigated peptide, the amount of internal energy deposited, how the energy was introduced, the charge state, and other factors (7). Low-energy dissociation conditions as observed in ion trap CID mainly generate fragment ions containing sequence-specific amino acid information about the investigated peptides (8). This occurs because the energy deposited during this fragmentation method primarily facilitates the fragmentation of precursor ions yielding single peptide bond fragmentation between individual amino acids (9).
With faster activation methods, such as beam-type/quadrupole CID (10), generated fragments can undergo further collisions. Multiple bonds can thereby be fragmented, giving rise to internal sequence ions, which in combination with regular b- and y-type cleavage produce specific amino-immonium ions (11). These immonium ions appear in the very low m/z range of the MS/MS spectrum, and for the majority of naturally occurring amino acids such immonium ions are unique for that particular residue (12, 13). Exceptions for this are the leucine/isoleucine and lysine/glutamine pairs, which produce immonium ions with the same chemical mass. Overall, immonium ions can confirm the presence of certain amino acid residues in a peptide, whereas information regarding the position or the stoichiometry of these amino acid residues cannot be ascertained. Because tryptic peptides on average contain 9 to 12 amino acids, they frequently contain many different residues; as a result, the analytical information hidden in the regular amino acid immonium ions might be limited. However, immonium ions can be used to support peptide sequence assignment during proteomic database searching (14).
Contrary to the 20 naturally occurring residues, many amino acids can be modified by various post-translational modifications (PTMs), and these PTM-bearing residues can themselves generate unique immonium ions—the so-called diagnostic ions. The two most prominent examples are phosphorylation of tyrosine and acetylation of lysine residues (15), which generate diagnostic ions at m/z = 216.0424 and m/z = 126.0917, respectively. Thus, the presence of these unique ions in a MS/MS spectrum can unequivocally identify the sequenced peptide as harboring a given PTM. Evidently, knowledge regarding modification-specific diagnostic ions is of great importance for the identification and validation of modified peptides in MS-based proteomics (16, 17). Additionally, such PTM-specific information can be informative in targeted proteomics approaches facilitating MS/MS precursor ion scanning (18) and become valuable in post-acquisition analysis involving extracted ion chromatograms for specific m/z values. Moreover, information regarding diagnostic ions can be a powerful addition to analytical approaches such as selected reaction monitoring, a targeted technique that relies on ion-filtering capabilities to comprehensively study peptides and PTMs (19).
Currently only a minor subset of modified amino acids has been investigated for diagnostic ions, primarily because of the lack of unbiased methods for mapping such ions in large-scale proteomics experiments. The identification of diagnostic ions is a labor-intensive endeavor, requiring manual interpretation of large numbers of MS/MS spectra for proper validation of low-mass fragmentation ions. As a result, most studies on diagnostic ions have been performed on a few selected synthetic peptides, as the interrogation of larger biological datasets has not been feasible (15, 20).
Here we describe a proteomic approach utilizing a novel algorithm based upon binning of tandem mass spectra for fast and automated mapping of analogously occurring product ions. The developed algorithm is completely independent of instrument type and fragmentation technique employed, but it performs more favorably under experimental conditions that augment the generation of immonium ions. As a result, the performance of the algorithm is benchmarked on data derived from LTQ Orbitrap Velos and Q Exactive mass spectrometers, which exhibit improved HCD performance (21–23). HCD has proven to be a powerful fragmentation technique, particularly for PTM analysis (24, 25), as no low mass detection cutoff is observed as compared with fragmentation experiments on ion trap mass spectrometers (26). Moreover, the beam-type energy deposited during HCD fragmentation allows for improved generation of both immonium and other sequence-related ions relative to CID (27, 28). Additionally, HCD experiments are performed at very high resolution, yielding high mass accuracy (<10 ppm) on all detected fragment ions, which allows the algorithm to utilize very narrow mass binning and hence easily determine the exact chemical composition of any novel detected ions.
Briefly, the algorithm takes all significantly identified MS/MS spectra and bins them together in discrete mass bins. As commonly occurring ions, such as immonium and diagnostic ions, will have same chemical composition and consequently the same m/z, they will cluster in the same mass bins, whereas sequence-specific fragment ions will scatter across the binned mass range. For validation of the presented approach, we mapped known and novel diagnostic ions from a variety of PTM-bearing amino acids, demonstrating the sensitivity and specificity of the method. Moreover, we demonstrate that mass spectral binning additionally can be employed for automated mapping of composition-specific neutral losses from large-scale proteomic experiments.
EXPERIMENTAL PROCEDURES
Sample Preparation
For acetylation and phosphorylation analysis via spectral immonium ion detection, publicly available datasets were downloaded and used (24, 29, 30). For additional analysis, rat liver tissues were prepared as previously described (31). Briefly, 50 μg of whole cell extract was loaded onto a one-dimensional SDS-PAGE gel, and proteins were extracted, reduced by 1 mm dithiothreitol, alkylated with 5.5 mm chloroacetamide (32), and digested using modified sequencing-grade trypsin according to standard in-gel protocols (1). Peptide samples were subsequently concentrated using a sample concentrator and acidified with 0.1% trifluoroacetic acid before being desalted on reverse phase C18 StageTips (33).
Mass Spectrometric Analysis
All MS experiments were performed on a nanoscale HPLC system (EASY-nLC, Thermo Fisher Scientific) connected to either a hybrid LTQ-Orbitrap Velos (Thermo Fisher Scientific) or a Q Exactive mass spectrometer equipped with a nanoelectrospray source (Proxeon Biosystems). Each peptide sample was autosampled and separated in a 15-cm analytical column (75-μm inner diameter) in-house packed with 3-μm C18 beads (Reprosil Pur-AQ, Dr. Maisch, Ammerbuch-Entringen, Germany) with a 2-h gradient from 5% to 40% acetonitrile in 0.5% acetic acid. The effluent from the HPLC was directly electrosprayed into the mass spectrometer.
Identification of Peptides and Proteins by MaxQuant
All raw data analysis was performed with the MaxQuant software suite (34) supported by the Andromeda search engine (35). Data were searched against a concatenated target/decoy (forward and reversed) version of the corresponding IPI database (version 3.68) (both human and rat database versions were employed in this study). We followed the step-by-step protocol of the MaxQuant software suite to generate MS/MS peak lists that were filtered to contain at most six peaks per 100-Da interval prior to the Andromeda database search. The mass tolerance for searches was set to maximum values of 7 ppm for peptide masses and 20 ppm for HCD fragment ion masses. Data were searched with carbamidomethylation as a fixed modification and protein N-terminal acetylation and methionine oxidation as variable modifications. In addition, to each database search the specific PTM investigated for diagnostic ions was added as a variable modification. A maximum of two missed cleavages was allowed. We required strict tryptic specificity, and protease specificity was set to LysC only. Peptide assignments were statistically evaluated in a Bayesian model on the basis of sequence length and Mascot score. We accepted only peptides and proteins with a false discovery rate of less than 1%, estimated on the basis of the number of accepted reverse hits. Protein sequences of common contaminants such as human keratins and proteases used were added to the database.
RESULTS
Description of Algorithm
For unbiased mapping of commonly occurring fragment ions, we developed an algorithm referred to as SPectral Immonium Ion Detection (SPIID) based upon binning of tandem mass spectra. Currently three different types of analyses are implemented in the presented algorithm. First, a histogram of fragment spectra reveals unique fragment ions, such as immonium ions, common to a collection of tandem mass spectra of peptides. Second, normalization of unmodified MS/MS spectra against modified spectra reveals diagnostic ions associated with a specific modification. Third, precursor MH+ alignment of MS/MS spectra enables the systematic study of composition-specific neutral losses.
The basic principle of the approach relies on the knowledge that immonium ions and PTM-specific diagnostic ions will appear at the same m/z in MS/MS spectra, whereas sequence-specific fragment ions appear at discrete m/z values in the investigated mass range. As a result, creating a histogram of all observed fragmentation ions across a number of MS/MS spectra will result in common ions (immonium and diagnostic ions) clustering into unique mass bins, whereas backbone fragment ions (b- and y-ions) will scatter across the entire bin range (Fig. 1A). To demonstrate the spectral mass binning approach, we binned MS/MS spectra derived from a large-scale analysis of standard HeLa lysates (23). The generated histogram depicts all commonly occurring fragment ions throughout the binned MS/MS range (Fig. 1B). The strongest peaks present in the histogram correspond to known immonium ions for amino acids such as lysine/glutamine (m/z 101.0709), histidine (m/z 110.0713), phenylalanine (m/z 120.0808), tyrosine (m/z 136.0757), and tryptophan (m/z 159.0917). Because the dataset is derived from a HeLa lysate digested with trypsin, the majority of peptide sequences contain either a lysine or an arginine at the C-terminal position (36). Following this, the spectral binning approach identified the trypsin-specific y1 ions for lysine (m/z 147.1128) and arginine (m/z 175.1190). Notably, the abundance of these two mass bins was very similar, confirming that an equal share of the investigated MS/MS spectra harbored either a lysine or an arginine y1 ion.
Fig. 1.

A, experimental description of spectral binning by SPIID. All acquired MS/MS spectra are binned into mass bins, yielding a histogram in which analogous fragment ions such as immonium ions are located in same mass bins. B, histogram representing frequently occurring ions from a tryptic digest, primarily represented by amino-specific immonium ions and tryptic y1 ions. C, very low bin sizes can be used for HCD-generated MS/MS spectra, as shown for the mass accuracy of the lysine y1 ion by spectral binning. To demonstrate this, we specifically for this analysis binned with a mass tolerance of ±0.0005 Da. D, number of theoretical chemical isomers at various mass accuracies. E, list of the most commonly observed tryptic fragment ions measured with high resolution after HCD fragmentation.
With HCD fragmentation providing high-accuracy fragment ions, a low level of noise in the established histograms is ensured (Fig. 1B). To demonstrate the achieved mass accuracy, we analyzed more than 24,000 lysine-containing y1 ions in our dataset, which revealed an overall mass deviation of 0.00103 Th (Fig. 1C). At such mass accuracies, the chemical composition of the observed ions can easily be determined (Fig. 1D). With an overall mass accuracy of 0.001 Da, a maximum of five chemical compositions can be assigned to each mass bin up to m/z 300 (green distribution). In contrast to this, the number of chemical compositions that fit to each mass bin increases dramatically when the mass accuracy is increased to 0.02 Da, whereas at 0.5 Da a unique chemical composition can be deduced only at masses below 25 Da (red distribution).
Mapping Diagnostic (Reporter) Ions with SPIID
Although not a strict requirement for the interrogation of immonium ions, the SPIID analysis most informatively is applied to data files already matched against a protein sequence database using standard proteomics search engines (6). The search engine strategy follows standard approaches employed for proteomics PTM analysis and allows for significant dentification of unmodified and PTM-modified peptide sequences (37). Prior to operation of the SPIID algorithm, all search engine results must therefore be extracted and loaded directly into SPIID through the graphical interface (see the description below and a more detailed description in the supplemental material). The SPIID algorithm first divides all significantly identified MS/MS spectra into two peptide groups depending on whether the identified MS/MS spectrum belongs to a modified or a non-modified peptide. For the identification of novel diagnostic ions, this is an important prerequisite for SPIID analysis, as the reliability of identified diagnostic ions directly relates to the overall reliability of peptide identifications by the search engine. Thus, only highly significant data identified with a false discovery rate of less than 1% should be submitted for subsequent SPIID analysis. It should be noted that SPIID is able to simultaneously handle several PTM analyses derived from a single proteomics experiment.
In the next step each modified spectrum is further subdivided into specific PTM categories (e.g. lysine-acetylation-containing spectra are separated into a separate subcategory), and for each PTM subcategory all MS/MS spectra are mass binned across the preset m/z range, thereby creating fragment-specific histograms for each analyzed category (as shown in Fig. 2C). The binning histogram is generated arbitrarily from a starting m/z mass value, and this is followed by the generation of discrete mass bins, which can be defined by the user. The size of the mass bin can be varied accordingly but is preset to 40 ppm because of the high-accuracy fragment ions generated by HCD. The default value works well in our hands but can easily be adjusted in the SPIID software interface. Notably, the bin width is dependent on the mass accuracy of the instrumentation employed and can be affected by instrument-dependent factors such as calibration status and resolution settings.
Fig. 2.

A, experimental description of normalized spectral binning by SPIID for investigation of diagnostic ions. B, SPIID analysis of diagnostic ions for phospho-tyrosine, revealing the known reporter ion. C, SPIID analysis of diagnostic ions for lysine acetylation revealing the known reported ion. D, comparison of Andromeda score distributions when searching acetyl lysine-containing peptides without (black bars) or with (white bars) diagnostic ion information. E, diagnostic ion analysis by SPIID for SUMO-containing peptides revealed novel and distinct reporter ions. F, diagnostic ion analysis by SPIID for lysine formylation revealed a distinct reporter ion.
Because this initial binning of MS/MS spectra only represents generally observed ions, an additional layer of filtering is added to reveal the modification-specific diagnostic ions. To this end, each PTM-specific histogram is normalized and compared to a base histogram. This base histogram is generated by binning all MS/MS spectra belonging to unmodified MS/MS spectra, and in the normalization process the base histogram is subtracted from the PTM-specific histogram (Fig. 2A). This normalization step ensures that all commonly observed fragmentation ions, such as amino acid immonium ions and protease-specific ions, are alleviated, as these ions occur in both modified and unmodified peptides. The normalized PTM histogram in the end yields all unique mass peaks related to the investigated PTM-bearing peptides, and as a result, the identified masses correspond to PTM-specific diagnostic ions (Fig. 2A).
Validation of SPIID Algorithm
For validation of the algorithm, we tested SPIID on datasets enriched for tyrosine phosphorylation and lysine acetylation, two well-characterized PTMs that both generate a unique and analytically important diagnostic ion (38, 39). The phosphorylation dataset contained in total 277 identified MS/MS spectra belonging to tyrosine phosphorylation, while the acetylation dataset contained 611 MS/MS spectra identified as acetylated lysine. Within the two datasets, 18,421 and 11,518 MS/MS spectra were identified as unmodified, respectively. Running the SPIID algorithm on these datasets identified prominent diagnostic ions at the correct m/z masses corresponding to the known immonium ions of tyrosine phosphorylation (Fig. 2B) and lysine acetylation (Fig. 2C). Because HCD fragmentation generates high-accuracy fragment ions, and because the two diagnostic ions were derived from a large number of MS/MS spectra, the exact masses could easily be determined (m/z 216.042 and m/z 126.091, respectively), in full agreement with the known masses for these ions.
Notably, the SPIID analysis revealed that analytical caution should be taken when using diagnostic ions as markers for lysine acetylation. Our group previously reported that N-terminal acetylation of peptides can occur as a chemical artifact in samples prepared using one-dimensional SDS gels (16, 40). Moreover, acetylation is a PTM that commonly occurs on protein N termini. Consequently, localization of the acetylation group can be difficult when lysine residues are located N-terminally in peptide sequences. In such instances regular MS/MS sequencing cannot determine whether the acetylation group is located on the N terminus of the peptide or the side-chain of the lysine residue. As a result, our results demonstrate that the diagnostic ion at m/z = 126.091 is a significant marker for true lysine side-chain acetylation, as only acetylation on the side-chain of lysine residues can generate this particular fragment ion (41). In contrast to this, the previously reported diagnostic ion for lysine acetylation (m/z = 143) can be generated from both the lysine side-chain and peptide N-terminal acetylations (42). Moreover, peptide sequences harboring N-terminal GL, GI, or VA amino acid combinations may upon higher energy collisions generate an a2 ion with an identical mass. In summary, our SPIID analysis confirmed that only the m/z 126.091 ion should be considered for mapping of lysine-acetylation-containing peptides.
The analytical value of immonium and diagnostic ions has previously been demonstrated in various de novo approaches (43, 44) and shown to restrict the search space in proteomics experiments (14). For that reason, we made a rudimentary assessment of the analytical advantages of a single diagnostic ion in a proteomics experiment. To this end, we analyzed a proteomic acetylation dataset with and without the PTM-specific diagnostic ion included in the data search (Fig. 2D). The analysis revealed a shift toward higher Andromeda scores, confirming previous observations of improved identification significance upon the addition of diagnostic ions. Overall, the addition of a single diagnostic ion to the database searched increased the median Andromeda score distribution from 86.2 to 89.6, corresponding to an increase in significance (p value) by a factor of 2.2.
Diagnostic Ions of PTMs
Having established that SPIID is a sensitive, fast, and accurate approach for the identification of diagnostic ions in large-scale proteomics experiments, we next employed the algorithm for the analysis of other diagnostic ions. To this end, we decided to first look at whether mass spectrometric analysis of lysine ubiquitylation holds any unique diagnostic ions. Previous analysis reported that the diGly remnant does not generate any diagnostic ion, whereas the miscleaved LeuArgGlyGly would give rise to b2 and b4 diagnotisc ions derived from side-chain cleavage of the ubiquitin tag. However, this analysis was based upon investigation of only a single synthetic peptide (45). In agreement with earlier observations, no diagnostic ion for the diGly ubiquitin remnant was observed when using SPIID with various large-scale ubiquitin diGly datasets (46–48). Instead the two prominent diagnostic ions for peptides bearing a miscleaved ubiquitin tag (LeuArgGlyGly) were confirmed as previously described (supplemental Fig. S2). Next we extended our analysis of diagnostic ions to include PTMs such as lysine formylation and SUMOylation. For the SUMO analysis we analyzed 14 MS/MS spectra containing SUMO2 enriched substrates derived from the SUMO paralog strategy where an arginine mutation is introduced at the sixth position from the C terminus (47, 49). When this mutation is introduced, cell lysates digested with trypsin will render a five-amino-acid signature tag (QQTGG-) at the SUMO modification site. As was observed for lysine ubiquitylation, SPIID identified several diagnostic ions belonging to side-chain fragmentation of the SUMO tag (Fig. 2E). Although these diagnostic ions do not have a unique chemical composition, their abundance in the SPIID analysis was quite pronounced, meaning these ions hold strong analytical information. This information can be particularly useful for future validation of SUMO-containing MS/MS spectra.
Lysine formylation is structurally similar to lysine acetylation and methylation, with lysine dimethylation even having the same nominal mass as lysine formylation (mass difference ΔM = 0.03638 Da). Because all investigated data were acquired with high-resolution mass spectrometry capable of mass measurement within a low p.p.m. window, distinguishing the mass difference between lysine formylation and lysine dimethylation was straightforward. This eliminated the risk of any dimethylated peptides being falsely identified as lysine formylation (50). SPIID analysis of 103 lysine formylation MS/MS spectra revealed a very strong diagnostic ion at m/z = 112.0795, corresponding to the formation of a six-membered pyridine-like ring through the elimination of ammonia (Fig. 2F). This ion formation is similar to the structure of the lysine acetylation diagnostic ion and reflects the most stable form of these molecules (41).
As a final experiment, we prepared a rat liver sample for proteomic analysis (as detailed in “Experimental Procedures”) by loading 100 μg of the protein lysate onto a one-dimensional SDS-PAGE gel, cutting the gel into 20 equally sized fractions, and then measuring each sample on the LTQ Orbitrap Velos. The resulting RAW files were searched with a broad range of PTMs, and using the SPIID algorithm we investigated the presence of diagnostic ions for these various PTMs. Table I shows for which PTMs a diagnostic ion was identified, the exact mass of that ion, and a chemical composition.
Table I. PTM enriched immonium HCD fragments.
| Formylated lysine | 112.0756 (C6H9NO + H+) |
|---|---|
| Acetylated lysine | 126.0913 (C7H11NO + H+) |
| Oxidated methionine | None found |
| Phosphorylated tyrosine | 216.0420 (C8H10NO4P + H+) |
| Phosphorylated serine | None found |
| Phosphorylated threonine | None found |
| Hydroxyproline | None found |
| Kynurenine | 163.0866 (C9H10N2O3 + H+) |
| GG-lysine | None found |
| LRGG-lysine | |
| b4 | 384.2354 (C16H29N7O4 + H+) |
| b2 | 270.1925 (C12H23N5O2 + H+) |
| QQTGG-lysine | |
| b5 | 472.2151 (C18H29N7O8 + H+) |
| b5* | 455.1885 (C18H26N6O8 + H+) |
| b5° | 454.2045 (C18H27N7O7 + H+) |
| b4* | 398.1670 (C16H23N5O7 + H+) |
| b4°* | 380.1564 (C16H21N5O6 + H+) |
| b3 | 358.1721 (C14H23N5O6 + H+) |
| b3* | 341.1455 (C14H20N4O6 + H+) |
| b3°* | 323.1349 (C14H18N4O5 + H+) |
| b2 | 257.1244 (C10H16N4O4 + H+) |
| b2° | 239.1139 (C10H14N4O3 + H+) |
| c1 | 146.0924 (C5H11N3O2 + H+) |
| c1° | 128.0818 (C5H9N3O + H+) |
PTMs investigated for diagnostic ions. Analysis confirmed currently known diagnostic ions while identifying novel ions for lysine formylation, kynurenine, and SUMO.
Mapping Fragmentation-specific Neutral Losses with SPIID
Having established that SPIID is capable of mapping composition-specific fragment ions such as immonium and diagnostic ions, we next wanted to investigate the algorithm's ability to perform more advanced spectral analyses. In recent years the analytical utility and diagnostic value of neutral losses induced by CID, electron transfer dissociation, or electron capture dissociation have been realized (51–53). A similar catalog of commonly observed neutral losses from HCD has also been described (54). However, to our knowledge no commonly available software tool has been described for the easy extraction of such neutral loss information. To demonstrate the ability of SPIID to extract such information, we first used high-resolution isotope spacing to deconvolute and deisotope each detected fragment ion to its singly charged counterpart for all HCD-generated MS/MS spectra. Next, all fragment ions were aligned to the parent mass (MH+) of the individual MS/MS spectra (Fig. 3A). All MS/MS spectra were then converted to negative m/z values that could easily, and in a single step, be interrogated for common neutral losses through spectral binning by SPIID (Fig. 3B). The results demonstrated that HCD generally did not induce many neutral losses relative to CID and electron capture dissociation/electron transfer dissociation, although a strong water loss and losses corresponding to intact amino acids were observed, with the latter associated with y-ion formation through regular backbone fragmentation. Next we investigated the same HCD-induced neutral losses in a phosphopeptide enriched sample by interrogating a sample derived from SCX and TiO2 enriched tryptic peptides (55, 56). Because of the chemical nature of the peptide phosphogroup, it easily detaches during collision-induced fragmentation, generating neutral losses corresponding to various types of phospo-groups, such as HPO3 and H3PO4. As expected, our SPIID analysis revealed prominent losses of phospho-groups, followed additionally by loss of H2O and combined losses (e.g. H2O + H3PO4 = H5PO5) (Fig. 3C).
Fig. 3.

A, experimental description of spectral binning by SPIID for interrogation of neutral losses in MS/MS spectra. All MS/MS spectra were first normalized to their individual MH precursor mass and subsequently binned by SPIID. B, neutral loss analysis of regular tryptic digest revealed strong neutral loss of H2O and amino acids. C, neutral loss analysis of SCX-fractionated phosphopeptide sample revealed strong losses of H2O and variations of phospho-losses. D, neutral loss analysis of flow-through fraction from SCX-prepared phospho-sample revealed distinct losses of phospho- and possible ADP- or ATP-related species.
Usually the majority of phosphopeptides from an SCX-based phosphoproteomics experiment are retained on the SCX column, whereas multiphosphorylated peptides typically are present in the flow-through (57). However, the flow-through samples are often hampered by overall low identification rates. Thus, in order to investigate potential compositional differences between SCX fractions, we performed a neutral loss analysis of all acquired spectra from the SCX flow-through sample using SPIID (Fig. 3D). The SCX flow-through analysis generated distinct neutral phosphorylation losses (1×H3PO4 = −97.98 Da; 2×H3PO4 = −195.96 Da; 3×H3PO4 = −293.93 Da; 4×H3PO4 = −391.91) and combinations with loss of water, in full agreement with the flow-through sample largely containing multiphosphorylated peptides. Still, very large losses were distinctly observed in this fraction (−427.03 Da, −507.00 Da, and −609.06 Da), which could correspond to the loss of ADP, ATP, and phospho-ribose+ADP, respectively). Whether these losses are related to peptide species remains to be determined, but they could also correspond to the flow-though fraction being particularly enriched in metabolic species, which could additionally explain why poorer identification rates often are observed for this fraction. Proteomic investigations into the content of the SCX flow-through samples during phospho-enrichment experiments have started to emerge (57); however, whether such detailed analysis will be able to pinpoint the origin of observed losses remains to be seen.
Collectively, these results additionally demonstrate the flexibility of SPIID and the neutral loss differences between samples and highlight that different neutral loss configurations should be included in database search strategies dependent upon sample type. Notably, the SPIID analysis can additionally be extended to investigate individual fragment-ion-specific neutral losses for the mapping of novel ion species, as previously demonstrated by the increased abundance of an unusual x-ion fragment for phosphorylated serine or threonine residues (58).
How to Operate SPIID
The graphical user interface of SPIID is shown in Fig. 4A. The input required for SPIID is high-resolution MS/MS peak lists following the standard Mascot generic format (MGF). In the MGF file, each MS/MS spectrum is listed as a pair of mass and intensity values delimited by “BEGIN IONS” and “END IONS” statements (59), and is commonly regarded the most used file format for storing MS/MS data (60). The MGF input files can be generated through standard proteomics software tools such as Mascot Distiller, MassMatrix (61), Raw2MSM (62), Pyteomics (63), or msconvert (ProteoWizard) (64). Notably, the apl peak lists generated by MaxQuant are also supported. First (i) the user loads the MGF file into the SPIID program by clicking “Add MGF file.” This can be followed by optional (ii) loading of a specificity file containing information regarding the desired grouping and the raw files and scan numbers. The next step (iii) is the optional choice of which spectra to use as background determinants in the analysis; typically these would be all non-modified spectra, but other options are made available as well. The last step (iv) before processing can begin is determining the bin size and start/stop m/z values for the analysis. Finally (v), pressing the “Process” button will initiate the SPIID analysis, and the output will be depicted as illustrated in Fig. 4B. The top pane of the output window portrays the binned histogram for all modified MS/MS spectra being analyzed, and the middle pane portrays a similar histogram derived from all unmodified MS/MS spectra. The bottom pane of the output window depicts the final normalized diagnostic ion histogram, generated by subtraction of the two upper histograms (modified histogram minus unmodified histogram). The entire right side of the output window allows for changing the visualized output style according to various parameters such as font size, color, histogram annotation, depicted m/z range, etc. This ensures flexibility for the end user and allows for visual optimization of data output. The final diagnostic ion histogram can be exported to several file formats.
Fig. 4.
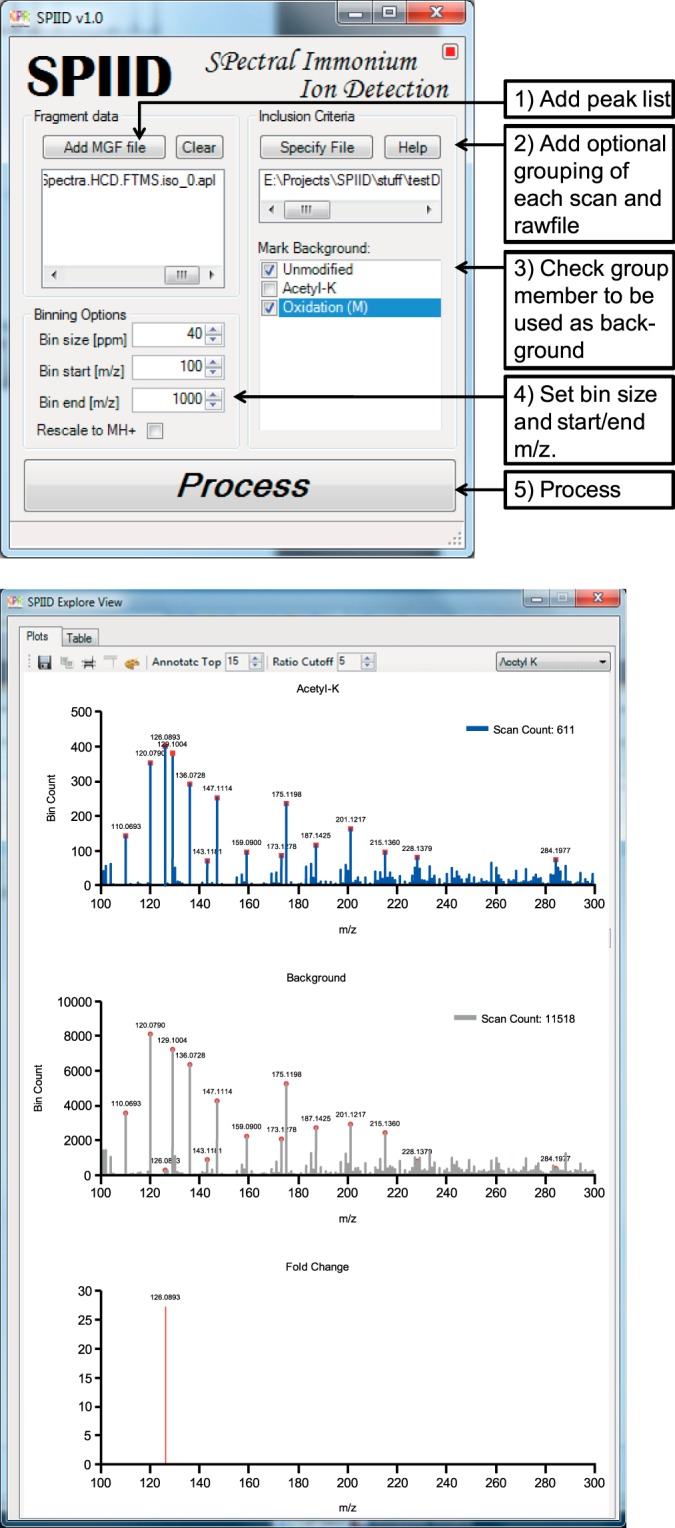
A, graphical view of the SPIID software graphical user interface and description of the operating procedure. B, graphical visualization of the output window.
CONCLUSION
The low mass region or MS/MS spectra contain immonium and other types of peptide-specific ions that can aid in identification of the peptide sequence content (65) or be employed for post-acquisition data analysis. Here we introduce the concept of binning high-resolution MS/MS spectra for the identification of immonium/diagnostic ions. Evaluation of the developed algorithm showed fast, accurate, and sensitive identification of diagnostic ions associated with a wide variety of PTMs. The algorithm's ability to confirm known diagnostic ions, such as those associated with tyrosine phosphorylation and lysine acetylation, and the identification of certain novel ones proves the accuracy of the presented approach. In this study SPIID was applied to several PTM carrying peptides, as well as for mapping of neutral fragment losses, but the algorithm should work equally well on all types of ions able to generate fragmentation spectra (66). Overall we believe that the presented approach for global mapping of diagnostic fragment ions will be a valuable tool for researchers working in the field of proteomics (67, 68). Moreover, we envision that the presented algorithm can be applied to specialized areas of proteomics as well. These could, for example, entail experiments using novel chemical labeling strategies, where the configuration of specific ions for improved data analysis could be highly advantageous, or the development of database search engines for the adaption of optimal scoring systems.
The SPIID algorithm and software program are provided with the manuscript as supplemental material. The file is called SPIID and can be installed without any system requirements on a standard Windows-compatible PC. A detailed description of how to operate the algorithm can be found in the supplemental material.
Supplementary Material
Acknowledgments
We thank members of the NNF-CPR for fruitful discussions and careful reading of the manuscript, and we thank the members of our department for the availability of proteomics datasets.
Footnotes
Author contributions: C.D.K., J.V.O., and M.L.N. designed research; C.D.K. performed research; C.F., A.J.H., J.V.O., and M.L.N. contributed new reagents or analytic tools; C.D.K., J.V.O., and M.L.N. analyzed data; C.D.K. and M.L.N. wrote the paper.
* The work carried out in this study was in part supported by the Novo Nordisk Foundation Center for Protein Research, the Lundbeck Foundation, and the PRIME-XS project (Grant No. 262067), funded by the European Union 7th Framework Program.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CID
- collision-induced dissociation
- HCD
- higher-energy collisional activation dissociation
- MGF
- Mascot generic format
- MS/MS
- tandem mass spectrometry
- PTM
- post-translational modification
- SCX
- strong cation exchange
- SPIID
- spectral immonium ion detection.
REFERENCES
- 1. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2007) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 2. Choudhary C., Mann M. (2010) Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 11, 427–439 [DOI] [PubMed] [Google Scholar]
- 3. Cody R. B., Amster I. J., McLafferty F. W. (1985) Peptide mixture sequencing by tandem Fourier-transform mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 82, 6367–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hunt D. F., Yates J. R., Shabanowitz J., Winston S., Hauer C. R. (1986) Protein sequencing by tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 83, 6233–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roepstorff P., Fohlman J. (1984) Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601. [DOI] [PubMed] [Google Scholar]
- 6. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 7. Steen H., Mann M. (2004) The ABC's (and XYZ's) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 5, 699–711 [DOI] [PubMed] [Google Scholar]
- 8. Louris J. N., Cooks R. G., Syka J. E. P., Kelley P. E., Stafford G. C., Todd J. F. J. (1987) Instrumentation, applications, and energy deposition in quadrupole ion-trap tandem mass spectrometry. Anal. Chem. 59, 1677–1685 [Google Scholar]
- 9. Biemann K. (1988) Contributions of mass spectrometry to peptide and protein structure. Biomed. Environ. Mass Spectrom. 16, 99–111 [DOI] [PubMed] [Google Scholar]
- 10. Wells J. M., McLuckey S. A. (2005) Collision-induced dissociation (CID) of peptides and proteins. Methods Enzymol. 402, 148–185 [DOI] [PubMed] [Google Scholar]
- 11. Johnson R. S., Martin S. A., Biemann K., Stults J. T., Watson J. T. (1987) Novel fragmentation process of peptides by collision-induced decomposition in a tandem mass spectrometer: differentiation of leucine and isoleucine. Anal. Chem. 59, 2621–2625 [DOI] [PubMed] [Google Scholar]
- 12. Barber M., Bordoli R. S., Garner G. V., Gordon D. B., Sedgwick R. D., Tetler L. W., Tyler A. N. (1981) Fast-atom-bombardment mass spectra of enkephalins. Biochem. J. 197, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barber M., Bordoli R. S., Sedgwick R. D., Tetler L. W. (1981) Fast atom bombardment mass spectrometry of two isomeric tripeptides. Organic Mass Spectrom. 16, 256–260 [Google Scholar]
- 14. Hansen T. A., Kryuchkov F., Kjeldsen F. (2012) Reduction in database search space by utilization of amino acid composition information from electron transfer dissociation and higher-energy collisional dissociation mass spectra. Anal. Chem. 84, 6638–6645 [DOI] [PubMed] [Google Scholar]
- 15. Falick A. M., Hines W. M., Medzihradszky K. F., Baldwin M. A., Gibson B. W. (1993) Low-mass ions produced from peptides by high-energy collision-induced dissociation in tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 4, 882–893 [DOI] [PubMed] [Google Scholar]
- 16. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 17. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. (2010) 3, ra3. [DOI] [PubMed] [Google Scholar]
- 18. Steen H., Kuster B., Fernandez M., Pandey A., Mann M. (2002) Tyrosine phosphorylation mapping of the epidermal growth factor receptor signaling pathway. J. Biol. Chem. 277, 1031–1039 [DOI] [PubMed] [Google Scholar]
- 19. Unwin R. D., Griffiths J. R., Leverentz M. K., Grallert A., Hagan I. M., Whetton A. D. (2005) Multiple reaction monitoring to identify sites of protein phosphorylation with high sensitivity. Mol. Cell. Proteomics 4, 1134–1144 [DOI] [PubMed] [Google Scholar]
- 20. Harrison A. G., Young A. B., Schnoelzer M., Paizs B. (2004) Formation of iminium ions by fragmentation of a2 ions. Rapid Commun. Mass Spectrom. 18, 1635–1640 [DOI] [PubMed] [Google Scholar]
- 21. Olsen J. V., Schwartz J. C., Griep-Raming J., Nielsen M. L., Damoc E., Denisov E., Lange O., Remes P., Taylor D., Splendore M., Wouters E. R., Senko M., Makarov A., Mann M., Horning S. (2009) A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics 8, 2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Michalski A., Damoc E., Hauschild J. P., Lange O., Wieghaus A., Makarov A., Nagaraj N., Cox J., Mann M., Horning S. (2011) Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 10, M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelstrup C. D., Young C., Lavallee R., Nielsen M. L., Olsen J. V. (2012) Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J. Proteome Res. 11, 3487–3497 [DOI] [PubMed] [Google Scholar]
- 24. Frese C. K., Altelaar A. F. M., Hennrich M. L., Nolting D., Zeller M., Griep-Raming J., Heck A. J. R., Mohammed S. Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. (2011) 10, 2377–2388 [DOI] [PubMed] [Google Scholar]
- 25. Savitski M. M., Lemeer S., Boesche M., Lang M., Mathieson T., Bantscheff M., Kuster B. Confident phosphorylation site localization using the Mascot Delta Score. Mol. Cell. Proteomics 10, M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwartz J. C., Senko M. W., Syka J. E. (2002) A two-dimensional quadrupole ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. (2002) 13, 659–669 [DOI] [PubMed] [Google Scholar]
- 27. Olsen J. V., Macek B., Lange O., Makarov A., Horning S., Mann M. (2007) Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 4, 709–712 [DOI] [PubMed] [Google Scholar]
- 28. Hunt D. F., Buko A. M., Ballard J. M., Shabanowitz J., Giordani A. B. (1981) Sequence analysis of polypeptides by collision activated dissociation on a triple quadrupole mass spectrometer. Biomed. Mass Spectrom. 8, 397–408 [DOI] [PubMed] [Google Scholar]
- 29. Francavilla C., Rigbolt K. T., Emdal K. B., Carraro G., Vernet E., Bekker-Jensen D. B., Streicher W., Wikstrom M., Sundstrom M., Bellusci S., Cavallaro U., Blagoev B., Olsen J. V. (2013) Functional proteomics defines the molecular switch underlying FGF receptor trafficking and cellular outputs. Mol. Cell 51, 707–722 [DOI] [PubMed] [Google Scholar]
- 30. Weinert B. T., Wagner S. A., Horn H., Henriksen P., Liu W. R., Olsen J. V., Jensen L. J., Choudhary C. (2011) Proteome-wide mapping of the Drosophila acetylome demonstrates a high degree of conservation of lysine acetylation. Sci. Signal. 4, ra48. [DOI] [PubMed] [Google Scholar]
- 31. Lundby A., Lage K., Weinert B. T., Bekker-Jensen D. B., Secher A., Skovgaard T., Kelstrup C. D., Dmytriyev A., Choudhary C., Lundby C., Olsen J. V. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nielsen M. L., Vermeulen M., Bonaldi T., Cox J., Moroder L., Mann M. (2008) Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 5, 459–460 [DOI] [PubMed] [Google Scholar]
- 33. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 34. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 35. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. (2011) 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 36. Olsen J. V., Ong S. E., Mann M. (2004) Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3, 608–614 [DOI] [PubMed] [Google Scholar]
- 37. Yates J. R., Ruse C. I., Nakorchevsky A. (2009) Proteomics by mass spectrometry: approaches, advances, and applications. Annu. Rev. Biomed. Eng. 11, 49–79 [DOI] [PubMed] [Google Scholar]
- 38. Steen H., Kuster B., Fernandez M., Pandey A., Mann M. (2001) Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode. Anal. Chem. 73, 1440–1448 [DOI] [PubMed] [Google Scholar]
- 39. Kim J. Y., Kim K. W., Kwon H. J., Lee D. W., Yoo J. S. (2002) Probing lysine acetylation with a modification-specific marker ion using high-performance liquid chromatography/electrospray-mass spectrometry with collision-induced dissociation. Anal. Chem. 74, 5443–5449 [DOI] [PubMed] [Google Scholar]
- 40. Nielsen M. L., Savitski M. M., Zubarev R. A. (2006) Extent of modifications in human proteome samples and their effect on dynamic range of analysis in shotgun proteomics. Mol. Cell. Proteomics 5, 2384–2391 [DOI] [PubMed] [Google Scholar]
- 41. Zhang K., Yau P. M., Chandrasekhar B., New R., Kondrat R., Imai B. S., Bradbury M. E. (2004) Differentiation between peptides containing acetylated or tri-methylated lysines by mass spectrometry: an application for determining lysine 9 acetylation and methylation of histone H3. Proteomics 4, 1–10 [DOI] [PubMed] [Google Scholar]
- 42. Trelle M. B., Jensen O. N. (2008) Utility of immonium ions for assignment of epsilon-N-acetyllysine-containing peptides by tandem mass spectrometry. Anal. Chem. 80, 3422–3430 [DOI] [PubMed] [Google Scholar]
- 43. Spengler B. (2004) De novo sequencing, peptide composition analysis, and composition-based sequencing: a new strategy employing accurate mass determination by Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 703–714 [DOI] [PubMed] [Google Scholar]
- 44. Chi H., Sun R. X., Yang B., Song C. Q., Wang L. H., Liu C., Fu Y., Yuan Z. F., Wang H. P., He S. M., Dong M. Q. (2010) pNovo: de novo peptide sequencing and identification using HCD spectra. J. Proteome Res. 9, 2713–2724 [DOI] [PubMed] [Google Scholar]
- 45. Parker C. E., Mocanu V., Warren M. R., Greer S. F., Borchers C. H. (2005) Mass spectrometric determination of protein ubiquitination. Methods Mol. Biol. 301, 153–173 [DOI] [PubMed] [Google Scholar]
- 46. Danielsen J. M., Sylvestersen K. B., Bekker-Jensen S., Szklarczyk D., Poulsen J. W., Horn H., Jensen L. J., Mailand N., Nielsen M. L. (2011) Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell. Proteomics 10, M110.003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., Choudhary C. (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteomics 10, M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sylvestersen K. B., Young C., Nielsen M. L. (2013) Advances in characterizing ubiquitylation sites by mass spectrometry. Curr. Opin. Chem. Biol. 17, 49–58 [DOI] [PubMed] [Google Scholar]
- 49. Galisson F., Mahrouche L., Courcelles M., Bonneil E., Meloche S., Chelbi-Alix M. K., Thibault P. (2011) A novel proteomics approach to identify SUMOylated proteins and their modification sites in human cells. Mol. Cell. Proteomics 10, M110.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wisniewski J. R., Zougman A., Mann M. (2008) Nepsilon-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 36, 570–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia Q., Lee M. V., Rose C. M., Marsh A. J., Hubler S. L., Wenger C. D., Coon J. J. (2011) Characterization and diagnostic value of amino acid side chain neutral losses following electron-transfer dissociation. J. Am. Soc. Mass Spectrom. 22, 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Falth M., Savitski M. M., Nielsen M. L., Kjeldsen F., Andren P. E., Zubarev R. A. (2008) Analytical utility of small neutral losses from reduced species in electron capture dissociation studied using SwedECD database. Anal. Chem. 80, 8089–8094 [DOI] [PubMed] [Google Scholar]
- 53. Martin D. B., Eng J. K., Nesvizhskii A. I., Gemmill A., Aebersold R. (2005) Investigation of neutral loss during collision-induced dissociation of peptide ions. Anal. Chem. 77, 4870–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Michalski A., Neuhauser N., Cox J., Mann M. (2012) A systematic investigation into the nature of tryptic HCD spectra. J. Proteome Res. 11, 5479–5491 [DOI] [PubMed] [Google Scholar]
- 55. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 56. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villen J., Li J., Cohn M. A., Cantley L. C., Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nie S., Dai J., Ning Z. B., Cao X. J., Sheng Q. H., Zeng R. (2010) Comprehensive profiling of phosphopeptides based on anion exchange followed by flow-through enrichment with titanium dioxide (AFET). J. Proteome Res. 9, 4585–4594 [DOI] [PubMed] [Google Scholar]
- 58. Kelstrup C. D., Hekmat O., Francavilla C., Olsen J. V. (2011) Pinpointing phosphorylation sites: quantitative filtering and a novel site-specific x-ion fragment. J. Proteome Res. 10, 2937–2948 [DOI] [PubMed] [Google Scholar]
- 59. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 60. Deutsch E. W. (2012) File formats commonly used in mass spectrometry proteomics. Mol. Cell. Proteomics 11, 1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu H., Freitas M. A. (2009) MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 9, 1548–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 63. Goloborodko A. A., Levitsky L. I., Ivanov M. V., Gorshkov M. V. (2013) Pyteomics—a Python framework for exploratory data analysis and rapid software prototyping in proteomics. J. Am. Soc. Mass Spectrom. 24, 301–304 [DOI] [PubMed] [Google Scholar]
- 64. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nielsen M. L., Bennett K. L., Larsen B., Moniatte M., Mann M. (2002) Peptide end sequencing by orthogonal MALDI tandem mass spectrometry. J. Proteome Res. 1, 63–71 [DOI] [PubMed] [Google Scholar]
- 66. Robbe C., Capon C., Coddeville B., Michalski J. C. (2004) Diagnostic ions for the rapid analysis by nano-electrospray ionization quadrupole time-of-flight mass spectrometry of O-glycans from human mucins. Rapid Commun. Mass Spectrom. 18, 412–420 [DOI] [PubMed] [Google Scholar]
- 67. Mann M., Jensen O. N. (2003) Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 [DOI] [PubMed] [Google Scholar]
- 68. Jungmichel S., Rosenthal F., Altmeyer M., Lukas J., Hottiger M. O., Nielsen M. L. (2013) Proteome-wide identification of poly(ADP-ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 52, 272–285 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.