

Abstract
Quantifying the similarity of spectra is an important task in various areas of spectroscopy, for example, to identify a compound by comparing sample spectra to those of reference standards. In mass spectrometry based discovery proteomics, spectral comparisons are used to infer the amino acid sequence of peptides. In targeted proteomics by selected reaction monitoring (SRM) or SWATH MS, predetermined sets of fragment ion signals integrated over chromatographic time are used to identify target peptides in complex samples. In both cases, confidence in peptide identification is directly related to the quality of spectral matches. In this study, we used sets of simulated spectra of well-controlled dissimilarity to benchmark different spectral comparison measures and to develop a robust scoring scheme that quantifies the similarity of fragment ion spectra. We applied the normalized spectral contrast angle score to quantify the similarity of spectra to objectively assess fragment ion variability of tandem mass spectrometric datasets, to evaluate portability of peptide fragment ion spectra for targeted mass spectrometry across different types of mass spectrometers and to discriminate target assays from decoys in targeted proteomics. Altogether, this study validates the use of the normalized spectral contrast angle as a sensitive spectral similarity measure for targeted proteomics, and more generally provides a methodology to assess the performance of spectral comparisons and to support the rational selection of the most appropriate similarity measure. The algorithms used in this study are made publicly available as an open source toolset with a graphical user interface.
In “bottom-up” proteomics, peptide sequences are identified by the information contained in their fragment ion spectra (1). Various methods have been developed to generate peptide fragment ion spectra and to match them to their corresponding peptide sequences. They can be broadly grouped into discovery and targeted methods. In the widely used discovery (also referred to as shotgun) proteomic approach, peptides are identified by establishing peptide to spectrum matches via a method referred to as database searching. Each acquired fragment ion spectrum is searched against theoretical peptide fragment ion spectra computed from the entries of a specified sequence database, whereby the database search space is constrained to a user defined precursor mass tolerance (2, 3). The quality of the match between experimental and theoretical spectra is typically expressed with multiple scores. These include the number of matching or nonmatching fragments, the number of consecutive fragment ion matches among others. With few exceptions (4–7) commonly used search engines do not use the relative intensities of the acquired fragment ion signals even though this information could be expected to strengthen the confidence of peptide identification because the relative fragment ion intensity pattern acquired under controlled fragmentation conditions can be considered as a unique “fingerprint” for a given precursor. Thanks to community efforts in acquiring and sharing large number of datasets, the proteomes of some species are now essentially mapped out and experimental fragment ion spectra covering entire proteomes are increasingly becoming accessible through spectral databases (8–16). This has catalyzed the emergence of new proteomics strategies that differ from classical database searching in that they use prior spectral information to identify peptides. Those comprise inclusion list sequencing (directed sequencing), spectral library matching, and targeted proteomics (17). These methods explicitly use the information contained in empirical fragment ion spectra, including the fragment ion signal intensity to identify the target peptide. For these methods, it is therefore of highest importance to accurately control and quantify the degree of reproducibility of the fragment ion spectra across experiments, instruments, labs, methods, and to quantitatively assess the similarity of spectra. To date, dot product (18–24), its corresponding arccosine spectral contrast angle (25–27) and (Pearson-like) spectral correlation (28–31), and other geometrical distance measures (18, 32), have been used in the literature for assessing spectral similarity. These measures have been used in different contexts including shotgun spectra clustering (19, 26), spectral library searching (18, 20, 21, 24, 25, 27–29), cross-instrument fragmentation comparisons (22, 30) and for scoring transitions in targeted proteomics analyses such as selected reaction monitoring (SRM)1 (23, 31). However, to our knowledge, those scores have never been objectively benchmarked for their performance in discriminating well-defined levels of dissimilarities between spectra. In particular, similarity scores obtained by different methods have not yet been compared for targeted proteomics applications, where the sensitive discrimination of highly similar spectra is critical for the confident identification of targeted peptides.
In this study, we have developed a method to objectively assess the similarity of fragment ion spectra. We provide an open-source toolset that supports these analyses. Using a computationally generated benchmark spectral library with increasing levels of well-controlled spectral dissimilarity, we performed a comprehensive and unbiased comparison of the performance of the main scores used to assess spectral similarity in mass spectrometry.
We then exemplify how this method, in conjunction with its corresponding benchmarked perturbation spectra set, can be applied to answer several relevant questions for MS-based proteomics. As a first application, we show that it can efficiently assess the absolute levels of peptide fragmentation variability inherent to any given mass spectrometer. By comparing the instrument's intrinsic fragmentation conservation distribution to that of the benchmarked perturbation spectra set, nominal values of spectral similarity scores can indeed be translated into a more directly understandable percentage of variability inherent to the instrument fragmentation. As a second application, we show that the method can be used to derive an absolute measure to estimate the conservation of peptide fragmentation between instruments or across proteomics methods. This allowed us to quantitatively evaluate, for example, the transferability of fragment ion spectra acquired by data dependent analysis in a first instrument into a fragment/transition assay list used for targeted proteomics applications (e.g. SRM or targeted extraction of data independent acquisition SWATH MS (33)) on another instrument. Third, we used the method to probe the fragmentation patterns of peptides carrying a post-translation modification (e.g. phosphorylation) by comparing the spectra of modified peptide with those of their unmodified counterparts. Finally, we used the method to determine the overall level of fragmentation conservation that is required to support target-decoy discrimination and peptide identification in targeted proteomics approaches such as SRM and SWATH MS.
EXPERIMENTAL PROCEDURES
Yeast Culture, Protein Isolation, Digestion, Phospho-enrichment, and Dephosphorylation
The yeast strain BY4741 MATa his3Δ leu2Δ met15Δ ura3Δ was grown in triplicate in 100 ml of S.D. medium until an OD600 of 0.7. The cells were harvested by adding 100% of ice-cold tricholoroacetic acid (6.25% final concentration) and centrifugation at 1500 × g for 5 min at 4 °C. The cell pellet was washed twice with 10 ml of ice-cold acetone. The acetone was removed and the final cell pellet (1 volume) was resolubilized in 1 volume of lysis buffer (8 m urea, 0.1 m NH4HCO3, and 5 mm EDTA). The cells were disrupted by glass bead beating (5 times 5 min at 4 °C). The five supernatants were pooled and the total protein amount was estimated by BCA Protein Assay Kit (Thermo, Rockford, US). Three batches of 3 mg of yeast proteins were reduced with 12 mm dithiotreitol at 37 °C for 30 min and alkylated with 40 mm iodoacetamide at room temperature in the dark for 30 min. Samples were diluted with 0.1 m NH4HCO3 to a final concentration of 1 m urea and the proteins were digested with sequencing grade porcine trypsin (Promega, Madison, WI, USA) to a final enzyme:substrate ratio of 1:100. The protein digest was stopped with formic acid to a final concentration of 1% and the peptides were desalted using reverse phase Sep-Pak tC18 cartridges (Waters, Milford, MA, USA). The peptides were concentrated using a vacuum centrifuge and stored at −20 °C. For the phospho-enrichments, 3 mg of digested peptides were incubated with 1.25 mg of TiO2 resin (GL Sciences Inc. Tokyo, Japan) essentially as previously reported (34). The peptides were desalted using reverse phase MicroSpin columns (Nest Group Inc. Southborough, MA). The peptides were concentrated using a vacuum centrifuge and stored at −20 °C. For the dephosphorylation reaction, a batch of 30–50 μg of phosphorylated peptides were desalted using reverse phase MicroSpin columns as before except that the two final washes were performed with water and the elution with 300 μl of 50% ACN in water, without any acid. The eluted peptides were concentrated using a vacuum centrifuge. The peptides were reconstituted in 150 μl of 100 mm NaCl, 50 mm Tris-HCl, 10 mm MgCl2, 1 mm dithiothreitol at pH 7.9 and incubated with 5 μl of Alkaline Phosphatase (New England Biolabs, Ipswhich, MA, USA) for 3 h at 37 °C. The resulting solution was then acidified using 50% TFA and the peptides desalted again using the reverse phase cartridges MicroSpin Column. The eluted peptides were concentrated using a vacuum centrifuge. The concentration of each peptide sample was adjusted to 1 μg/μl (with 2% ACN, 0.5% FA) according to UV absorption at 280 nm measured using a NanoDrop ND-1000 photometer.
For supplemental Fig. 8, the samples originated from the same yeast strain, but were generated for a time course osmotic shock experiment that will be the subject of a separate publication (Selevsek, N. et al. submitted). The samples were prepared essentially using the same protocol, except that an osmotic shock treatment was triggered by addition of 0.4 m salt at time point 0. The samples used for this paper consisted in (1) five technical replicates of the time point 0, injected either consecutively or several days apart (see below), and in (2) three biological replicates of time point 0 (injected consecutively) plus the first biological replicate of time point 60 min and 120 min (injected several days apart).
LC-MS/MS Acquisition Set-ups
The different peptide samples were analyzed either on a Thermo LTQ Orbitrap XL, on a Thermo Obritrap Elite (both Thermo Scientific, San Jose, CA, USA) or on a 5600 TripleTOF mass spectrometer (ABSciex, Concord, Ontario, Canada) under typical acquisition conditions for each machine. For the 5600 TripleTOF shotgun and SWATH MS acquisition, the chromatographic separations of peptides was performed on a NanoLC-2Dplus HPLC system (Eksigent, Dublin, CA, USA) coupled to a 75 μm diameter fused silica emitter, packed with 20 cm of Magic C18 AQ 3 μm resin (Michrom BioResources, Auburn, CA, USA). Peptides (1 μl injections containing 1 μg peptide amount) were loaded on the column from a cooled (4 °C) Eksigent autosampler at 300 nl/min of an isocratic 98% Buffer A (2% acetonitrile, 0.1% formic acid) and 2% Buffer B (98% acetonitrile, 0.1% formic acid). Peptides were separated at a flow rate of 300 nl/min with a 120 min linear gradient of 2% to 35% Buffer B. For the 5600 TripleTOF shotgun experiments the mass spectrometer was operated with a “top20” method, where a 500 ms survey scan (TOF-MS) was collected from which the top 20 ions were selected for subsequent automated MS/MS measurements where each MS/MS event consisted of a 150 ms scan. The selection criteria for parent ions included an intensity threshold of 250 counts per second and a charge state greater than or equal to 2+. Once a precursor ion was fragmented by MS/MS its mass and the mass of its isotopes were excluded for a period of 15 s. Ions were isolated using “unit” quadrupole resolution and fragmented in the collision cell using the collision energy equation (0.0625 x m/z - 3.5) with an additional collision energy spread of +/− 15 eV within the 150 ms accumulation time to mimic SWATH fragmentation conditions. In the instances where there were less than 20 parent ions per survey scan which met the selection criteria, those ions which did were subjected to longer extended MS/MS accumulation times to maintain a constant total cycle time of 3.5 s. For the 5600 TripleTOF SWATH-MS experiments the mass spectrometer was operated in a looped product ion mode. Using an isolation width of 25 Da, a set of 32 overlapping windows (1 Da overlap) was constructed covering the mass range 400 to 1200 Da. The collision energy for each window was determined based on the appropriate collision energy for a 2+ ion centered in the respective window (equation: 0.0625 x m/z - 3.5) with a collision energy spread of ± 15 eV. An accumulation time of 100 ms was used for each fragment ion scan and for the survey scans acquired at the beginning of each cycle, resulting in a total cycle time of 3.3 s. The sequential precursor isolation window set-up was as follows: [400–425], [424–450], [449–475], [474–500]…, [1174–1200], with an effective (100%) transmission of ∼25 Da and ∼0.3 Da attrition on either side of the isolation window.
For the LTQ Orbitrap XL data acquisition, the peptide separation was carried out with a Proxeon EASY-nLC II liquid chromatography system connected to an RP-HPLC column (75 μm x 10 cm) packed with Magic C18 AQ (3uM) resin, running a linear gradient from 95% solvent A (0.1% formic acid, 2% acetonitrile) and 5% solvent B (98% acetonitrile, 0.1% formic acid) to 35% solvent B over 90 min at a flow rate of 300 nl/min. The data acquisition mode was set to obtain one high-resolution MS scan in the Orbitrap (60,000 resolution at 400 m/z). The 10 most abundant ions from the MS scan were fragmented by collision induced dissociation (CID) and MS/MS fragment ion spectra were acquired in the linear ion trap (LTQ) at 35% normalized collision energy. Charge state screening was enabled and unassigned or singly charged ions were rejected. The dynamic exclusion window was set to 30s and limited to 500 entries.
For the Thermo Orbitrap Elite shotgun acquisition, 1 μg of sample was separated on a Thermo Easy-nLC 1000 HPLC system using a 50 cm long, 75 μm diameter ID PepMap column (Thermo, particle size 3 μm) with a 120 min gradient from 5% B to 35% B at a flow rate of 300 nl/min. Solvents were A: 3% acetonitrile, 0.1% formic acid in water; B: 3% water, 0.1% formic acid in acetonitrile. MS/MS spectra were acquired using ion trap-CID at 30% normalized collision energy (NCE) or using collision cell CID (HCD) using 30 or 35% NCE by sequencing the top 15 most abundant precursors per cycle. Resolution settings were: MS1 = 120,000 and MS2 = normal resolution for trap-CID and MS1 = 60,000 and MS2 = 15,000 for HCD. In both modes, precursors of charge state +1 and precursors of unknown charge state were excluded from fragmentation. Precursor isolation width was 2 m/z in all cases and dynamic exclusion was enabled for 30 s. MS1 scans were set to a maximum of 1,000,000 counts and a maximum fill time of 200 ms. MS2 trap CID scans were set to a maximum of 10,000 counts and a maximum fill time of 100 ms. MS2 OT HCD scans were set to a maximum of 50,000 counts and a maximum fill time of 100 ms. It should be noted that for all the data presented in this paper (except supplemental Fig. S8), the samples were acquired as consecutive technical replicate injections, in order to minimize potential variations in instrument stability that might have obscured the intrinsic performance of the machine in recording reproducible fragment ion spectra. For supplemental Fig. S8 however, the samples were either acquired as consecutive injections or separated by several days of measurement (see supplemental Fig. S8 caption for details).
Generation of Spectral Library and Database Searching
The shotgun LTQ Orbitrap XL, Orbitrap Elite (.raw) and TripleTOF (.wiff) data files were converted to centroided mzXML using the Proteowizard converter (35) and searched with Sorcerer-SEQUEST (TurboSequest v4.0.3rev11 running on Sage-N Sorcerer v4.0.4) using the Saccharomyces cerevisiae yeast SGD database (release 03 Febr. 2011, containing 6717 yeast protein entries, concatenated with 6717 corresponding “tryptic peptide pseudo-reverse” decoy protein sequences). For the search, we allowed for semi-tryptic digests and up to two missed cleavages per peptide, and we used carbamidomethylation as fixed modification on cysteine and oxidation as variable modification on methionine. The Sequest search results were converted to pep.xml format and further processed using PeptideProphet (TPP version 4.5.2) (36). The search results were sorted by decreasing PeptideProphet probability and filtered at 1% false discovery rate (FDR) by decoy counting at the peptide spectrum matches (PSM) level. The complete list of peptide identifications for the various instrument modes can be found in the supplemental Tables S1. Consensus spectra libraries were built independently for each file with SpectraST (v. 4, included in TPP version 4.5.2) using the peptides identified above 1% FDR for the replicate experiment (for the intrinsic comparison, for example, Fig. 2 and 3) or for the instrument mode (for the portability analysis, Fig. 4 and 6). For the high mass accuracy MS/MS measurements (e.g. Elite HCD and TripleTOF), the CID-QTOF option was used when running SpectraST to improve the quality of consensus generation. For the TripleTOF and Elite HCD shotgun results, the peptide library files were further converted into an input assay list using an in-house developed python script. The script used the SpectraST .sptxt library as input and retrieved the top 6–100 most intense (singly or doubly charged) y or b fragment ions for each spectra with m/z above 400 and outside of the range of the 25 Da swath/precursor fragmentation window and whose m/z matched the theoretical fragment ion masses within 0.05 Da tolerance. For the phospho-peptide libraries, y and b fragment ions with neutral losses (-80,-98) were also exported, when those obeyed the same selection rules as above. For the dephosphorylated peptide assays used to query for phospho-modified peptides counterparts, we used only the transitions until the modification site, with the same relative intensities and retention time and we modified the mass of the precursors accordingly to the mass of the PTM. The complete sets of assay lists used in this study can be found in the supplemental Tables S2. It should be noted that though the assay list contains between 6 and 100 transitions for the peptides, the targeted peptides were scored and identified using the six most intense transitions from that assay list (see below).
Fig. 2.
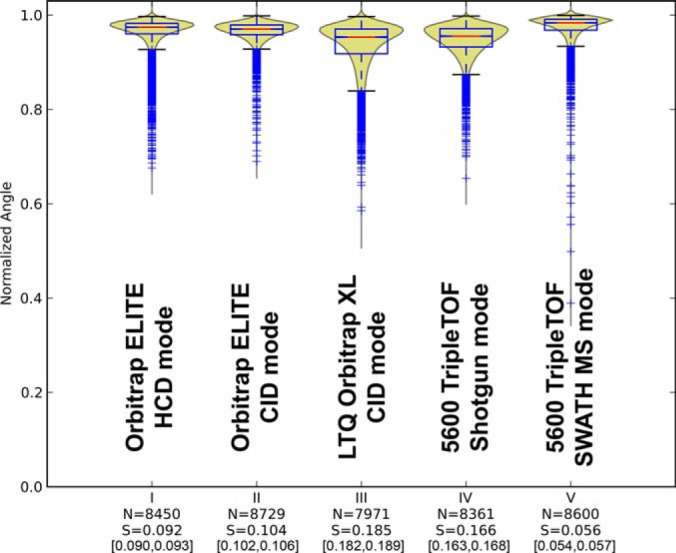
Conservation of fragmentation for technical replicates of yeast naked peptides commonly identified in (I) Orbitrap ELITE HCD, (II) Orbitrap ELITE CID, (III) LTQ Orbitrap XL CID, (IV) TripleTOF shotgun, and (V) TripleTOF SWATH MS. “N” indicates the number of comparisons underlying each violin plot and “S” the level of benchmarked perturbation (Fig. 1) that would best match the corresponding distribution (see supplemental Fig. S7). The 95% bootstrap confidence interval of the S-score is also reported between square brackets. Note that, though only commonly identified peptides were used to generate those plots, the number of comparisons “N” may slightly vary between instrument modes. This is because the comparisons were performed pairwise between the technical triplicates but a given peptide may sometimes be identified only in two out of the three measurements for some modes.
Fig. 3.
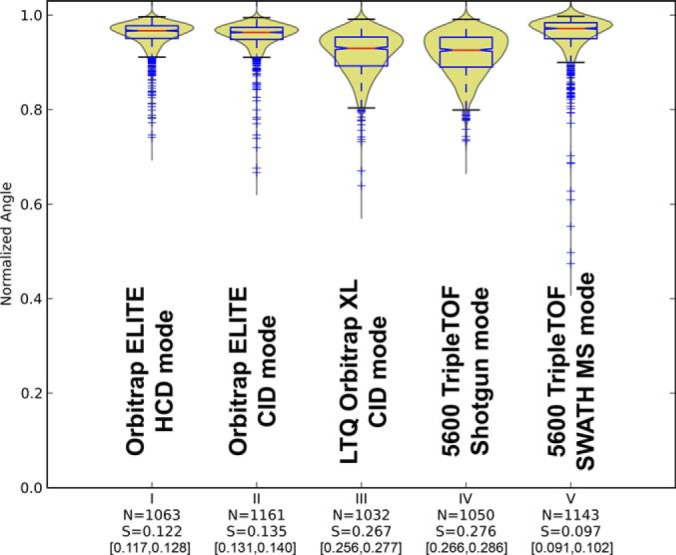
Conservation of fragmentation for technical replicates of yeast phosphopeptides identified in common in (I) Orbitrap ELITE HCD, (II) Orbitrap ELITE CID, (III) LTQ Orbitrap XL, (IV) TripleTOF shotgun, and (V) TripleTOF SWATH MS.
Fig. 4.
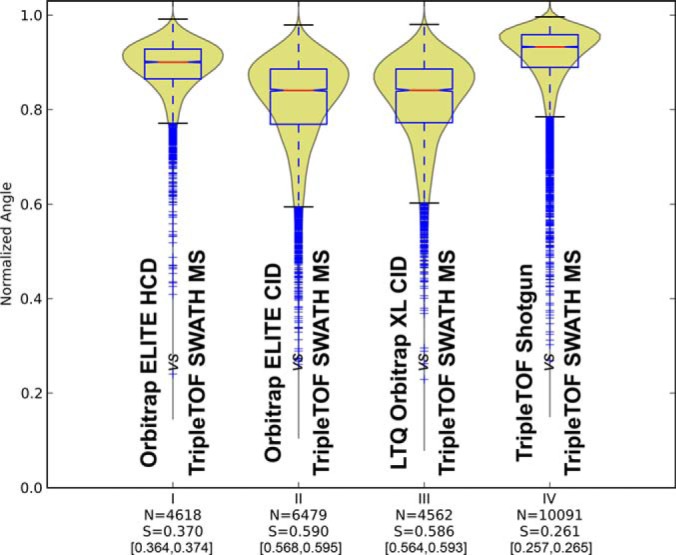
Cross-device similarities for the peptide fragmentation patterns observed in (I) Orbitrap ELITE HCD, (II) Orbitrap ELITE CID, (III) LTQ Orbitrap XL, and (IV) TripleTOF shotgun, toward the fragment intensities of the peptides obtained by SWATH MS data extraction used as common reference.
Fig. 6.
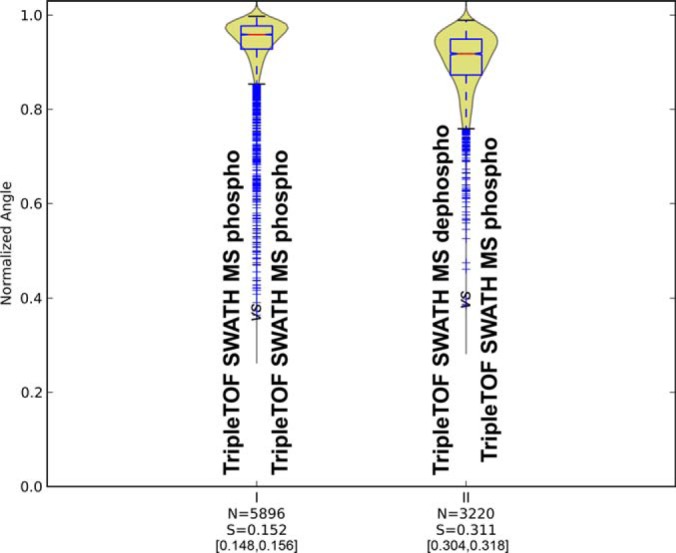
(I) Conservation of fragmentation for technical replicates of yeast phosphopeptides measured in TripleTOF SWATH mode (II) Spectral similarities between samples of dephosphorylated phosphopeptides and phosphopeptides measured in TripleTOF SWATH mode.
SWATH- MS Targeted Data Extraction
Targeted data extraction of the SWATH data was performed with the ABSciex PeakView Software (RC.2.0.3). The peak group scores in PeakView are calculated by mapping each individual mini-score toward a corresponding local background score distribution using 200 decoys (human, scrambled sequences) per time/swath bin (10 min/25 Da) [Navarro, P. in preparation]. As for the targets, the decoys retention times were centered in the middle of the extraction window. The number of decoys retrieved/identified from those background estimates does not matter because it is the rank of the mini-scores of the targets among the local decoys mini-scores that is used for downstream analysis. For this, the background was trained on the first SWATH MS replicate file for each sample type (naked, phosphorylated, and dephosphorylated peptide analyses) using six decoy transitions, to match the number of transitions used for the targets (see below). We operated PeakView to extract the six most intense transitions of the peptides present in the assay lists described above with a 15 min time window and 50 ppm MS/MS tolerance. These six most intense transitions were used to score each targeted peptide, and as many corresponding decoy-transition assays which were generated by pseudo-reversing (keeping the C-terminal residue) the sequence of the targeted peptides. False discovery rate was determined essentially as for shotgun by sorting the highest scored peak group for each assay by decreasing score and counting the number of decoy assays until 5% FDR is reached [Navarro, P. in preparation]. This first stage of data extraction represents essentially the “standard” workflow for peptide identification by SWATH MS targeted data extraction. To correct for cases when a top ranked identified peptide might be inconsistent across the different replicates, we used an in-house python script to identify the 2 min wide cluster populated with the highest number of identified peak groups (with score above 1E6) across replicates. The peak groups present in that most populated cluster were re-ranked to peak group 1 and were therefore considered as identification evidences of the targeted peptides. The complete lists of peptides identified by SWATH MS targeted data extraction can be found in the supplemental Tables S3. Once the peptide is identified using its six most intense transitions, its exact/experimental retention time of identification is known for each of the individual SWATH MS measurements. At this point, we diverged from the standard SWATH MS targeted extraction workflow to trigger a second round of data extraction for each identified peak group (re-)ranked 1 from extraction stage 1 at its exact experimental retention time in that SWATH MS run within a 1.5 min time window, using this time the maximum number of transitions available for that peptide. We use this 2nd stage extraction to retrieve high quality fragment ion intensities for the 6–100 transitions integrated for each high quality peak group. Indeed, if one would try to extract 100 transitions for some targets in the stage 1 already, the identification score for those peptides would be very difficult to compare with those of peptides identified from six transitions extraction. The 2nd stage extraction is therefore a method to retrieve as many fragment ion intensities as possible for each peak group, independently of the peak group identification score, because we know already from the 1st stage extraction that this peak group represents the right peptide identification at the right retention time. At this 2nd extraction stage PeakView generates a pseudo-SpectraST sptxt file containing the integrated area for all the extracted peptide transitions for each replicate SWATH MS measurement. These files are then used as “SWATH-grade” peptide fragment intensity measurements that can be processed with the same set of python scripts for the similarity comparison (intrinsic comparison or portability analysis).
Importantly, in order to avoid any bias for the computation of the cross-similarity, we decided to turn off the fragmentation intensity pattern score (that evaluates in PeakView the similarity of the extracted transitions to that of the assay library) during the whole series of SWATH MS data extraction. In this case, the peptide identification thus relies solely on the other mini-scores such as the transitions chromatographic peak co-elution/shape, consistency of the retention time, mass accuracy of the extracted fragments in the MS/MS spectra at the apex of the peak, etc. [Navarro, P. in preparation]. Though this impacted on the performance of the identification, it ensured that the retrieved peptides were not only the ones with high similarity to that of the library assay (which would have clearly biased our analysis) but instead had a wide range of relative fragment ion intensity unrelated to that of the assay library used.
Spectral Similarity Measures and Spectral Comparisons
a. Generation of Randomized/Perturbed Mass Spectra
For randomizing a mass spectrum with N top peaks (six for all data presented in this study) with increasing and decreasing peak intensities as result of randomization, we define a value
As unidirectional increases/decreases over a large subset of peaks, signified by high values of δ, in spectral randomization would create a bias toward low strength of randomization, we sample δ from a normal distribution of weights favoring low values of δ.
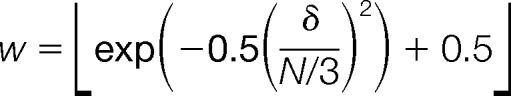 |
Using δ, all N-permutations of {inc,Dec} selected with replacement are generated. For each simulation, a randomization frame is selected from the set of defining the direction of randomization for each peak
For a given peak with initial intensity Ii, the randomized intensity Ir is defined as
Where, r denotes the strength of perturbation and is sampled from a uniform distribution with , with n showing the total number of different randomization strengths, and rcent the central randomization strength at the corresponding randomization step. For the data presented in this study, we used 20 randomization steps, yielding the following upper and lower randomization bounds:
b. Spectral Normalization and Comparison
Preprocessing operations such as spectral intensity scaling prior to normalization have been subject of previous studies (18, 29, 32) and will not be discussed here. For our analysis, we used square root intensity transformation and normalized intensity vectors to unit length as suggested elsewhere (18, 29, 32). For an input spectrum SI with N top peaks, the normalized output spectrum SOi is calculated as follows:
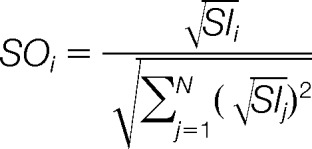 |
Two square root-transformed and normalized spectra (S1,S2) are then compared as follows, yielding similarities denoted as λ, depending on the used similarity measure:
Dot product:
Normalized spectral contrast angle*:
Spectral correlation**:
Bray-Curtis distance*:
Euclidean distance*:
For ensuring comparability, all measures used for spectral comparison were mapped to the result space [0,1] with value 1 denoting the highest similarity. As the spectral vectors were confined to unit length with strictly positive values, results were by default confined to the result space [0,1] except for Spectral Correlation (**), where the result range [-1,1] was mapped to the correct range by shifting and scaling. The correct behavior for increasing/decreasing similarity was achieved in distance measures (*) by subtracting the result from 1.
c. Generation of Reference Benchmark Distributions
Using an experimentally obtained dataset of diverse spectral intensity profiles (10,000 peptides), we generated using each seed peptide, a set of randomized spectra (100 per seed) as described in subsection a. Then we compared each seed pairwise to each 100 randomized spectra using the preprocessing approach and similarity measures described in subsection b. We repeated this procedure for each of the 20 perturbation ranges, obtaining 20 similarity score distributions with 1,000,000 samples. These distributions are used both to evaluate the sensitivity and descriptive power of the tested similarity measures; and as a reference benchmark to map experimental result comparisons to a population similarity score.
d. Generation of Experimental Similarity Score Datasets
High-confidence mass spectra from experimentally obtained datasets are compared as described in subsection b provided that the following criteria are fulfilled:
Each spectral dataset originating from one run of an instrument, contains one unique (consensus) mass spectrum for a given peptide sequence with a given precursor charge. This is accomplished by creating a consensus library of the search results of each run.
For naked peptide or PTM-PTM comparisons, the assigned peptide sequences including PTM locations are equal. For naked-PTM cross-comparisons, the naked base sequences of both peptides are equal.
The peptides carry equal precursor charges.
The peptides have at least N commonly identified peptide fragments (N = 6 for the presented results) with charge Zf < Zprecursor that obey the following: (1) The fragments cannot be the precursor itself or any of its neutral loss fragments, (2) the fragments cannot contain a heavy isotope, (3) the fragments are either of type y or b, (4) for PTM carrying peptides' comparisons to naked peptides, the fragments need to be in the shared region described on supplemental Fig. S13.
For peptide pairs fulfilling this condition, the top N common peptides are designated and compared as follows:
For both mass spectra, all initial peak intensities are normalized so that .
Following intensity normalization, nonmatching or invalid fragments are discarded using the rules described above.
The list of remaining valid fragments are merged and ordered with respect to mean normalized intensity coming from the two compared spectra.
Top N fragments of the ordered list are used in the spectral similarity analysis using the previously described similarity measures.
Finally, the filtered spectra are compared pairwise as described in subsection b. For a data set with R replicates, a maximum of such pairwise comparisons can be generated for a peptide with a given sequence and precursor charge (3 for r = 3). Similarity scores are then used to describe the population similarities of the compared groups such as replicates from the same machine or cross-comparisons.
e. Mapping Experimentally Obtained Similarity Score Distributions to the Reference Benchmark
We cross-compared experimentally acquired score distributions with reference benchmark distributions as follows:
The Kolmogorov-Smirnov statistic is used to create a pseudo-similarity score D = 1 − D between the experimentally acquired scores and the benchmark reference at each perturbation level.
- The experimental-benchmark cross-comparison similarities D at 20 perturbation levels are fitted to a lognormal or gamma model, where the model which gives the smaller norm of residuals, that is, fit error, is selected. The perturbation level yielding the maximum of the fitted function is the estimation for the S-score. The S-score maps the experimentally obtained similarities to a hypothetical perturbation level, could generate the given experimental similarity distribution. The following equations are used for the lognormal and gamma models for x defined as the D at each perturbation level:
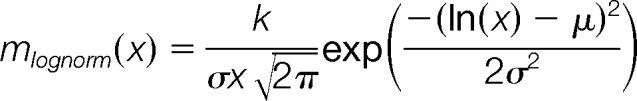
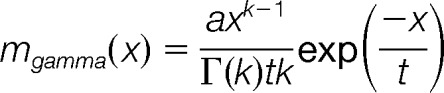
Finally, we established bootstrap confidence intervals for the S-score estimates by resampling both the experimentally obtained similarities and the reference benchmarks similarities at each perturbation level. During resampling, we reduced the benchmark dataset 10-fold so that each perturbation level has 100,000 reference similarities, in order to reduce the computational workload. The reported S-score is the score obtained by using the full, original benchmark and the original experimental similarity score dataset, whereas the reported 95% confidence intervals correspond to the 2.5th and 97.5th percentiles of the simulated a bootstrapped distribution of 1000 S-scores.
Peptide Physicochemical Properties and Their Effect on Fragmentation Variability
Peptide properties were calculated in nonmodified peptides using Biopython v1.62. GRAVY was calculated as described by Kyte et al. (37). The LOWESS estimation was performed in Python-Statsmodels using default settings (frac = 0.67, it = 3).
RESULTS
Using a Benchmark of Simulated Spectra Set of Controlled Dissimilarity to Assess the Performance of Spectral Similarity Measures
So far, the performance of methods to assess spectral similarity has mostly been performed by using empirical data, for example, by comparing score distributions of database searches for spectra from similar or dissimilar peptide identifications (29, 32) or by assessing the recall characteristics of the (correct) peptide identifications by spectral library searches (18, 20, 24, 26, 29, 32). To test the sensitivity of spectral similarity measures to controlled changes between two compared spectra we extend the testing concept of Wan et al. (25) and stochastically generated in silico a set of benchmark spectra using experimental MS/MS spectra as seeds and systematically perturbing the intensities of their six most intense signals. By varying the strength of the perturbation, denoted by S (0≤ S ≤ 1), simulated spectra were generated with well-controlled levels of dissimilarity to the seed. A total benchmark spectral library of twenty million spectra was thus generated, with 1,000,000 spectra (100 spectra per peptide from a sample of 10,000 seed spectra) for each of the 20 levels of perturbation (see Material & Methods for details regarding the intensity transformation and normalization preprocessing steps and the supplemental Figs. S1–S4 for methodological details). Using the described benchmarked perturbation spectra set, the performance of any spectral similarity measure can be objectively tested stepwise across the range of 0 to 100% perturbation for the relative fragment ion intensities.
Fig. 1 shows the response of the three most commonly used similarity measures, dot product, spectral correlation, and normalized spectral contrast angle, toward the systematic perturbations of our benchmark spectral library. The results indicate that the normalized spectral contrast angle shows the best performance among the compared similarity measures, with a consistent decreasing trend over the whole range of perturbation strength and with small variations and minimal overlap for each perturbation step (Fig. 1C). In comparison, the dot product displayed a relatively weak response to stochastic perturbations (Fig. 1A), reporting scores below 0.8 only for the strongest perturbation levels (80–100%). Spectral correlation, on the other hand, generated a large number of low scores even for low perturbation levels, as highlighted by the prevalence of outliers (Fig. 1B). Furthermore, its score distribution presented important overlaps between adjacent spectral perturbation levels. The performance assessment for two additional geometric distance measures, Bray-Curtis distance and Euclidean distance, using the same benchmarked perturbation spectra set approach can be seen in the supplemental material (supplemental Fig. S5). Both measures showed an overall good performance. Based on these results, we decided to use the normalized spectral contrast angle as the similarity measure of choice for the remainder of this study.
Fig. 1.

Response of (A) dot product, (B) spectral correlation, and (C) normalized spectral contrast angle to increasing perturbations. The violin plots combine the kernel density estimation of the score distributions with the statistical features shown by the overlaid box plots. The blue tails denote individual outlier scores at each perturbation level.
Analysis of the Intrinsic Fragmentation Variability for Mass Spectrometers Operating in Shotgun and Targeted Acquisition Modes
To assess the level of peptide fragmentation variability generated upon collision induced dissociation (CID), we performed consecutive technical triplicate injections of a yeast tryptic digest on several instruments: a Thermo LTQ Orbitrap XL in CID mode, a Thermo Orbitrap Elite in data dependent CID or HCD mode, and an ABSciex 5600 TripleTOF instrument operated in DDA shotgun or DIA SWATH MS mode. Each MS injection was searched and representative peptides consensus MS/MS spectra were built independently for each run using spectra above 1% FDR. We then used the six most intense fragments common in the consensus spectra to compute the fragmentation similarity score for the peptides identified in the various technical replicates of a given instrument mode (see Experimental Procedures). This allows essentially to assess the intrinsic fragmentation variability inherent to each instrument mode. Fig. 2 shows the normalized spectral contrast angle score distributions for the peptides commonly identified across all instruments modes. The data indicates that peptide fragmentation conservation is highest for the TripleTOF 5600 in SWATH MS mode, closely followed by the Orbitrap Elite both in CID and HCD modes. It is worth noting that if the same analysis was performed not only for the commonly identified peptides, but for all the peptides above 1% FDR for each instrument mode, overall the same trends were observed (supplemental Fig. S6).
The qualitative comparison of the score distributions obtained from technical replicates in each instrument provided a first hint about the relative consistency of fragment ion pattern obtained on the various instrument modes. We next attempted to quantify the respective levels of peptide fragmentation variability intrinsically produced by each instrument mode. This was achieved by assessing the similarity between the experimental fragmentation score distributions for each instrument mode to that of each computed benchmark perturbation level using the Kolmogorov-Smirnov statistic. The fitting of a lognormal/gamma model (supplemental Fig. S7) yielded a direct estimation of the equivalent fragment intensity perturbation level S for each instrument mode. The absolute levels of fragmentation variability could therefore be estimated to vary between 5.6% (SWATH MS) and 18.5% (LTQ Orbitrap XL) on average, depending on the instrument mode used in this study.
To assess the fragmentation reproducibility across longer time periods and across more divergent sample sets, we extended our analysis to compare (1) technical replicate injections (same sample) recorded consecutively, (2) technical replicate injections (same sample) but recorded several days apart, (3) biological replicate injections (different samples but same time point/biological condition) recorded consecutively, and (4) biological samples injections (different biological conditions) and recorded several days apart. The results are shown on supplemental Fig. S8 and show slightly worse fragmentation reproducibility for the biological replicates compared with the technical replicates. However, under the acquisition used here, the data do not show any noticeable effect of the day of acquisition on the quality of the results. In other words, the peptide spectra recorded by technical replicate injections even across several days still retain the same reproducibility as when acquired back-to-back.
We used the same approach to analyze the level of fragmentation conservation for phosphorylated peptides. We analyzed in triplicate a yeast tryptic digest enriched for phosphopeptides using the same instrument modes as described above and performed the spectral similarity comparison, this time also considering fragments with neutral loss (-80/-98 Da) if detected among the six most intense peptide fragments. The results (Fig. 3 and supplemental Fig. S10) show overall similar trends for the instruments performance for phosphorylated peptides as for naked peptides (Fig. 2), with a slightly lower overall fragmentation fidelity for phosphopeptides. These data indicate that for the instrument modes tested phosphorylated peptides do not show significantly lower fragmentation reproducibility, and that the −80/−98 Da neutral loss fragments (present between 76 and 87% of the cross phosphopeptides spectral comparisons depending on the instrument mode) displayed also relatively good levels of conservation during fragmentation.
To confirm that these results were solely acquisition-dependent and not biased by specific properties of peptides, we next investigated the effect of physicochemical properties of peptides on fragmentation variability. We computed (1) the molecular weight, (2) the isoelectric point, (3) the GRAVY (Grand average of Hydropathy) index, (4) peptide instability, and (5) the presence of proline in the peptides (because this amino acid usually yields atypical fragmentation patterns with highly prominent cleavage at the N-terminal side of Pro). We then established the effect/influence of these properties on the measured conservation of fragmentation using the data from the TripleTOF instrument in shotgun mode (supplemental Fig. S6, column 4). The results (supplemental Fig. S9) show that peptide physicochemical properties have no major effect on the fragmentation variability with the possible exception of low molecular weight peptides which show slightly higher fragmentation reproducibility.
Assessment of the Transferability of Fragment Ion Assays from Shotgun to Targeted Proteomic Analyses
Targeted proteomics via SRM or SWATH-MS (33) critically depends on a high degree of fragmentation conservation because the relative signal intensity of fragment ion signals (transitions) is an important subscore in identification algorithms (31). It is therefore important to know the level of conservation between the spectra used to generate the reference assay (usually originating from shotgun acquisition performed with the same or another instrument) and the signals that will actually be measured/extracted for the targeted peptide. Throughout this study, we decided to focus our analysis on SWATH MS targeted data extraction because too many SRM measurements would have been required to yield the sample size necessary for proper statistical comparisons. However, because of the conceptual similarity of SWATH-MS and SRM as XIC-based peptide identification methods using a defined number of transitions to query peptides, it is very likely that most of the methodology and conclusions described hereafter will also apply to SRM.
Figs. 2 and 3 have already demonstrated the high levels of fragmentation reproducibility of SWATH MS analysis as a targeted proteomics technique. To test the transferability of reference fragmentation assays for SWATH MS targeted data extraction, we performed cross-device similarity analyses between the Orbitrap Elite in HCD and CID mode, LTQ Orbitrap XL, and TripleTOF shotgun mode using this time the intensities of the fragment ion signals from the TripleTOF SWATH MS measurements as reference for each comparison (Fig. 4). The results show that the TripleTOF shotgun peptide fragmentation and Orbitrap Elite HCD peptide fragmentation provided the most similar fragmentation to that observed for SWATH MS targeted data analysis, with 26 and 37% fragmentation divergence respectively. These results are consistent with the intrinsic fragmentation reproducibility observed for the different instrument designs and operation modes (Fig. 2) and follow expectations of fragmentation similarity between quadrupole and beam-type devices (Orbitrap Elite in HCD mode and TripleTOF) as opposed to resonance excitation in ion traps (LTQ Orbitrap XL and Orbitrap Elite in CID mode) (38, 39). To ensure that this result was not biased by the fact that the SWATH MS peptides were initially identified using a TripleTOF shotgun library, we performed the same analysis using peptides identified in SWATH MS extraction using fragments, this time originating from the Orbitrap Elite HCD identifications. The results show that whether the original assay library used to trigger SWATH extraction is generated by TripleTOF (Fig. 4) or Orbitrap Elite HCD (supplemental Fig S11), the overall similarity results retain the same trend: the closest fragmentation patterns to SWATH MS in terms of relative intensities originate from the TripleTOF shotgun, followed by the Orbitrap Elite in HCD mode.
Fragmentation Conservation as a Discriminating Feature for Peptide Identification in Targeted MS/MS
Although it is important for targeted MS/MS to know the extent of fragmentation variability between the reference assays and the transition signals measured from targeted peptides, it is even more important to know which level of fragmentation variability is actually acceptable before it starts penalizing confidence of the peptide identification.
To answer this question, we re-evaluated the performance of each spectral similarity measure as a relative fragment intensity score able to efficiently discriminate between target and decoy signals during SWATH MS targeted data analysis. Two sets of assays were used: First, a reference “target” assay library that contained the mass and relative intensity information for the six most intense fragment ions of the peptides identified over 1% FDR in the TripleTOF 5600 shotgun experiments; and second, a “decoy” assay library that was generated in silico by pseudo-reversing the amino acid sequence for each corresponding target peptide. Each decoy assay therefore contained the same experimental relative intensities as the target assay, but associated with “wrong” fragment ion masses (originating from the pseudo-reverse peptide fragment sequence). The masses of the target and decoy shotgun assays were then used to extract fragment ion chromatograms in the SWATH MS datasets and their initial relative intensities were assessed pairwise to those of the corresponding SWATH MS integrated fragment ion signals as reference, using one of the spectral similarity measures. As above, the resulting target and decoy spectral similarity score distributions can be matched to those of the corresponding benchmarked perturbation libraries, allowing the estimation of absolute levels of fragment dissimilarity for each condition.
The results are shown in Fig. 5 and supplemental Fig. S12. As expected, for all scores, the target shotgun assays displayed relatively good levels of fragmentation similarity (equivalent to 10–28% perturbation depending on the score used) compared with that of the SWATH MS integrated fragment signals. In contrast, the relative fragment ion intensities of the decoy assays displayed a quasi-random match (equivalent to 100% perturbation) compared with that of the corresponding SWATH MS integrated signals. Overall, all similarity measures achieved good discrimination between the target and decoy score distributions except spectral correlation, which attributed quite early relatively high scores to decoy spectra (Fig. 5B). Interestingly and despite the compressed response anticipated from Fig. 1, the dot product achieved equivalent performance in separating targets and decoys compared with the normalized spectral contrast angle, after remapping the raw score values onto the corresponding perturbation benchmark spectra set. Using one of those two similarity measures, very few decoy assays displayed high similarity scores compared with their corresponding target assays. Therefore, in principle, any instrument offering overall intrinsic fragmentation reproducibility and assay transferability performance above this threshold (35% in the case of the normalized spectral contrast angle) should support effective peptide identification in SWATH MS targeted extraction via relative fragment intensity. Higher fragmentation reproducibility, of course, would be expected to further increase the discrimination between targets and decoys and would thus strengthen again the power of such fragmentation similarity scores for peptide identifications.
Fig. 5.

Target-decoy separation characteristics of (A) dot product, (B) spectral correlation, (C) normalized spectral contrast angle. The blue curves denote the target assay similarities whereas the red curves denote the decoy assay similarities in a TripleTOF shotgun/SWATH mode comparison. The lognormal/gamma fit parameters as well as the fit errors (norm of residuals) are reported for each fit.
Conservation of the Fragmentation Between Nonmodified and Phosphorylated Peptides and New Strategies to Extract Modified Peptides Using Nonmodified Reference Assays
As a final application example of our tool, we sought to assess the possibility to identify by targeted mass spectrometry modified peptides using assays originating from their nonmodified counterpart. We questioned whether a conserved fragmentation pattern can be observed between nonmodified and phosphorylated peptides and whether the level of similarity between the shared fragments of the two species would support the reliable probing and identification of the modified peptide in targeting data sets. For this, we considered independently two distinct fragment regions of a modified peptide and focused on the y and b fragments preceding the site of the first modification on the modified peptide sequence that are shared with its “naked” counterpart (supplemental Fig. S13). We used those shared fragments with equal masses to probe how well their relative intensities are conserved between the modified and nonmodified peptides and whether those can be used to search for the modified peptide starting from a library of spectra of nonmodified peptides. We tested this hypothesis by analyzing a sample of phospho-enriched yeast tryptic peptides that were either analyzed in their phosphorylated form or, following phosphatase treatment, in their naked form. As low-abundant proteins make up a large subset of the yeast phosphoproteome, we used this strategy to increase the number of “naked” peptide assays in our spectral library that would match to the modified peptides already identified in the phospho-enriched samples. Fig. 6 shows the fragmentation similarity between a set of nonmodified (phosphatase treated) peptides with their corresponding modified peptide counterparts. Although the intrinsic variability of the phosphopeptide fragmentation within replicates of SWATH MS extractions was around 15%, the fragmentation conservation for the shared fragments dropped to 31% equivalent perturbation. However, this fragmentation conservation was just enough to support targeted extraction of the phospho-enriched SWATH MS data using the nonmodified peptide assays and we indeed could retrieve 1177 phosphopeptides out of the 2758 originally identified by SWATH MS extraction using the phospho shotgun library (supplemental Tables S3). Considering that only 1462 peptide assays (1843 precursors) with at least six shared transitions could actually be generated for those 2758 phosphopeptides using the dephosphorylated peptide identifications, the 1177 phosphopeptides identified constitute a 80% success rate for the method. Interestingly, we also generated shared phosphopeptide assays not only for the sequences already validated using the shotgun phosphopeptide library but also for any peptide sequence identified after dephosphorylation. By doing so, we could actually retrieve by SWATH MS data extraction 87 novel phosphopeptides (5% FDR) that failed to be identified by shotgun analysis.
Open Source Software Package Available
The methodology used during this study to assess the fragmentation performance of MS instruments or to compare peptide fragmentation processes in general may also be of interest to other research groups and applied to questions different from the ones investigated in this paper. We therefore packaged the algorithms used throughout our analyses into a graphical user interface. This software package generates as a first step the benchmarked spectra set of increasing perturbation strength for the similarity measures of interest. As described in this study, this benchmarked spectra set are used as a reference to translate the nominal values of the various similarity measures into absolute estimates of percentage of fragment intensity perturbation. The package allows the assessment of fragmentation similarity among and across spectral libraries provided in the open source SpectraST .sptxt format. We also devised a flexible analysis workflow for peptides carrying PTMs that can be fully user-defined. The details of usage of the software package and some case studies are provided in the supplementary materials. We believe that this toolset will be useful for many users in academic or clinical settings as well as for MS manufacturers or high-end users for tuning and testing intrinsic fragmentation performance of mass spectrometers or for testing the portability of their assays between instruments.
DISCUSSION
Performance of the Similarity Measures
With recent advances in spectral library searching and targeted proteomics analyses, the use of the relative fragment ion intensities to enhance confidence in peptide identification has gained in importance. However, several critical questions remain to be answered about the conservation of peptide fragmentation characteristics. For example, which similarity measure should be used to most sensitively assess similarity of the measured/extracted targeted fragment ion intensities versus those of the reference assays contained in the library? Which level of fragmentation variation is really reflected by the nominal values of those measures? Which level of fragmentation perturbation is still acceptable (for each similarity measure) to actually support proper peptide identification?
To answer those questions, we first evaluated the response of some widely used similarity measures toward perturbations of a set of spectra generated in silico, where increasing levels of dissimilarity to a seed spectrum were simulated. It is important to mention that the benchmark spectra set is not supposed to simulate the complex phenomenon of MS/MS peptide fragmentation. On the contrary, it is used here as a purely mathematical reference for interpreting similarity scores. With this methodology, a proper similarity measure that aims at accurately quantifying spectral similarities should present the following characteristics: (1) consistent decrease of similarity scores upon increasing levels of perturbation, (2) distribution of scores in narrow bands with small variance for each perturbation step, and (3) little overlap of the score distributions upon increasing perturbation strengths. According to our results, the normalized spectral contrast angle showed overall a good performance (Fig. 1C), with a consistent decreasing trend over the range of perturbation and with small variations and minimal overlap for each perturbation step.
It is especially worth mentioning that dot product nominal values only decreased from 1 to 0.95 for spectral perturbation levels up to 60% (0≲ S ≲0.6), whereas normalized spectral contrast angle values dropped from 1 to 0.8 for the same range of perturbations. Researchers should therefore be aware that seemingly “high” nominal values of the regular dot product such as those reported in the literature (e.g. 0.96 (30) or 0.95 (22)) do not actually necessarily guarantee high degrees of spectral similarity. It is also worth mentioning that Skyline, a popular SRM analysis software tool, originally used the regular dot product raw values to score the similarity of the relative fragment intensities of the targets toward the reference library assays (23). However, the software developers decided to switch to the normalized spectral contrast angle as main scoring function for their software (Brendan MacLean, personal communication) based on the preliminary results of this study showing that high dot product values may be generated even for loosely similar spectra and therefore may appear misleading as such without mapping to its perturbation benchmark set (see below). The same cautionary remarks also apply to the spectral correlation measure, which in comparison showed a large overlap of the distributions between each perturbation level (Fig. 1B). Therefore here again, it may be difficult to interpret the real significance of “high” nominal score values (e.g. 0.98) that could be produced by basically any level of perturbation between 10 and 100% perturbation, because of the broad score distribution overlaps between the perturbation steps.
The performance of the similarity measures was also compared as identification score classifiers for their ability to discriminate target peptides and decoy assays score distributions during targeted proteomics analysis (31). There all investigated spectral similarity measures except for spectral correlation, performed well by properly classifying targets when those were less than 35% dissimilar to that of their reference library assays (Fig. 5C). It is worth noting that the dot product, although showing a more “compressed” response to increasing perturbations in the benchmark (Fig. 1A), still yielded a proper measure of similarities in a population level, when its benchmark set was used as a reference to convert the raw values/distribution into the equivalent percentage of spectra perturbation. To quantify this however, the function used for the goodness of fit needs to be sensitive enough for capturing the information de-emphasized by the cosine term of the dot product. The Kolmogorov-Smirnov statistic used throughout the study to map the experimental similarity measure distributions onto the perturbation benchmark sets allowed to estimate an accurate goodness of fit (S-score), whereas a binned histogram similarity failed to properly map the response of the dot product onto its benchmark reference, although it showed equal performance when assessing the other similarity measures (results not shown). These findings underline again the importance of not relying on raw score values as similarity threshold estimates but rather on comparing the similarity measure used onto its benchmarked reference score response and the corresponding goodness of fit. Spectral correlation showed the weakest discrimination in separating target and decoy populations with decoys overlapping with the target distribution even for relatively high scores (Fig. 5B). Interestingly, the spectral correlation is the similarity measure implemented in mProphet to score the fragment intensity correlation of the targeted peptides to that of the reference assays (31). Despite the poor target-decoy separation performance that we report here, fragment intensity correlation was reported as one of the most discriminating parameters in the mProphet study (31). Thus, we foresee that an even higher performance in peptide identification can be expected for the scoring of relative fragment ion intensity when using a more sensitive and discriminating similarity measure.
It is important to note that the S-scores evaluated by Kolmogorov-Smirnov statistic span a wide range of numeric values throughout this study with (1) technical replicates from the latest generation mass spectrometers yielding high similarities and acting as positive controls (0.056 ≤ S ≤ 0.166, Fig. 2), (2) target-(pseudo-reversed) decoy comparisons yielding low similarities and acting as negative controls (S≈1, Fig. 5 and supplemental Fig. S12), and (3) cross-instrument analyses yielding intermediate values depending on the compared pair of instruments (0.261 ≤ S ≤ 0.590, Fig. 4). This wide range of response indicates that the perturbation spectra set can indeed capture (dis)similarities encountered in real life similarity analysis and as such be used to provide equivalent perturbation level estimates. The confidence in the perturbation level estimates (S-scores) was further assessed by bootstrapping and showed very good consistency by falling in very narrow confidence intervals, especially for larger population size datasets (e.g. Fig. 5 and supplemental Fig. S12).
Taken together, these results strongly suggest that only peptide assays with a difference in relative fragment ion intensities of less than 35% to the experimental targeted recordings should be used for targeted proteomic measurements. This does not mean that the targeted peptides will fail to be identified if the assays are more than 35% different than the measurements. In fact, the other scores used to identify the peptides (e.g. co-elution of the transitions, co-elution with its corresponding isotopically labeled reference peptide) may still suffice to pinpoint the peptide identification. However, our results indicate that, beyond 35% fragmentation divergence to the reference assay, the scoring of the relative fragment ion intensities of the targeted peptide may severely impair the overall identification performance. Confirmation of the compatibility of a particular device configuration or assay library-acquired sample match to this criterion can be done by the toolset presented in the supplemental materials by analyzing replicates from the same instrument or across different instruments for peptide fragmentation similarity, depending on application (see the toolset manual in supplementary materials).
Though the normalized spectral contrast angle was selected in this study to illustrate the methodology and for estimating the conservation of the top six most intense fragments in targeted proteomics, it is important to note that the other similarity measures should not be unequivocally disregarded and may still be very relevant to use when sensitive discrimination of fine dissimilarities is counterproductive. This would be the case, for example, when the fragment intensities of the reference spectra are not inputted at all such as with in silico generated spectra during database search, or when they are expected to present low fidelity such as is when fragment ion intensities originating from other fragmentation modes or MS instruments or from theoretical fragmentation predictors are used. The focus of this study was specifically on targeted proteomics, and it is therefore beyond the scope of this study to perform exhaustive performance analysis when large number of fragments have to be matched as is the case of extensive MS/MS spectra matching such as during database or spectral library searching. But we hope that the assessment methodology described here using a benchmarked spectra set of controlled dissimilarity will provide new insights for the rational evaluation and selection of the similarity measure that would most optimally perform for each dedicated task.
Evaluation of the Reproducibility of Mass Spectrometric Fragmentation Measurements
Having established a 35% fragment intensity divergence as the threshold for efficient peptide identification in targeted proteomics, we sought to determine what was actually the intrinsic fragmentation variability encompassed in spectral libraries generated from various instrument acquisition methods. Modern mass spectrometers are expected to produce highly reproducible recordings of the peptide fragmentation events. However, because each instrument measurement may intrinsically encompass a certain level of fluctuation, we aimed to quantitatively assess the fragmentation variability that actually affects the experimental peptide collision recordings. We thus calculated the normalized spectral contrast angle similarity scores for the six most intense fragment ions of peptides that were recorded during technical replicate analyses of the same sample using a given instrument mode. For those comparisons, we did not perform on purpose any specific tuning (regarding acquisition times or ion trap filling) to artificially improve the ion statistics of the instruments. We rather sought to operate the instruments as close as possible to their regular acquisition set-up for proteomic measurements and used consecutive technical replicate injections to minimize external biases (sample reproducibility, changes in instrument performance) that might otherwise have obscured the assessment of the instrument fragmentation reproducibility.
By matching the experimental fragmentation score distributions of each instrument mode to the perturbation benchmark spectra set, it became apparent that the SWATH MS targeted extraction analysis exhibited the highest level of peptide fragmentation conservation with only 5% equivalent fragmentation variability between replicates, followed by the Orbitrap Elite HCD consensus spectra which entailed 9% variability between replicates (Fig. 2). With this quantitative measure, it is also easier to appreciate the qualitative jump in fragmentation fidelity achieved between the LTQ Orbitrap XL and the Orbitrap Elite in CID mode, where the intrinsic fragmentation variability decreased by almost a factor of two from 18% to 10% between those two consecutive generations of instruments (40). The result is essentially the same for nonmodified peptides (Fig. 2) as for phosphorylated peptides (Fig. 3) which produce more complex fragmentation patterns, with variable prominence of neutral loss fragments, depending on the type of collision used (e.g. ion trap excitation versus quadrupole fragmentation/HCD). The analyzed peptide pool's effect on our results has been shown to be insignificant (supplemental Fig. S9), with exception of very short or very low-pI peptides. The molecular weight's effect on the scores can be explained by the limited number of matching fragments for shorter peptide, which limits the number of comparison combinations and may therefore improve the fragmentation reproducibility. The use of a whole-proteome yeast peptide library minimizes the bias caused by such outliers that may otherwise be present when using smaller sample size such as synthetic peptide libraries.
The greater peptide fragmentation fidelity observed for the SWATH MS mode can easily be explained by the fact that the fragment ion signals originate from peak areas integrated over the chromatographic elution of the fragment ions during SWATH MS targeted data extraction which is in contrast to stochastic “snapshot” fragmentation recordings of DDA which are inherently more prone to fluctuations in the ion statistics and to chemical noise.
Overall, our results suggest that the state-of-the-art shotgun solutions are capable of acquiring reproducible peptide fragmentation intensity patterns, within less than 35% intrinsic variation, which thus in principle supports high-quality spectral library generation and efficient peptide identification in targeted proteomics. As the instruments continue to improve, we foresee even a further gain in the quality of the spectra library generated and in the value of using the conservation of the fragmentation intensities as discriminating score for even more effective peptide identification.
As such, the methodology presented here may actually be used to answer several questions such as (1) what is the intrinsic performance expected for the MS mode used (Fig. 2), (2) what is the stability performance for that MS mode across several days/weeks of analysis—quality control of the machine performance basically—(supplemental Fig. S8), (3) what is the quality of the biological replicates generated for one experiment, using the similarity of MS/MS spectra as a proxy, and (4) how divergent are different biological samples (as assessed by the onset of interferences/cofragmenting peptides). This matrix of quality control parameters could easily be established and/or run on preexisting shotgun data present in any lab to estimate the quality of a given dataset before downstream analysis.
Highly reproducible peptide fragmentation patterns do not only facilitate peptide identification by targeted proteomics, but also enhance the overall quantification accuracy of those peptides. As fragmentation events are governed by physicochemical phenomena and are expected to be conserved, the source of the variability in spectral intensities may arise from spray instability, condition fluctuations in the collision cell or errors in the ion statistics at the detector level (41) which directly propagate into errors in label-free quantification approaches. Increased fragmentation pattern reproducibility offered by newer generation of mass spectrometers will continue to improve quantification accuracy of mass spectrometry-based proteomics experiments. The results presented in this study confirm the superiority of an XIC-based method, SWATH-MS over shotgun approaches in the assessment of peptide fragmentation intensities, which is in line with the established position of SRM in the field as the most accurate mass spectrometry method for peptide quantification. We believe that this study and the accompanying open-source tool will support the assessment of experimental variables including device, acquisition settings, data analysis parameters, and their effects on accurate peptide quantification especially in performance-critical applications such as medical diagnostics.
Portability of Peptide Fragmentation Data for Targeted Proteomics
The intrinsic fragmentation reproducibility of different instruments is only one part of the equation. Indeed, some instruments may appear intrinsically more reproducible than others, but for the purpose of library generation for targeted proteomics it is actually more important to know which one yields the fragmentation patterns most closely related to those of the targeted measurement that will actually be performed. To address this question, we carried out a cross-device comparison between each instrument in shotgun mode and the SWATH targeted data extraction. We found that, though the Orbitrap Elite HCD was the instrument producing intrinsically the most reproducible peptide fragmentation recordings in shotgun mode, the assays originating from the TripleTOF 5600 shotgun acquisition still yielded the most similar fragmentation patterns (26% divergence, Fig. 4) to that of the fragment ion intensities extracted from the SWATH MS files. Because this portability measure encompasses the intrinsic fragmentation variability of the instrument as well, these results indicate that assays acquired on the TripleTOF 5600 in shotgun mode yield highest similarity of the fragmentation to that of the TripleTOF 5600 in SWATH MS mode and should warrant highest relative intensity score discrimination and peptide identification during targeted analysis.
The relatively low transferability of the assays between instruments is somewhat contradictory to the results of previous studies where the authors assumed that a dot product value of 0.95 or 0.98 was indicative of high fragmentation similarity between the instruments they tested. However such nominal value of dot product is actually indicative of already more than 50% fragmentation divergence (Fig. 1) which, as we showed above, may not warrant efficient peptide identification. Therefore we re-emphasize here the importance of carefully quantifying the nominal values of those similarity measures and of using conditions to generate spectral libraries that will be as close as possible to the targeted measurements. To take full advantage of the peptide fragmentation similarity as a powerful discriminant score for peptide identification, we therefore advocate to acquire, when possible, instrument specific libraries and/or to purposely tune the fragmentation parameters (e.g. collision gas pressure or collision energy equations) of the MS instrument used for targeted proteomics to acquire spectra with closest similarity to those of reported in publicly available spectral libraries (e.g. Peptide Atlas (42) and SRM Atlas (43)). If properly used,we expect that well-performing spectral similarity measures (such as the normalized spectral contrast angle used to obtain the data presented in this study), together with a reference set of benchmarked spectra with controlled dissimilarities, should help to standardize fragmentation diagnostics analyses of high-end MS devices and to assess or devise optimal targeted proteomic acquisition parameters to best take advantage of pre-existing spectral libraries generated with other instrument acquisition set-ups.
Using Nonmodified Peptide Fragmentation Patterns to Identify Their Modified Counterparts with Targeted Proteomics
Compared with nonmodified peptides, the analysis of peptides with PTMs remains challenging for targeted proteomics because of the more difficult generation of the reference assays. Though the modified peptides can in principle also be chemically synthesized on a large scale to generate templates for library generation (44), this task remains so far more technically challenging and costly than for nonmodified-peptides (43). Hence, we asked whether it would be possible to use the fragmentation patterns of “naked” peptide to generate assays for targeting peptides with PTMs. The concept of identifying a modified peptide by targeting fragment ion signals from its nonmodified counterpart was already exemplified by our group with oxidized methionine peptides in a former publication (33). However, whether this strategy can be applicable on a larger scale and for peptides with more labile modifications such as phosphorylation has not yet been quantitatively demonstrated. By computing the normalized spectral contrast angle scores for the common fragments of peptides identified both in their nonmodified and phosphorylated forms, we found a reasonable degree of fragmentation conservation (31% divergence) between both species. This value supports phosphopeptide identifications using the relative fragment intensities of their nonmodified counterparts. The similarity of the common transitions shared by nonmodified and phosphopeptides would be just at the eligibility threshold of the SWATH MS assay “portability” (35%). However, as the MS instrumentations continues to improve, there is no reason to believe that the lower fragmentation reproducibility reported for the phosphopeptides in SWATH mode (∼10%, Fig. 3) may not reach the levels observed for the nonmodified peptides (∼5%, Fig. 2) and therefore yield even higher recovery of phosphopeptide identifications using nonmodified libraries.
We therefore applied this strategy to mine a phospho-enriched sample using fragmentation assays from de-phosphorylated peptides. The results led to the confident identification of around half of the phosphopeptides compared with what would have been identified from a phospho-specific spectral library. The lower yield of this approach can be largely explained by the low number of accessible assays for peptides for which the modification is close to the peptide termini and for which either the entire y or b ion series are not common and cannot be used for relative intensity scoring. Also, because we used the retention time of the nonmodified peptide to trigger the data extraction of the modified form, it may happen that the phosphorylated peptide fell outside the extraction window. However, it is interesting to note that this method could also retrieve completely novel phosphopeptides that were not identified in shotgun experiments. Together with the flexible targeted data extraction capabilities of SWATH MS datasets, we now implemented this strategy as a preliminary screening method for the discovery of modified peptides forms especially for those which are not known or for which the modified peptide assays are not readily accessible. Because this strategy works for phosphorylated peptides, despite their complex fragmentation characteristics (45), we expect that it may be extended to other types of PTMs particularly those with a more stable link to the peptide backbone.
It is important to note once again that the benchmark mapping methodology presented here does not aim at simulating the fragmentation behavior of the peptides, modified or nonmodified. We used the similarity measure and its associated perturbation benchmark set to assess whether there is significant/sufficient similarity between those experimentally acquired spectra (nonmodified and modified) to use the assays of one species to query for the other in targeted proteomics. Fig. 5 showed that less than 35% fragmentation divergence is necessary for efficient separation of targets and decoys by targeted proteomics. That result was obtained by mapping the targets and decoy distribution onto the perturbation benchmark set of the similarity score of choice (normalized spectral contrast angle in this case). And it is with the same perturbation benchmark that we estimate the similarity between the modified and nonmodified peptides. Both results are thus mapped/scaled to the same perturbation benchmark and are therefore directly comparable. Hence the conclusion that, if the PTM does not induce more than 35% fragmentation divergence between the common transitions of the nonmodified and modified peptides, the assays of one species may be used to query the other by targeted proteomics.
Overall these results also open new perspectives for targeted proteomics and may soften recurring criticisms regarding the requirement of extensive “PTM specific” libraries for targeting modified peptides. As shown here, such libraries may not always be necessary when modified peptides of sufficient fragmentation similarity to that of the nonmodified peptides are encountered. In those cases indeed, the naked peptide assays may actually prove sufficient to mine and identify the modified peptide form.
Concluding Remarks
The presented results suggest that properly tested spectral similarity measures, together with a reference set of benchmarked spectra with controlled dissimilarities, enable objective assessment of the fragmentation performance of mass spectrometers and constitute a robust and sensitive measure to score and identify peptides in targeted proteomics. We hope that the methodology and toolset presented here will help scientists in implementing standardization protocols and in assessing the portability of their assays across instruments and throughout time to gain optimal peptide identification and quantification in research and clinical settings.
Supplementary Material
Acknowledgments
We thank Witold Wolski (ETH Zurich/SyBIT) and Brendan MacLean (University of Washington) for the stimulating discussions about similarity measures and their comments regarding the transformation/scaling of the fragment ion spectra relative intensities prior to the use of the similarity measures.
Footnotes
Author contributions: U.H.T., L.C.G., and R.A. designed research; U.H.T., L.C.G., and A.M. performed research; U.H.T., L.C.G., A.M., P.N., and A.L. contributed new reagents or analytic tools; U.H.T. and L.C.G. analyzed data; U.H.T., L.C.G., A.M., A.L., and R.A. wrote the paper.
* This work was in part funded by the European Union 7th Framework Programme, PRIME-XS project Grant Agreement Number 262067, by advanced ERC grant (#233226) Proteomics v3.0 to RA, and the PhosphonetPPM project of SystemsX.ch, the Swiss initiative for systems biology.
 This article contains supplemental Figs. S1 to S13 and Tables S1 to S3.
This article contains supplemental Figs. S1 to S13 and Tables S1 to S3.
1 The abbreviations used are:
- SRM
- Selected Reaction Monitoring
- LC-MS/MS
- Liquid Chromatography-Tandem Mass Spectrometry
- SWATH MS
- Sequential Windowed Acquisition of all Theoretical Mass Spectra
- PTM
- Post-Translational Modification
- CID
- collision induced dissociation
- HCD
- higher-energy collision induced dissociation
- XIC
- extracted ion chromatogram
- GRAVY
- grand average of hydropathy
- pI
- isoelectric point.
REFERENCES
- 1. Paizs B., Suhai S. (2005) Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548 [DOI] [PubMed] [Google Scholar]
- 2. Steen H., Mann M. (2004) The ABC's (and XYZ's) of peptide sequencing. Nat. Rev. Mol. Cell Bio. 5, 699–711 [DOI] [PubMed] [Google Scholar]
- 3. Nesvizhskii A. (2007) Protein Identification by Tandem Mass Spectrometry and Sequence Database Searching. In: Matthiesen R., ed. Mass Spectrometry Data Analysis in Proteomics, pp. 87–119, Humana Press; [DOI] [PubMed] [Google Scholar]
- 4. Zhang Z. (2004) Prediction of low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 76, 3908–3922 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Z. (2005) Prediction of low-energy collision-induced dissociation spectra of peptides with three or more charges. Anal. Chem. 77, 6364–6373 [DOI] [PubMed] [Google Scholar]
- 6. Zhang Z. (2011) Prediction of collision-induced-dissociation spectra of peptides with post-translational or process-induced modifications. Anal. Chem. 83, 8642–8651 [DOI] [PubMed] [Google Scholar]
- 7. Li W., Ji L., Goya J., Tan G., Wysocki V. H. (2011) SQID: an intensity-incorporated protein identification algorithm for tandem mass spectrometry. J. Proteome Res. 10, 1593–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Craig R., Cortens J. P., Beavis R. C. (2005) The use of proteotypic peptide libraries for protein identification. Rapid Commun. Mass Sp.: RCM 19, 1844–1850 [DOI] [PubMed] [Google Scholar]
- 9. Desiere F., Deutsch E. W., King N. L., Nesvizhskii A. I., Mallick P., Eng J., Chen S., Eddes J., Loevenich S. N., Aebersold R. (2006) The PeptideAtlas project. Nucleic Acids Res. 34, D655–D658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones P., Cote R. G., Martens L., Quinn A. F., Taylor C. F., Derache W., Hermjakob H., Apweiler R. (2006) PRIDE: a public repository of protein and peptide identifications for the proteomics community. Nucleic Acids Res. 34, D659–D663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Frohlich F., Walther T. C., Mann M. (2008) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 12. Riffle M., Eng J. K. (2009) Proteomics data repositories. Proteomics 9, 4653–4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fenyo D., Eriksson J., Beavis R. (2010) Mass spectrometric protein identification using the global proteome machine. Methods Mol. Biol. 673, 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thakur S. S., Geiger T., Chatterjee B., Bandilla P., Frohlich F., Cox J., Mann M. (2011) Deep and Highly Sensitive Proteome Coverage by LC-MS/MS Without Prefractionation. Mol. Cell. Proteomics 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farrah T., Deutsch E. W., Hoopmann M. R., Hallows J. L., Sun Z., Huang C. Y., Moritz R. L. (2013) The state of the human proteome in 2012 as viewed through PeptideAtlas. J. Proteome Res. 12, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 18. Stein S. E., Scott D. R. (1994) Optimization and Testing of Mass-Spectral Library Search Algorithms for Compound Identification. J. Am. Soc. Mass Spectr. 5, 859–866 [DOI] [PubMed] [Google Scholar]
- 19. Beer I., Barnea E., Ziv T., Admon A. (2004) Improving large-scale proteomics by clustering of mass spectrometry data. Proteomics 4, 950–960 [DOI] [PubMed] [Google Scholar]
- 20. Frewen B. E., Merrihew G. E., Wu C. C., Noble W. S., MacCoss M. J. (2006) Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal. Chem. 78, 5678–5684 [DOI] [PubMed] [Google Scholar]
- 21. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 22. Sherwood C. A., Eastham A., Lee L. W., Risler J., Vitek O., Martin D. B. (2009) Correlation between y-Type Ions Observed in Ion Trap and Triple Quadrupole Mass Spectrometers. J. Proteome Res. 8, 4243–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yen C. Y., Houel S., Ahn N. G., Old W. M. (2011) Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library. Mol. Cell. Proteomics 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wan K. X., Vidavsky I., Gross M. L. (2002) Comparing similar spectra: from similarity index to spectral contrast angle. J. Am. Soc. Mass Spectr. 13, 85–88 [DOI] [PubMed] [Google Scholar]
- 26. Tabb D. L., MacCoss M. J., Wu C. C., Anderson S. D., Yates J. R. (2003) Similarity among tandem mass spectra from proteomic experiments: Detection, significance, and utility. Anal. Chem. 75, 2470–2477 [DOI] [PubMed] [Google Scholar]
- 27. Wang J. A., Perez-Santiago J., Katz J. E., Mallick P., Bandeira N. (2010) Peptide Identification from Mixture Tandem Mass Spectra. Mol. Cell. Proteomics 9, 1476–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yates J. R., 3rd, Morgan S. F., Gatlin C. L., Griffin P. R., Eng J. K. (1998) Method to compare collision-induced dissociation spectra of peptides: potential for library searching and subtractive analysis. Anal. Chem. 70, 3557–3565 [DOI] [PubMed] [Google Scholar]
- 29. Liu J., Bell A. W., Bergeron J. J. M., Yanofsky C. M., Carrillo B., Beaudrie C. E. H., Kearney R. E. (2007) Methods for peptide identification by spectral comparison. Proteome Sci. 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Graaf E. L., Altelaar A. F., van Breukelen B., Mohammed S., Heck A. J. (2011) Improving SRM assay development: a global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra. J. Proteome Res. 10, 4334–4341 [DOI] [PubMed] [Google Scholar]
- 31. Reiter L., Rinner O., Picotti P., Huttenhain R., Beck M., Brusniak M. Y., Hengartner M. O., Aebersold R. (2011) mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 8, 430–435 [DOI] [PubMed] [Google Scholar]
- 32. Wolski W. E., Lalowski M., Martus P., Herwig R., Giavalisco P., Gobom J., Sickmann A., Lehrach H., Reinert K. (2005) Transformation and other factors of the peptide mass spectrometry pairwise peak-list comparison process. BMC bioinformatics 6, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gillet L. C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics: MCP 11, O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bodenmiller B., Aebersold R. (2010) Quantitative analysis of protein phosphorylation on a system-wide scale by mass spectrometry-based proteomics. Method Enzymol. 470, 317–334 [DOI] [PubMed] [Google Scholar]
- 35. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M. Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 37. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 38. Wells J. M., McLuckey S. A. (2005) Collision-induced dissociation (CID) of peptides and proteins. Method Enzymol. 402, 148–185 [DOI] [PubMed] [Google Scholar]
- 39. Lau K. W., Hart S. R., Lynch J. A., Wong S. C., Hubbard S. J., Gaskell S. J. (2009) Observations on the detection of b- and y-type ions in the collisionally activated decomposition spectra of protonated peptides. Rapid Commun. Mass Sp.: RCM 23, 1508–1514 [DOI] [PubMed] [Google Scholar]
- 40. Second T. P., Blethrow J. D., Schwartz J. C., Merrihew G. E., MacCoss M. J., Swaney D. L., Russell J. D., Coon J. J., Zabrouskov V. (2009) Dual-pressure linear ion trap mass spectrometer improving the analysis of complex protein mixtures. Anal. Chem. 81, 7757–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zubarev R. A., Makarov A. (2013) Orbitrap mass spectrometry. Anal. Chem. 85, 5288–5296 [DOI] [PubMed] [Google Scholar]
- 42. Deutsch E. W., Lam H., Aebersold R. (2008) PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. Embo. Rep. 9, 429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Picotti P., Rinner O., Stallmach R., Dautel F., Farrah T., Domon B., Wenschuh H., Aebersold R. (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 7, 43–46 [DOI] [PubMed] [Google Scholar]
- 44. Marx H., Lemeer S., Schliep J. E., Matheron L., Mohammed S., Cox J., Mann M., Heck A. J., Kuster B. (2013) A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 31, 557–564 [DOI] [PubMed] [Google Scholar]
- 45. Boersema P. J., Mohammed S., Heck A. J. (2009) Phosphopeptide fragmentation and analysis by mass spectrometry. J. Mass Spectrom. 44, 861–878 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.