

Abstract
Arginine phosphorylation is an emerging post-translational protein modification implicated in the bacterial stress response. Although early reports suggested that arginine phosphorylation also occurs in higher eukaryotes, its overall prevalence was never studied using modern mass spectrometry methods, owing to technical difficulties arising from the acid lability of phosphoarginine. As shown recently, the McsB and YwlE proteins from Bacillus subtilis function as a highly specific protein arginine kinase and phosphatase couple, shaping the phosphoarginine proteome. Using a B. subtilis ΔywlE strain as a source for arginine-phosphorylated proteins, we were able to adapt mass spectrometry (MS) protocols to the special chemical properties of the arginine modification. Despite this progress, the analysis of protein arginine phosphorylation in eukaryotes is still challenging, given the great abundance of serine/threonine phosphorylations that would compete with phosphoarginine during the phosphopeptide enrichment procedure, as well as during data-dependent MS acquisition. We thus set out to establish a method for the selective enrichment of arginine-phosphorylated proteins as an initial step in the phosphoproteomic analysis. For this purpose, we developed a substrate-trapping mutant of the YwlE phosphatase that retains binding affinity toward arginine-phosphorylated proteins but cannot hydrolyze the captured substrates. By testing a number of active site substitutions, we identified a YwlE mutant (C9A) that stably binds to arginine-phosphorylated proteins. We further improved the substrate-trapping efficiency by impeding the oligomerization of the phosphatase mutant. The engineered YwlE trap efficiently captured arginine-phosphorylated proteins from complex B. subtilis ΔywlE cell extracts, thus facilitating identification of phosphoarginine sites in the large pool of cellular protein modifications. In conclusion, we present a novel tool for the selective enrichment and subsequent MS analysis of arginine phosphorylation, which is a largely overlooked protein modification that might be important for eukaryotic cell signaling.
Post-translational modification of proteins is an important signal transduction mechanism used by all living cells. Reversible protein phosphorylation is involved in almost every aspect of cellular regulation, such as growth, proliferation, and survival, in response to both intracellular and extracellular stimuli. The broad impact of protein phosphorylation has been revealed thanks to the development of efficient phosphopeptide enrichment techniques and sensitive MS instrumentation, which ultimately enabled large-scale phosphoproteomic analyses (1). However, most studies have been limited to the analysis of serine, threonine, and tyrosine phosphorylations, as they are most amenable to the currently available methodologies. Although phosphorylation of histidine, arginine, and lysine residues was described decades ago, their distribution remains elusive, as these modifications are acid-labile and thus incompatible with standard MS methods. The difference in stability results from the fact that in phosphoarginine, phosphohistidine, and phospholysine residues the phosphate is attached to a nitrogen atom (N-phosphorylation), forming an acid-labile phosphoramidate bond (2). In comparison, in phosphoserine, phosphothreonine, and phosphotyrosine residues the phosphate moiety is attached to a hydroxyl group (O-phosphorylation), yielding an acid-stable phosphoester bond (2). Acidic conditions are inherent in most phosphoproteomic protocols. Therefore, our group established a TiO2-based phosphoproteomic workflow compatible with the analysis of acid-labile phosphorylations (3). We used the developed methodology to study protein arginine phosphorylation in the Gram-positive bacterium Bacillus subtilis, revealing the central role of phosphoarginine in the regulation of the bacterial stress response (3).
To this end it has been shown that arginine phosphorylation is an important but largely unexplored protein modification. Evidence of protein arginine phosphorylation in a number of organisms was obtained mainly in the 1970s and 1980s (reviewed in Refs. 2 and 4). For example, in a series of studies, Wakim and co-workers partially purified a chromatin-bound kinase that displayed in vitro arginine phosphorylating activity toward histone H3. Such a kinase could be isolated from both mouse leukemia cells (5) and quiescent rat heart endothelial cells (6). However, the identity of the kinase could not be determined, nor were these studies ever followed up with research using modern MS-based techniques. The first protein arginine kinase described in detail was McsB, occurring in B. subtilis and related Gram-positive bacteria (7). McsB acts during stress conditions by negatively regulating the activity of the transcriptional repressor CtsR, which controls the expression of class III heat shock genes, including chaperones and proteases, as well as the mcsB gene itself (7, 8). Besides CtsR, McsB has been shown to phosphorylate at least 40 different proteins in vivo in a stress-dependent manner (3). Strikingly, McsB does not display any homology to known serine/threonine or tyrosine protein kinases, but it does to phosphagen kinases that phosphorylate the guanidinium group of free arginine or creatine, which are used as short-term energy storage molecules. As a consequence, sequence homology searches cannot distinguish McsB-like protein arginine kinases from metabolic enzymes. In B. subtilis, the activity of McsB is counteracted by the protein-arginine phosphatase YwlE (9, 10). Despite sequence and structural homology to protein-tyrosine phosphatases (PTPs),1 YwlE is very specific toward phosphoarginine. A single amino acid substitution (threonine instead of isoleucine) in the highly conserved PTP signature motif localized at the active site establishes a polarity filter that is responsible for the pronounced phosphoarginine selectivity (9). Bioinformatic and biochemical analysis identified a bona fide arginine phosphatase, CG31469 of Drosophila melanogaster, harboring the characteristic threonine filter residue (9). Thus, arginine phosphorylation may be a widespread and highly overlooked protein modification.
In addition to the acid sensitivity of the phosphoramidate bond, another important caveat for the analysis of arginine phosphorylation in higher eukaryotes is the marked abundance of O-phosphorylations, which may hide the presence of other, less represented phosphorylations. For instance, it has been proposed that more than 100,000 O-phosphorylation sites may be present in the phosphoproteome of human cells (11). Currently, conventional phosphoproteomic studies are able to identify over 10,000 phosphopeptides in eukaryotic cell lines (12). Such abundant phosphorylations could compete with phosphoarginine during the phosphopeptide enrichment procedure, as well as get preferentially selected during data-dependent MS acquisition. Here we present a method to enrich samples for arginine phosphorylation prior to MS analysis. For this purpose, we developed a YwlE phosphatase trapping mutant that can be used to pull down arginine-phosphorylated proteins from cell extracts. The resulting reduction in sample complexity should greatly facilitate the identification of arginine phosphorylation in eukaryotic samples.
EXPERIMENTAL PROCEDURES
B. subtilis Cultures
A B. subtilis strain carrying a ywlE::specR chromosomal insertion (3) was grown in liquid Luria–Bertani medium. Cultures grown overnight at 37 °C were diluted in 2 l of fresh medium to an A600 of 0.05 and incubated at 37 °C under 200 rpm orbital shaking until an A600 of 0.8 was reached. To induce heat stress and therefore increase McsB activity (3), the incubator temperature was raised to 48 °C over a 15-min time span, and the culture was then kept at this temperature for another 30 min. Cells were harvested via centrifugation at 4000 rpm for 30 min and washed with buffer (25 mm Tris/HCl, pH 7.5, 100 mm NaCl). Dry pellet aliquots were stored at −80 °C.
DNA Construct Design
Previously published plasmids were used for both B. subtilis and Geobacillus stearothermophilus YwlE expression (9). They corresponded to a pET21a(+) (Novagen, Darmstadt, Germany) vector containing the full-length ywlE gene fusioned to a C-terminal hexa-histidine tag. The mutant YwlE versions were generated using the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). All constructs were verified by DNA sequence analysis.
Protein Expression and Purification
YwlEG.ste was overexpressed in the Escherichia coli BL21 (DE3) strain. Cells were grown at 37 °C with 200 rpm orbital shaking in Luria–Bertani medium supplemented with 100 mg/ml ampicillin until exponential phase (A600 of 0.6 to 0.8) was reached. Expression was then induced with 0.5 mm isopropyl 1-thio-β-D-galactopyranoside for 3 h. Cells were harvested via centrifugation at 4000 rpm for 30 min, washed with buffer A (25 mm Tris/HCl, pH 7.5, 300 mm NaCl), and stored dry at −80 °C. For the production of proteins containing the artificial amino acids Bpa and DiZPK, the YwlEG.ste expression vectors containing the site-specific amber codon mutations were transformed into E. coli BL21 (DE3) strains carrying the plasmids encoding for the orthogonal tRNA and synthetase pair responsible for the incorporation of the unnatural amino acids. Plasmids pDULE-pBpa (13) and pSUPAR-Mb-DiZPK-RS (14) were used for addition of the Bpa and DiZPK amino acids, respectively. Expression conditions were the same, except that cultures were supplemented with tetracyclin 25 μg/ml and 1 mm Bpa (Bachem, Bubendorf, Switzerland) or 34 μg/ml chloramphenicol and 1 mm DiZPK (kindly provided by Dr. Chen).
For purification, cell pellets were resuspended in buffer A containing 1 mg/ml lysozyme, 0.1 mm PMSF, and 10 μg/ml DNase, incubated on ice for 30 min, and disrupted by sonication. Lysates were cleared via centrifugation at 20,000 rpm at 4 °C for 30 min and loaded on a Ni2+-nitrilotriacetate column (HisTrap HP, GE Healthcare LifeSciences) equilibrated in buffer A. Washes were performed using a stepwise imidazole gradient, typically at 25 and 50 mm concentrations. The His-tagged proteins were eluted with 250 mm imidazole and concentrated using Vivaspin devices (GE Healthcare LifeSciences). Buffer exchange was then performed using a HiPrep 26/10 Desalting column (GE Healthcare LifeSciences) equilibrated with 25 mm Tris/HCl, pH 7.5, 100 mm NaCl. When YwlEG.ste was purified for crystallization trials, the Ni2+-nitrilotriacetate elution fractions were applied to a Superdex-75 column (prep-grade; GE Healthcare LifeSciences) equilibrated with 20 mm Tris/HCl, pH 7.5, 100 mm NaCl. Protein aliquots were stored at −80 °C after the addition of 10% glycerol. The B. subtilis YwlE C7A mutant used for the comparison of B. subtilis and G. stearothermophilus YwlE trapping mutants (supplemental Fig. S2) was expressed and purified following the same protocol. Analysis of the YwlEG.ste oligomeric state via analytical gel filtration was performed using a Superdex 75 3.2/30 column (GE Healthcare LifeSciences) using 25 mm Tris/HCl, pH 7.5, 100 mm NaCl.
Crystallization and Structure Solution of YwlEG.ste
The YwlEG.ste crystals were grown via the sitting-drop vapor diffusion method in 96-well plates at 19 °C. YwlEG.ste was crystallized by mixing 100 nL of protein (80 mg/ml) with 100 nL of a reservoir solution containing 2 m ammonium sulfate and 100 mm Tris/HCl, pH 8.2. The crystals belonged to the monoclinic space group P21 and contained one YwlEG.ste dimer per asymmetric unit. For cryo-protection, the crystals were transferred to the crystallization solution supplemented with 25% glycerol and then flash-frozen in liquid nitrogen. The data for a single-wavelength anomalous dispersion experiment were collected at the Swiss Light Source (Beamline X06SA) at λ = 0.9762 Å using a Pilatus detector (Dectris, Baden, Switzerland). The diffraction data were indexed and integrated with MOSFLM (15) and scaled with SCALA (16). Selenium sites were located with SnB (17), crystallographic phasing was performed with Sharp (18), and solvent flattening was performed with SOLOMON (19). The resulting electron density map was of excellent quality, allowing unambiguous tracing of YwlE and bound phosphate and water molecules. The structure was built and refined with the programs ARP/wARP (20), O (21), COOT (22), and CNS (23), and the molecular illustrations were prepared with Pymol (Schrödinger Portland, OR, LLC). Data collection, phasing, and refinement statistics are summarized in supplemental Table S1.
YwlE-lysozyme Pull-downs
Arginine-phosphorylated lysozyme was used as a model protein for testing the binding of YwlE mutants to arginine phosphorylation. Lysozyme was chosen because of (1) its excellent stability, (2) its highly basic nature, which facilitates the purification of the phosphorylated form via cation exchange chromatography, and (3) its small size, which facilitates intact protein mass measurement for phosphorylation rate estimation. To obtain the pure phosphorylated form, 60 mg of lysozyme (Sigma) was phosphorylated in vitro with 10 mg of recombinant G. stearothermophilus McsB (purified as previously described (7)) in 50 mm Tris/HCl, pH 7.5, 50 mm KCl, 8 mm MgCl2, 1 mm ATP at 35 °C for 1 h. The reaction was cleared by centrifugation at 4000g for 20 min and applied to a 5-ml HiTrap Heparin HP column (GE Healthcare LifeSciences) equilibrated with 25 mm Tris/HCl, pH 7.5, 50 mm KCl. The phosphorylated and unphosphorylated forms were separated by a 50-min linear KCl gradient ranging from 50 mm to 1 m final concentration (supplemental Fig. S1A). For phosphorylation analysis, early eluting fractions were diluted with 70% acetonitrile (ACN), 0.1% formic acid, promptly transferred to static nano-electrospray ionization emitters (ES380, Proxeon, West Palm, FL), and directly infused into the LTQ-Velos Orbitrap mass spectrometer (Thermo Scientific) with a voltage of 800 V. Protein masses were acquired in Orbitrap mass analyzer at a resolution of 100,000 (supplemental Fig. S1B). The fractions presenting a highly pure (>99%) mono-phosphorylated form of lysozyme were kept for further use.
For pull-down experiments, YwlE was first bound to magnetic beads (Dynabeads His-Tag Isolation & Pulldown, Invitrogen) in 25 mm Tris/HCl, pH 7.5, 300 mm NaCl, 1 mm DTT. Bound protein was estimated by comparing the protein concentration in the supernatant before and after bead incubation, as measured via the Bradford method (Bio-Rad Protein Assay kit). Phosphoarginine binding was performed by incubating 7 μmol of bead-immobilized YwlE with 7 μmol of lysozyme in 25 mm Tris/HCl, pH 7.5, 100 mm KCl, 1 mm DTT for 1 h at 4 °C under gentle agitation. The beads were then washed three times with 25 mm Tris/HCl, pH 7.5, 150 mm NaCl, 1 mm DTT. Proteins were eluted by boiling the beads in SDS sample buffer, which allowed for direct analysis via SDS-PAGE. Alternatively, elution of YwlE-bound proteins was performed by incubating the beads in 100 mm Tris/HCl, pH 7.5, 100 mm NaCl containing 10 mm phospho-l-arginine trisodium salt (Toronto Research Chemicals, Canada) for 2 h at room temperature. The program ImageJ (24) was used for the quantification of Coomassie-stained protein bands in SDS-PAGE gels.
To test the photocrosslinking of the DiZPK- or Bpa-containing YwlEG.ste trapping mutants to an arginine-phosphorylated protein, equimolar amounts of the YwlEG.ste mutant and phosphoarginine or control lysozyme were incubated together at 12 μm concentration for 1 h at 4 °C. The samples were then exposed to UV light for 30 min (or as otherwise indicated) using a 100-W long-wave mercury lamp (UVP, Upland, CA). Covalent linking of YwlEG.ste to lysozyme was then analyzed via SDS-PAGE.
YwlEG.ste in Vitro Pull-downs Using B. subtilis ΔywlE Cell Extracts
Dry pellets corresponding to ∼1.5 L of B. subtilis culture were resuspended in 15 ml of 25 mm Tris/HCl, pH 7.5, 100 mm NaCl supplemented with 3 mg/ml lysozyme, 10 μg/ml DNase, 0.1 mm PMSF, and 1X Complete protease inhibitor mixture (Roche) and incubated on ice for 30 min. Cells were disrupted by sonication, and lysates were cleared via centrifugation at 20,000g for 30 min. Protein content was estimated using the Bradford method. Aliquots of 38 mg of protein extract were mixed with 1 mg of purified G. stearothermophilus YwlE (D117A, C9A, or C9A F39Bpa, in triplicate) in binding buffer (25 mm Tris/HCl, pH 7.5, 100 mm NaCl) at a final volume of 2 ml, resulting in a YwlEG.ste concentration of 30 μm. The binding of YwlEG.ste to phosphoarginine proceeded for 1 h at 4 °C. The equivalent to 6 mg of His-Tag Dynabeads (Invitrogen), equilibrated in binding buffer, was then added to each sample, and samples were incubated under agitation for 1 h at 4 °C. Beads were washed two times with 1 ml of binding buffer and resuspended in a final volume of 200 μl of the same buffer. An aliquot of 2% of the resulting suspension was boiled in SDS sample buffer and directly analyzed via SDS-PAGE. The remaining samples were reduced with 2 mm DTT at 56 °C for 30 min, alkylated with 10 mm iodoacetamide in the dark at room temperature for 30 min, and digested with 5 μg of Trypsin Gold (Mass Spectrometry Grade, Promega, Madison, WI) overnight at 37 °C. On-bead trypsin digestion completion was inspected based on retention time and UV intensity (214 nm) distribution upon RP-HPLC separation of a 0.1% aliquot of the resulting supernatants on a monolithic column (Ultimate Plus equipped with a PepSwift PS-DVB column, 5 cm × 200 μm, all Dionex/Thermo-Fisher Scientific). Each sample was purified from buffer reagents via reverse-phase C18 solid phase extraction at neutral pH (Strata-X 200 mg cartridge, Phenomenex, Torrance, CA). Lyophilized samples were resuspended in 20 μl H2O, and TiO2 phosphopeptide enrichment using 0.5 mg of Titansphere 5-μm resin (GL Sciences, Tokyo, Japan) was performed according to a recently published protocol optimized for acid-labile phosphorylations (3). Briefly, the critical binding step was performed for 30 min in a lactate/acetate solution adjusted to pH 4 (300 mg/ml lactic acid, 12.5% acetic acid, 0.2% heptafluorobutyric acid, 60% ACN), which is a condition in which no significant hydrolysis of phosphoarginine occurs (3). Removal of unphosphorylated acidic peptides was performed in fast washing steps using solvent A (200 mg/ml lactic acid, 75% ACN, 2% trifluoroacetic acid, 2% heptafluorobutyric acid), solvent B (200 mg/ml lactic acid, 75% ACN, 10% acetic acid, 0.1% heptafluorobutyric acid, pH 4, with NH3), and solvent C (80% ACN, 10% acetic acid). Finally, bound peptides were eluted with a 1% NH3 solution. Furthermore, 30 mg of the protein extract prepared for the pull-downs was processed according to the FASP protocol (25), and three 1-mg aliquots of the resulting solid-phase-extraction-purified tryptic digest were submitted to TiO2 enrichment using 1 mg of Titansphere resin.
Of note, we could not observe any in vitro binding of the YwlEG.ste trapping mutant to arginine-phosphorylated peptide samples (data not shown). This is consistent with the notion that phosphatases have a markedly reduced ability to use phosphopeptides as substrates (26). As a consequence, in the phosphoarginine enrichment methodology we report here, the TiO2 phosphopeptide enrichment step cannot be avoided.
LC-MS/MS
Reverse-phase separation of all peptide mixtures was achieved on an Ultimate 3000 RSLC nano-flow chromatography system (Thermo Scientific). As previously described (3), 0.5% acetic acid (pH 4.5 with NH3) was used as a loading solvent to prevent phosphoarginine hydrolysis during the removal of salts in the pre-column (PepMapAcclaim C18, 5 mm × 0.3 mm, 5 μm, Thermo Scientific). Peptide separation was achieved on a C18 separation column (PepMapAcclaim C18, 50 cm × 0.75 mm, 2 μm, Thermo Scientific) by applying a linear gradient from 2% to 35% solvent B (80% ACN, 0.08% formic acid) in 120 or 240 min (pull-down and shotgun samples, respectively) at a flow rate of 230 nL/min. Solvent A was 2% ACN, 0.1% formic acid. The separation was monitored via UV detection, and the outlet of the detector was directly coupled to the nano-electrospray ionization source (Proxeon Biosystems) for MS analysis.
The sample was infused into the LTQ Orbitrap Velos electron-transfer dissociation mass spectrometer (Thermo Scientific) using PicoTip nanospray emitter tips (New Objective, Woburn, MA) at a voltage of 1.5 kV. Peptides were analyzed in data-dependent fashion in positive ionization mode, applying two different fragmentation methods: collision-induced dissociation and electron-transfer dissociation. The survey scan was acquired at a resolution of 60,000, and the six most abundant signals with charge states equal to or higher than 2+ and exceeding an intensity threshold of 1500 counts were selected for peptide fragmentation analysis. For MS/MS experiments, precursor ions were isolated within a 2.1-Da window centered on the observed m/z. To prevent repeated fragmentation of highly abundant peptides, selected precursors were dynamically excluded for 30 s from MS/MS analysis. Collision-induced dissociation fragmentation was achieved at a normalized collision energy of 35% with additional activation of the dephosphorylated precursor at M-49, M-32.7, and M-98 amu in a standard multistage activation method. For electron-transfer dissociation, peptides were incubated with fluoranthene anions allowing for charge-state-dependent incubation times (90 ms for 3+ charged peptides), and the resulting peptide fragments were detected in the ion trap analyzer.
Data Analysis
Raw data were extracted using the Protein Discoverer software suite (version 1.4.0.288, Thermo Scientific) and searched against a combined forward/reversed database of B. subtilis (strain 168) UniProt Reference Proteome with common contaminants added (4455 entries in total) using MASCOT (version 2.2.07, Matrix Science, London, UK). Carbamidomethylation of cysteine was set as a fixed modification. Phosphorylation of serine, threonine, tyrosine, and arginine and oxidation of methionine were selected as variable modifications. Because tryptic cleavage is impaired at phosphorylated arginine, a maximum of two missed cleavage sites was allowed, whereas fully tryptic cleavage of both termini was required. The peptide mass deviation was set to 5 ppm; fragment ions were allowed to have a mass deviation of 0.8 Da. False discovery rates were assessed using the Percolator tool (27) within the Protein Discoverer package. The results were filtered for peptide rank 1 and high identification confidence, corresponding to a 1% false discovery rate. Low-scoring peptides (Mascot score ≤ 18) were excluded from the analysis when they were not further supported by additional high-scoring identifications in other replicates or experiments. In the rare cases in which a peptide was mapped to more than one protein sequence, both protein hits are reported. For reliable phosphorylation site analysis, all phosphopeptide hits were automatically reanalyzed by the phosphoRS software (28) within the Protein Discoverer software suite. We considered a phosphorylation site as localized when the reported phosphoRS probability was greater than 90%. Spectra reporting ambiguous phosphorylation localization (phosphoRS site probablilities < 90%) are reported separately in supplemental Tables S2–S5 and were excluded from further analysis. When multiple peptide-spectrum matches (PSMs) were obtained for the same phosphopeptide within one sample, we chose to present only the PSM presenting the best identification/localization score compromise. The multiple redundant PSMs were ranked according to their phosphoRS probability scores into three categories (90%–94%, 94%–97%, and 97%–100%); the PSM presenting the best Mascot score within the highest phosphoRS category achieved was reported. In case a phosphopeptide had multiple PSMs pointing to different phosphosite localizations, the corresponding spectra were manually inspected. PSMs presenting wrong or inconclusive localizations were thus excluded from the final list of phosphopeptides. Noteworthy, 82.5% of the localized arginine phosphorylations presented very high (>99%) probability scores. Multiply phosphorylated peptides (reported in separate tabs in supplemental Tables S1 to S4) were also excluded from the analysis, because they cannot be classified into “phosphorylation type” categories.
Quantitative Analysis
To better judge the differences in phosphoarginine content of the nine pull-down samples (D117A, C9A, and C9A F39Bpa, in triplicate), all identified phosphoarginine peptides were quantified according to their total area in time-intensity chromatograms from MS1 full scans. For this purpose, the software Skyline version 2.1.0.4936 (29) was used with standard data-dependent analysis MS1 filtering settings, except that an ion match tolerance of 0.01 Th was applied in the transition settings, and only scans within 10 min of the MS/MS identifications were considered. We manually inspected and corrected all peak assignments to guarantee the least interference and the selection of the correct peak. Only peaks that presented an isotope dot-product of 0.9 or higher (meaning that peaks had to present low interference and reliable similarity to the peptide under examination) were included in the final analysis. Missing values represent the absence of a defined peak containing an isotope dot product above 0.9, meaning the phosphopeptide under examination was below detection (or completely absent) in the respective sample. To avoid redundancy, when multiple charge states were detected, only the most abundant one was kept for analysis. In the nine cases where a phosphopeptide was found both with and without methionine oxidation, the corresponding total areas were summed. Overall, of the 136 phosphoarginine peptides identified in the pull-down experiments, we were able to quantify 132 (97%).
Data Availability
The mass spectrometry data from this study have been submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (30) and assigned the identifier PXD000560. The YwlEG.ste crystal structure is available at the RCSB Protein Data Bank with the accession code PDB 4PIC.
RESULTS
Development of an Arginine Phosphatase Trapping Mutant
“Substrate trapping phosphatases” refer to mutant enzymes that are catalytically inactive but retain their substrate binding capability. Typically, the catalytic step is blocked by mutating the active site residues implicated in substrate dephosphorylation. As a consequence, the substrate is trapped in the catalytic pocket of the phosphatase, and in most cases the enzyme–substrate interaction is sufficiently stable for co-purification of the complex. Trapping mutants have been widely used for the characterization of a number of protein-tyrosine phosphatases, as a method to isolate and identify their physiological substrates (31). The YwlE arginine phosphatase has pronounced structural and sequence homology to low-molecular-weight PTPs, employing a similar catalytic mechanism (9). The bacterial YwlE is a 17-kDa protein composed of a minimal phosphatase core and lacking additional structural motifs, such as, for example, extra substrate recognition domains. Accordingly, YwlE is a very promiscuous arginine phosphatase, in the sense that it specifically targets phosphoarginine residues regardless of the protein context. We thus reasoned that a YwlE trapping mutant could be used as a universal tool for isolating arginine-phosphorylated proteins.
Similar to classical protein-tyrosine phosphatases, the YwlE arginine phosphatase employs two critical active site residues to dephosphorylate its substrates: Cys-9 and Asp-117 in YwlEG.ste. The cysteine residue acts as a nucleophile attacking the phosphorous center of the substrate, leading to a covalent phosphoryl-cysteine intermediate (protein-arginine phosphatase–Cys•PO3) (32). In tyrosine phosphatase trapping mutants, the cysteine residue is typically substituted by a serine, impeding the initial step in the dephosphorylation reaction and leading to the formation of a stable noncovalent PTP-Ser•PO3-substrate complex (31). In YwlE, the catalytic aspartate acts (1) as a general acid protonating the leaving guanidinium group of the substrate and (2) as a general base activating a water molecule to hydrolyze the Cys-PO3 reaction intermediate, thereby releasing free phosphate and regenerating the active enzyme. Importantly, the aspartate (Asp-117) is located on the d-loop that closes over the phosphorylated residue upon substrate binding. In tyrosine phosphatases, substrate binding induces an 8- to 12-Å shift of the d-loop. This movement holds the substrate in place and brings the critical aspartate residue in close proximity to the substrate (32). In aspartate-mutated trapping phosphatases, the residue is usually mutated to an alanine, so that catalysis is blocked but the movement of the d-loop and consequent encapsulation of the substrate is maintained (31).
To obtain a substrate-trapping mutant of the YwlE arginine phosphatase, we generated three mutant versions of the highly active and stable protein from G. stearothermophilus, YwlEG.ste. These mutants were designed on the basis of recently reported crystal structures of the homologous B. subtilis phosphatase YwlEB.sub. Structural data of the YwlEB.sub C7S mutant incubated with free phosphoarginine (PDB 4KK4) showed that, in contrast to PTP Cys/Ser mutants, for YwlE the Cys/Ser mutation resulted in the formation of a phosphorylated serine residue, rather than a stable protein-arginine phosphatase–Ser•PO3-substrate complex (9). Although this YwlE mutant is barely active (33), because the active enzyme is not regenerated, it is not a suitable trapping mutant, as the unphosphorylated substrate is released. We therefore mutated the catalytic cysteine of YwlEG.ste to an alanine, thus completely abolishing the nucleophilic reactivity. Moreover, the high-resolution crystal structure of YwlEB.sub with a substrate-mimicking arginine captured in the active site (PDB 4KK3) revealed that the catalytic aspartate forms a hydrogen bond with the guanidinium group of the arginine substrate (9). Therefore, besides the classical D117A mutation, we also tested a D117N mutation, aiming to retain the hydrogen bond at this position.
To evaluate their substrate trapping capacity, we purified the His-tagged recombinant forms of the YwlEG.ste C9A, D117A, and D117N mutants and tested their binding to an arginine-phosphorylated protein via His-tag pull-downs. For this purpose, we produced a phosphoarginine model protein by phosphorylating chicken lysozyme in vitro with recombinant McsB arginine kinase and subsequently purifying the phosphorylated lysozyme from the unphosphorylated form via cation exchange chromatography (supplemental Fig. S1A). The obtained phosphoarginine model protein was 100% monophosphorylated, with the phosphorylation sites distributed among seven distinct arginine residues (supplemental Figs. S1B and S1C). Fig. 1 shows that the YwlEG.ste cysteine mutant—but, unexpectedly, not the two aspartate mutants—could stably bind to the arginine-phosphorylated substrate. Additionally, to further confirm the trapping effect of the Cys/Ala mutation, and to compare the substrate trapping efficiency of an alternative YwlE protein, we tested the respective mutation in the B. subtilis YwlE phosphatase. We also observed binding of YwlEB.sub C7A to the phosphoarginine protein, albeit with reduced efficiency (supplemental Fig. S2). Therefore, we employed the more efficient G. stearothermophilus YwlE protein in all subsequent trapping mutant pull-downs. Noteworthy, in both B. subtilis and G. stearothermophilus YwlE Cys/Ala mutants, the bound phosphoarginine lysozyme, but not the unphosphorylated control, could be partially released with free phosphoarginine incubation, demonstrating the specific binding of the arginine-phosphorylated lysozyme to the active site (supplemental Fig. S2). We thus conclude that the YwlE Cys/Ala mutant can efficiently trap arginine-phosphorylated proteins.
Fig. 1.
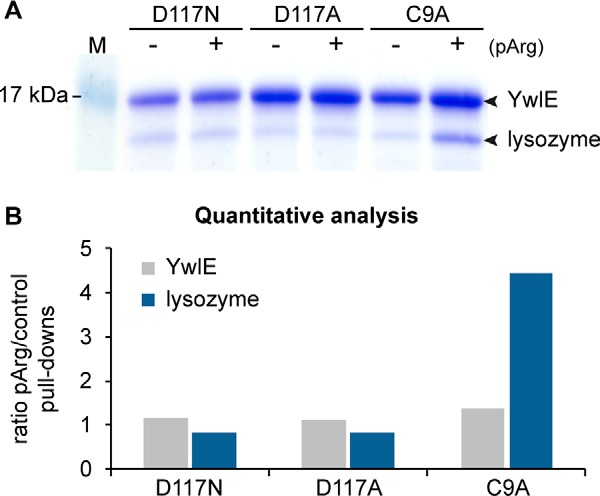
Testing of YwlEG.ste active site mutants for phosphoarginine binding. A, pull-down experiments were used to evaluate the binding of three His-tagged G. stearothermophilus YwlE mutants (YwlE D117N, D117A, and C9A) toward an arginine-phosphorylated model protein, lysozyme. Unphosphorylated lysozyme was used as a control for the assessment of unspecific binding. B, quantitative analysis of the gel bands presented in A, plotted as the protein ratios between phosphoarginine versus control pull-downs.
Generation of an Improved YwlEG.ste C9A Trapping Mutant
We next tested whether a specific cross-linking step could be used to covalently link the trapping YwlEG.ste to the captured substrates, aiming for improved purity and yields of arginine-phosphorylated proteins. For this purpose, we inserted an artificial amino acid residue, p-benzoyl-L-phenylalanine (Bpa), in the YwlEG.ste trapping mutant at strategic positions near the substrate binding site, allowing for UV-induced photocrosslinking of interacting proteins. We identified two suitable positions, F39 and F119 (Fig. 2A), and produced recombinant YwlEG.ste C9A mutants containing the Bpa residue using the amber codon suppression system in E. coli as previously described (13). To test for substrate cross-linking, we incubated the YwlEG.ste C9A F39Bpa and F119Bpa mutants with arginine-phosphorylated lysozyme and exposed them to UV light. SDS-PAGE analysis demonstrated that the mutant containing the Bpa amino acid at position 39, but not at position 119, could cross-link to the arginine-phosphorylated model protein (Fig. 2B). Although cross-linking seems to have been highly specific, as no cross-linking was observed in the unphosphorylated control, the overall amount of cross-linked YwlEG.ste-substrate was low. In contrast, His-tag pull-downs of the YwlEG.ste C9A F39Bpa mutant showed improved binding to phosphoarginine lysozyme relative to the plain YwlEG.ste C9A trapping mutant (supplemental Fig. S3A), demonstrating that cross-linking rather than substrate binding was inefficient. We tried to improve cross-linking yields by increasing UV exposure time (supplemental Fig. S3B), as well as by testing an alternative, less bulky artificial photocrosslinking amino acid, DiZPK (3-(3-methyl-3H-diazirine-3-yl)-propaminocarbonyl-Nε-l-lysine (14)), at the same position (supplemental Fig. S3C). However, as these attempts failed to increase the cross-linking efficiency, we discarded the use of the cross-linking step in the YwlE trapping mutant methodology.
Fig. 2.

Development of an improved YwlEG.ste trapping mutant. A, crystal structure of the G. stearothermophilus YwlE protein (PDB 4PIC). The top panel shows the dimer structure, with a phosphate group (in red/orange) occupying the catalytic site. The F39 residue contributing to dimer interaction is shown in dark red. The bottom panel shows the catalytic site of one YwlE molecule in detail, with the sites of Bpa incorporation (F119 and F39) represented in sticks. For reference, the catalytic residues Cys-9 and Asp-117 are also shown in yellow. The chemical structure of the Bpa unnatural amino acid is also presented in the inset in the bottom left corner. B, photocrosslinking of the Bpa-containing YwlEG.ste trapping mutants to a phosphoarginine model protein (lysozyme). Equimolar amounts of YwlEG.ste and lysozyme (at 12 μm concentration) were incubated for 1 h at 4 °C and exposed to UV light for 30 min. Unphosphorylated lysozyme was used as a negative control. Covalent linkage of the Bpa-containing YwlEG.ste to the model substrate was analyzed via SDS-PAGE. Cross-linked products are marked with a red asterisk. The identity of the indicated bands was confirmed by MS analysis. C, oligomerization analysis of YwlE. The G. stearothermophilus YwlE C9A trapping mutant and its Phe39 variants were analyzed for their oligomeric state distribution by means of analytical size exclusion chromatography. Monomeric species are indicated in the elution profiles (x-axis: elution volume in milliliters; y-axis: absorbance at 280 nm in mAU) with a blue background, and dimer and higher order oligomers are marked in light and dark gray, respectively.
Size exclusion chromatography revealed that purified YwlEG.ste was present in solution as a mixture of monomers, dimers, and higher order oligomers (trimers and tetramers). To evaluate the effect of YwlEG.ste oligomerization on substrate binding, we pursued a structural approach. We crystallized YwlEG.ste and solved the tridimensional structure of the dimeric protein at high resolution. The crystal structure of the YwlEG.ste dimer revealed that dimerization proceeds through the interface surrounding the catalytic site opening, thereby hindering the binding of substrates (Fig. 2A and supplemental Fig. S4). Accordingly, substrate binding—and, by extension, phosphoarginine protein trapping—could be influenced by YwlEG.ste oligomerization. As revealed by the YwlEG.ste structure, the F39 residue is a central component of the YwlE dimer interface, undergoing intimate contacts with the neighboring molecule (Fig. 2A and supplemental Fig. S5). Owing to this finding, we compared various YwlEG.ste C9A F39 mutants for their oligomerization state via analytical size exclusion chromatography. Fig. 2C shows that YwlEG.ste C9A and YwlEG.ste C9A F39A eluted in a similar manner, exhibiting a broad spectrum of different oligomers, whereas the YwlEG.ste C9A F39W mutant had fewer oligomeric species, being present predominantly as a dimer. Remarkably, size exclusion chromatography analysis of the YwlE C9A F39Bpa mutant, originally produced for photocrosslinking, revealed a purely monomeric form. We thus assume that the monomeric F39Bpa mutant is more efficient in binding to arginine-phosphorylated proteins, even if no UV cross-linking step is performed.
Proof-of-principle Experiment: YwlE Trapping Mutant Pull-down Using B. subtilis Cell Extracts
Next, we wanted to test whether the developed YwlE trapping mutant was able to pull down arginine-phosphorylated proteins at endogenous concentrations from complex samples. For this purpose, we used cell extracts from a B. subtilis ΔywlE strain that can accumulate a large number of arginine-phosphorylated proteins. We compared the number of arginine-phosphorylated peptides identified using the three different phosphatase mutants YwlEG.ste C9A, C9A F39Bpa, and D117A. The YwlEG.ste D117A mutant was used as a negative control, as no detectable binding to phosphoarginine-lysozyme was observed in the in vitro pull-downs (Fig. 1). C9A F39Bpa was used to evaluate the effect of disturbing YwlE oligomerization on trapping activity. To assess the technical reproducibility of the procedure, all experiments were done in triplicate using the same B. subtilis ΔywlE cell extract preparation. For comparison, the same cell extract was analyzed via “shotgun” phosphoproteomics (i.e. by digesting the total protein content with trypsin, enriching phosphopeptides with TiO2, and performing LC-MS/MS analysis). Fig. 3 shows that on average 23 phosphoarginine peptides could be identified in each YwlEG.ste C9A pull-down, a number that is markedly greater (p value = 0.01) than the number of arginine phosphorylations identified in each YwlEG.ste D117A pull-down (on average 9 per experiment). In total, the YwlEG.ste C9A pull-downs resulted in the identification of 35 unique phosphoarginine peptides mapped to 28 proteins (supplemental Table S2), whereas the D117A pull-downs resulted in 14 unique phosphoarginine peptides distributed over 12 proteins (supplemental Table S3). Noteworthy, the phosphoarginine peptides indentified in the D117A pull-downs could have resulted either from contaminants that were unspecifically bound to the magnetic beads or from a low trapping activity of this mutant. Importantly, only five phosphoserine/phosphothreonine/phosphotyrosine peptides could be identified in the YwlE C9A pull-downs, and two of those originated in proteins that were also arginine-phosphorylated. The results show that the YwlEG.ste C9A trapping activity can be used to selectively enrich arginine-phosphorylated proteins from cell extracts.
Fig. 3.

MS analysis of the YwlEG.ste trapping mutant pull-downs using B. subtilis cell extracts. A, the graphs depict the number of unique phosphopeptides identified in the pull-down experiments using the indicated YwlEG.ste mutants. For comparison, the number of phosphopeptides identified in shotgun phosphoproteomic analysis of the same B. subtilis ΔywlE cell extract is provided. The results obtained for each experimental replicate are shown. B, Venn diagrams demonstrate the overlap between the phosphoarginine identifications obtained in the pull-down experiments (top diagrams) or between the shotgun analysis and the YwlEG.ste C9A pull-downs (bottom diagrams). Numbers represent the number of arginine-phosphorylated peptides (left diagrams) or arginine-phosphorylated proteins (right diagrams) identified in each experiment. In this case, the datasets represent the sum of non-redundant identifications obtained in the three experimental replicates.
A strikingly greater number of phosphoarginine sites were identified in the YwlEG.ste C9A F39Bpa pull-downs: each replicate experiment revealed on average 87 phosphoarginine peptides, resulting in a total of 121 unique arginine phosphopeptides distributed over 68 different proteins (Fig. 3, supplemental Table S4). This finding indicates that the monomeric form of YwlEG.ste was indeed more efficient in capturing arginine-phosphorylated proteins. Additionally, we performed a quantitative analysis comparing the abundance of all phosphoarginine peptides identified in the pull-down experiments according to their MS1-based extracted ion chromatograms (supplemental Table S6). All nine pull-down samples (D117A, C9A, and C9A F39Bpa, in triplicate) were inspected for the presence of all phosphopeptides. The results are summarized in supplemental Fig. S6. One of the clear observations is that phosphoarginine peptides were present not only in greater numbers but also at higher intensities in the C9A F39Bpa pull-downs than in the D117A and C9A pull-downs (supplemental Fig. S6). Even though the peptides in the C9A samples were distributed in similar intensity ranges as for the D117A pull-downs, in the C9A experiments the phosphoarginine peptides were present in markedly greater numbers (supplemental Fig. S6). Accordingly, it is also evident that for most (83%) of the peak inspections in the three replicate pull-downs of the D117A negative control, the phosphoarginine peptide peak could not be detected, suggesting that the corresponding phosphoarginine peptides were not present in these samples. This further confirms that phosphoarginine could be efficiently enriched by the YwlE C9A proteins, and that the F39Bpa monomeric version was markedly more efficient in capturing phosphoarginine proteins. In regard to this, it should be noted that although the same amount of YwlEG.ste was incubated with the cell extract in all experiments, the final amount of YwlEG.ste C9A F39Bpa bound to the magnetic beads was less than for YwlEG.ste C9A and YwlE D117A (supplemental Fig. S7). A plausible explanation is that for YwlEG.ste C9A and D117A a single His-tag can tether multiple YwlE proteins (held together by oligomerization) to the magnetic beads, leading to an apparently greater bead binding capacity relative to the YwlEG.ste C9A F39Bpa. We therefore cannot exclude the possibility that for YwlEG.ste C9A F39Bpa a different population of contaminants could bind to the beads than in the D117A control. Most important, the proportion of phosphoarginine in the identified phosphopeptides was very high—96.7% on average. This number is markedly greater (p value = 0.005) than in the shotgun analysis, where phosphoarginine was present in only 85.3% of the identified phosphopeptides (supplemental Table S5). We thus presume that the improved binding properties, rather than elevated levels of contaminant proteins, accounted for the large number of phosphoarginine peptides indentified with the help of the YwlEG.ste C9A F39Bpa mutant. Moreover, even though triplicate analysis showed good technical reproducibility in all experiments (supplemental Fig. S8), the overlap between the shotgun analysis and the trapping mutant pull-downs was rather low (Fig. 3B), suggesting that the pull-downs were probably not as biased toward abundant phosphorylations as the shotgun analysis. Indeed, whereas the YwlEG.ste C9A F39Bpa pull-downs identified 10 arginine-phosphorylated transcriptional regulators (supplemental Table S4), representing a class of proteins that typically occur at low abundance, only 3 arginine-phosphorylated transcriptional regulators could be observed in the shotgun analysis (supplemental Table S5). This further evidences the advantages of performing selective enrichment of phosphoarginine in order to obtain a more comprehensive view of its occurrence in the cell.
DISCUSSION
Here we report the development of a specific phosphoarginine binder that can be used as an affinity tool to enrich arginine-phosphorylated proteins prior to MS analysis. The great advantages conferred by selective enrichment have been demonstrated for tyrosine phosphorylation analysis. Tyrosine phosphorylations are much less abundant than serine and threonine phosphorylations, typically representing only 1% to 2% of the phosphopeptide identifications in shotgun phosphoproteomic (global profiling) studies. Di Palma and co-workers compared the number of tyrosine phosphorylation identifications in two human cell lines using either a shotgun approach or selective phosphotyrosine enrichment by immunoprecipitation. Whereas deep phosphoproteome profiling via multidimensional strategies (Ti4+-IMAC followed by hydrophilic interaction liquid chromatography fractionation) could identify only about 300 phosphotyrosine peptides, targeted immunoaffinity enrichment enabled the identification of over 2000 tyrosine phosphorylations (12). This clearly showed that shotgun analysis is insufficient to provide a comprehensive view of underrepresented phosphorylations in eukaryotic samples, as a consequence of the marked masking effect of the predominant serine/threonine phosphorylations. We therefore expect that a specific enrichment method will also be essential for the identification of phosphoarginine proteins in various eukaryotic systems.
Previously, two phosphoarginine binders were reported in the literature. Hofmann et al. reported that the SH2 domain from Src kinase can bind to an arginine-phosphorylated peptide with a Kd of 550 ± 150 μm (34). However, the Kd toward the corresponding phosphotyrosine peptide was estimated as 0.13 ± 0.094 μm (34); therefore, it would be unsuitable for enriching arginine phosphorylations. Fuhrmann et al. reported a specific phosphoarginine antibody obtained in vitro by means of phage display technology (9). Although this antibody could specifically recognize phosphoarginine in purified proteins in dot-blot and ELISA experiments, which were fundamental for the identification of the YwlE arginine phosphatase, it failed to detect arginine phosphorylation in B. subtilis ΔywlE cell extracts (unpublished data). Immunization trials carried out in various animal systems failed to produce a selective antibody, even though phosphoarginine-containing proteins and peptides can be produced in large amounts. Presumably, the inherent lability of P-N linkages prevented obtaining antibodies against phosphoarginine via classical immunization methods that rely on antigen internalization and processing in endosomal/lysosomal compartments under acidic pH conditions (9). Therefore, antibody-based phosphopeptide enrichment procedures, such as those developed for phosphotyrosine (12), are unlikely to be implemented for phosphoarginine.
The phosphoarginine binder we report here consists of a trapping mutant derived from the bacterial YwlEG.ste arginine phosphatase. Phosphoproteomic analysis of B. subtilis lacking YwlE revealed the phosphorylation of at least 231 arginine residues, whereas no arginine phosphorylations could be identified in a B. subtilis wild-type strain (3). This clearly indicates that YwlE is a promiscuous phosphatase targeting arginine phosphorylation in any protein context. The observation that YwlE can bind to arginine phosphorylation sites introduced in a completely artificial model substrate, such as chicken egg lysozyme, further confirms the lack of protein substrate preference. Therefore, the bacterial YwlE phosphatase can be used as a general phosphoarginine binder to detect arginine phosphorylation in diverse biological systems. A recent study by our group revealed YwlE's pronounced specificity toward phosphoarginine, despite structural homology to tyrosine phosphatases. The high-resolution crystal structure of YwlE, together with a series of mutational studies, showed that such specificity can be explained by two independent selectivity filters (9). First, the depth of the binding pockets selects the size of the cognate protein modifications, in the case of YwlE allowing for dephosphorylation of larger phosphoresidues such as phosphotyrosine or phosphoarginine. Second, the presence of a threonine residue (T11) at the bottom of the substrate binding pocket serves as a “polarity filter” that senses the presence of the hydrophilic guanidinium group of the phosphoarginine residue. It is important to mention, though, that at slightly acidic pH levels YwlE has been reported to display some tyrosine dephosphorylation activity (35); therefore, selective phosphoarginine binding should always be performed in neutral to slightly alkaline conditions. In addition, the YwlE homolog of G. stearothermophilus was shown to be a remarkably stable protein that can be easily obtained in large amounts, with typical yields of 60 mg of YwlE protein per liter of E. coli expression culture. Therefore, the recombinant YwlEG.ste phosphatase is ideally suited for use as a phosphoarginine affinity tool. Another advantage is that arginine-phosphorylated proteins can be specifically eluted from the YwlE trap using free phosphoarginine or the competitive inhibitor vanadate, in situations where high purity of arginine-phosphorylated proteins is required.
Phosphatase trapping mutant pull-downs can be used in two different approaches, in vitro and in vivo (31). In the in vitro method, the purified phosphatase mutant is incubated with protein extracts containing the phosphorylated substrates, and the trap-substrate complex is subsequently isolated and analyzed. This method requires careful control of endogenous phosphatase activity in the protein extracts, which could potentially eliminate phosphorylation before trapping mutant binding is achieved. As general phosphatase inhibitors added to the sample would interfere with the trapping mutant binding, the in vitro method requires the inactivation of endogenous phosphatases prior to cell lysis. This implies that arginine phosphatases would have to be either genetically knocked out or in vivo inactivated. For cysteine-based phosphatases, such as YwlE, PTPs, and some general phosphatases, this can be easily achieved by applying the cell-permeable inhibitor pervanadate to the cell culture medium. Excess inhibitor can be washed out or inactivated with alkylating agents such as iodoacetamide, so that the inhibitor does not interfere with subsequent trapping mutant binding. The use of pervanadate also has the advantage of increasing endogenous phosphorylation levels, as was previously observed for pervanadate-treated B. subtilis (3). To date, YwlE and its T11-containing homologs represent the only described arginine phosphatase family. Remarkably, neither tyrosine phosphatases nor dual-specificity phosphatases exhibit arginine-dephosphorylating activity in vitro (9). Moreover, YwlE is solely responsible for all arginine dephosphorylating activity observed in B. subtilis cell extracts (9). This observation indicates that no other phosphatases in this bacterium, including serine/threonine and tyrosine phosphatase family members showing considerable conservation to catalytic units of eukaryotic phosphatases, can dephosphorylate phosphoarginine. Therefore, unless a specific phosphoarginine phosphatase is present in the sample, it should be possible to preserve the arginine modification during in vitro trapping mutant pull-downs. The major advantages of the in vitro approach are that it can be applied to any cell type, tissue, or organ sample and that it can be easily scaled up.
Alternatively, the major drawbacks of the in vitro method can be overcome by expressing the trapping mutant phosphatase in vivo. For tyrosine phosphatases, it has been observed that overexpression of the trapping mutant increases the net phosphorylation content in the cell, as the binding of the trap protects the phosphorylation sites from endogenous phosphatase activity (31). The same effect is to be expected for the YwlE trapping mutant. As a consequence, no previous knowledge of the endogenous arginine phosphatases, or control of their activity, would be required in the in vivo approach. The disadvantages of this method are that genetic manipulation is required in order to introduce the YwlE trapping mutant in the organism of interest and that the system used to incorporate the unnatural amino acid Bpa, required for the more efficient YwlE C9A F39Bpa trapping mutant, is currently limited to E. coli and some eukaryotic cell lines (13, 36).
Previous studies indicate that arginine phosphorylation may be present in very diverse organisms such as Granulosis virus (37) and mammals (5, 6). Most interesting, an unidentified arginine kinase has been described not only to be tightly associated with chromatin, but also to be capable of targeting histone H3 (5, 6), suggesting that phosphoarginine may play a novel role in epigenetics. This indicates that the development of efficient phosphoarginine analysis methods might lead to many novel and exciting findings in different fields of biological research.
CONCLUSIONS
In this work we present the development of a specific phosphoarginine binder consisting of a trapping mutant form of the YwlEG.ste arginine phosphatase. The YwlE-based phosphoarginine protein enrichment procedure can be used in combination with a pH-optimized phosphoproteomic workflow (3) to identify arginine phosphorylations in complex eukaryotic samples (Fig. 4) where the elevated numbers of O-phosphorylations would potentially obscure the presence of phosphoarginine. Proof-of-principle YwlE pull-down experiments demonstrated the efficient isolation of arginine-phosphorylated proteins from cell extracts, as well as significant reduction of serine, threonine, and tyrosine phosphorylations.
Fig. 4.
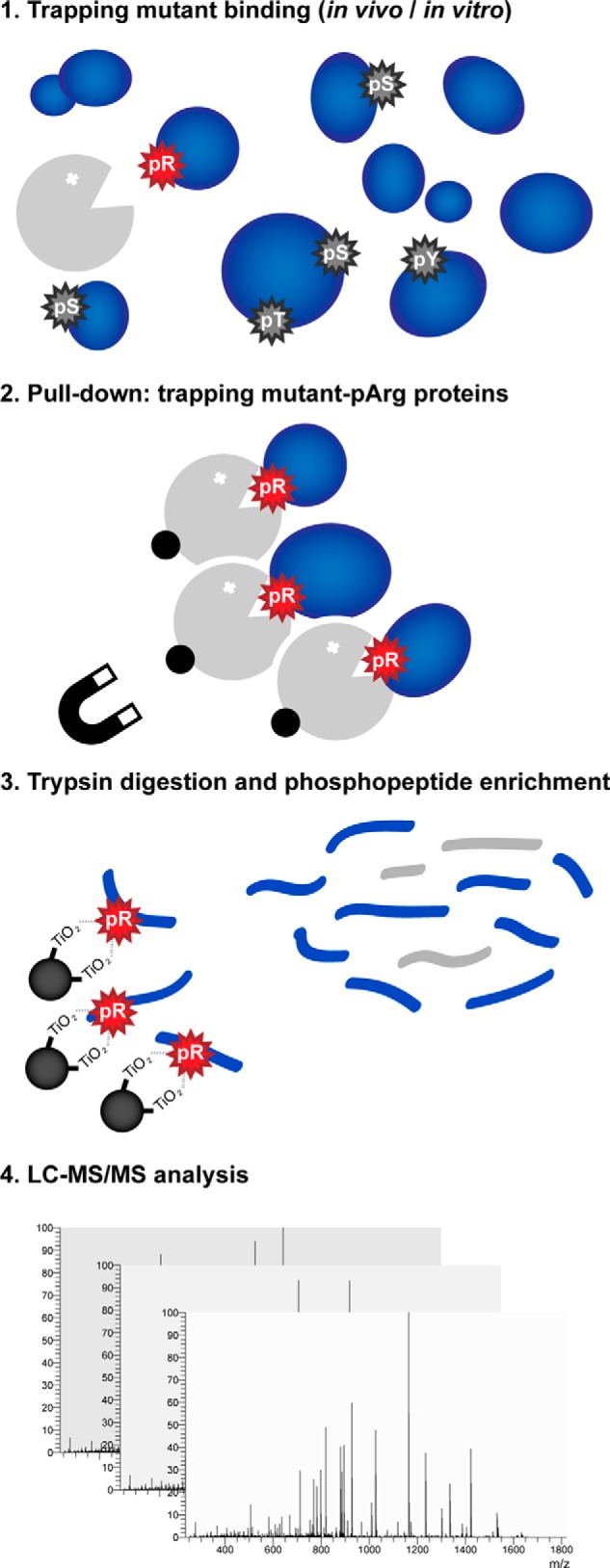
Proposed method for the investigation of arginine phosphorylation in complex samples. Global analysis of arginine phosphorylation can be performed by using the YwlEG.ste trapping mutant (in gray) to specifically bind arginine-phosphorylated proteins (step 1). The YwlEG.ste trap with bound phosphoarginine proteins can be isolated, thus eliminating the highly abundant O-phosphorylations that would hamper the identification of underrepresented phosphorylations (step 2). The arginine phosphorylation sites present in the resulting samples can then be identified via mass spectrometry, using a special TiO2-based phosphopeptide enrichment protocol optimized for acid-labile phosphorylations (3) (step 3) followed by LC-MS/MS analysis (step 4).
Supplementary Material
Acknowledgments
We thank Dr. Peng Chen for providing the DiZPK amino acid and the PRIDE Team for assistance with data deposition.
Footnotes
Author contributions: D.B.T., K.M., and T.C. designed research; D.B.T. and J.F. performed research; D.B.T. and J.F. analyzed data; D.B.T. and T.C. wrote the paper.
* The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013)/Grant Agreement No. 241548 - MeioSys and 262067 - PRIME-XS and from the Austrian Science Fund SFB F3402, P 22570-B09 (D.B.T.) and TRP 308-N15. The IMP is funded by Boehringer Ingelheim.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PTP
- protein-tyrosine phosphatase
- ACN
- acetonitrile
- PSM
- peptide-spectrum match
- Bpa
- p-benzoyl-L-phenylalanine
- DiZPK
- 3-(3-methyl-3H-diazirine-3-yl)-propaminocarbonyl-Nε-L-lysine
- TiO2
- titanium dioxide.
REFERENCES
- 1. Roux P. P., Thibault P. (2013) The coming of age of phosphoproteomics; from large data sets to inference of protein functions. Mol. Cell. Proteomics 12, 3453–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Besant P. G., Attwood P. V., Piggott M. J. (2009) Focus on phosphoarginine and phospholysine. Curr. Protein Pept. Sci. 10, 536–550 [DOI] [PubMed] [Google Scholar]
- 3. Schmidt A., Trentini D. B., Spiess S., Fuhrmann J., Ammerer G., Mechtler K., Clausen T. (2014) Quantitative phosphoproteomics reveals the role of protein arginine phosphorylation in the bacterial stress response. Mol. Cell. Proteomics 13, 537–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cieœla J., Frączyk T., Rode W. (2011) Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim. Pol. 58, 137–148 [PubMed] [Google Scholar]
- 5. Wakim B. T., Aswad G. D. (1994) Ca(2+)-calmodulin-dependent phosphorylation of arginine in histone 3 by a nuclear kinase from mouse leukemia cells. J. Biol. Chem. 269, 2722–2727 [PubMed] [Google Scholar]
- 6. Wakim B., Grutkoski P., Vaughan A., Engelmann G. (1995) Stimulation of a Ca2+-calmodulin-activated histone 3 arginine kinase in quiescent rat heart endothelial cells compared to actively dividing cells. J. Biol. Chem. 270, 23155–23158 [DOI] [PubMed] [Google Scholar]
- 7. Fuhrmann J., Schmidt A., Spiess S., Lehner A., Turgay K. R., Mechtler K., Charpentier E., Clausen T. (2009) McsB is a protein arginine kinase that phosphorylates and inhibits the heat-shock regulator CtsR. Science 324, 1323–1327 [DOI] [PubMed] [Google Scholar]
- 8. Kirstein J., Zühlke D., Gerth U., Turgay K., Hecker M. (2005) A tyrosine kinase and its activator control the activity of the CtsR heat shock repressor in B. subtilis. EMBO J. 24, 3435–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fuhrmann J., Mierzwa B., Trentini D. B., Spiess S., Lehner A., Charpentier E., Clausen T. (2013) Structural basis for recognizing phosphoarginine and evolving residue-specific protein phosphatases in Gram-positive bacteria. Cell Rep. 3, 1832–1839 [DOI] [PubMed] [Google Scholar]
- 10. Elsholz A., Turgay K., Michalik S., Hessling B., Gronau K., Oertel D., Mäder U., Bernhardt J., Becher D., Hecker M., Gerth U. (2012) Global impact of protein arginine phosphorylation on the physiology of Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 109, 7451–7456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choudhary C., Mann M. (2010) Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 11, 427–439 [DOI] [PubMed] [Google Scholar]
- 12. Di Palma S., Zoumaro-Djayoon A., Peng M., Post H., Preisinger C., Munoz J., Heck A. J. (2013) Finding the same needles in the haystack? A comparison of phosphotyrosine peptides enriched by immuno-affinity precipitation and metal-based affinity chromatography. J. Proteomics 91, 331–337 [DOI] [PubMed] [Google Scholar]
- 13. Farrell I. S., Toroney R., Hazen J. L., Mehl R. A., Chin J. W. (2005) Photo-cross-linking interacting proteins with a genetically encoded benzophenone. Nat. Methods 2, 377–384 [DOI] [PubMed] [Google Scholar]
- 14. Zhang M., Lin S., Song X., Liu J., Fu Y., Ge X., Fu X., Chang Z., Chen P. R. (2011) A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance. Nat. Chem. Biol. 7, 671–677 [DOI] [PubMed] [Google Scholar]
- 15. Sarmiento M., Puius Y. A., Vetter S. W., Keng Y. F., Wu L., Zhao Y., Lawrence D. S., Almo S. C., Zhang Z. Y. (2000) Structural basis of plasticity in protein tyrosine phosphatase 1B substrate recognition. Biochemistry 39, 8171–8179 [DOI] [PubMed] [Google Scholar]
- 16. Evans P. (2006) Scaling and assessment of data quality. Acta. Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 17. Weeks C. M., Miller R. (1999) The design and implementation of SnB version 2.0. J. Appl. Crystallogr. 32, 120–124 [Google Scholar]
- 18. Bricogne G., Vonrhein C., Flensburg C., Schiltz M., Paciorek W. (2003) Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta. Crystallogr. D Biol. Crystallogr. 59, 2023–2030 [DOI] [PubMed] [Google Scholar]
- 19. Abrahams J. P., Leslie A. G. W. (1996) Methods used in the structure determination of bovine mitochondrial F-1 ATPase. Acta. Crystallogr. D Biol. Crystallogr. 52, 30–42 [DOI] [PubMed] [Google Scholar]
- 20. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta. Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 22. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 23. Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta. Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 24. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wiśniewski J., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 26. Pinna L., Donella-Deana A. (1994) Phosphorylated synthetic peptides as tools for studying protein phosphatases. Biochim. Biophys. Acta 1222, 415–431 [DOI] [PubMed] [Google Scholar]
- 27. Käll L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 28. Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 29. Schilling B., Rardin M., MacLean B., Zawadzka A., Frewen B., Cusack M., Sorensen D., Bereman M., Jing E., Wu C., Verdin E., Kahn R., MacCoss M., Gibson B. (2012) Platform independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline. Application to protein acetylation and phosphorylation. Mol. Cell. Proteomics 11, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vizcaíno J. A., Côté R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Pérez-Riverol Y., Reisinger F., Ríos D., Wang R., Hermjakob H. (2013) The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blanchetot C., Chagnon M., Dube N., Halle M., Tremblay M. (2005) Substrate-trapping techniques in the identification of cellular PTP targets. Methods 35, 44–53 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Z. Y. (2003) Chemical and mechanistic approaches to the study of protein tyrosine phosphatases. Acc. Chem. Res. 36, 385–392 [DOI] [PubMed] [Google Scholar]
- 33. Fuhrmann J., Subramanian V., Thompson P. R. (2013) Targeting the arginine phosphatase YwlE with a catalytic redox-based inhibitor. ACS Chem. Biol. 8, 2024–2032 [DOI] [PubMed] [Google Scholar]
- 34. Hofmann F. T., Lindemann C., Salia H., Adamitzki P., Karanicolas J., Seebeck F. P. (2011) A phosphoarginine containing peptide as an artificial SH2 ligand. Chem. Commun. 47, 10335–10337 [DOI] [PubMed] [Google Scholar]
- 35. Musumeci L., Bongiorni C., Tautz L., Edwards R., Osterman A., Perego M., Mustelin T., Bottini N. (2005) Low-molecular-weight protein tyrosine phosphatases of Bacillus subtilis. J. Bacteriol. 187, 4945–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hino N., Okazaki Y., Kobayashi T., Hayashi A., Sakamoto K., Yokoyama S. (2005) Protein photo-cross-linking in mammalian cells by site-specific incorporation of a photoreactive amino acid. Nat. Methods 2, 201–206 [DOI] [PubMed] [Google Scholar]
- 37. Wilson M. E., Consigli R. A. (1985) Functions of a protein kinase activity associated with purified capsids of the granulosis virus infecting Plodia interpunctella. Virology 143, 526–535 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data from this study have been submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (30) and assigned the identifier PXD000560. The YwlEG.ste crystal structure is available at the RCSB Protein Data Bank with the accession code PDB 4PIC.