

Abstract
A combined therapy of sulindac sulfide and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a promising strategy for the treatment of cancer. Sulindac sulfide had been shown to induce the expression of death receptor 5 (DR5), a receptor for TRAIL, and sensitize cancer cells to TRAIL-induced apoptosis; however, the molecular mechanism underlying the upregulation of DR5 has not yet been elucidated. We demonstrate here that myeloid zinc finger 1 (MZF1) mediates the induction of DR5 by sulindac sulfide. Sulindac sulfide induced the expression of DR5 at the protein and mRNA levels in colon cancer SW480 cells. Furthermore, sulindac sulfide increased DR5 promoter activity. We showed that sulindac sulfide stimulated DR5 promoter activity via the −301 to −253 region. This region contained a putative MZF1-binding site. Site-directed mutations in the site abrogated the enhancement in DR5 promoter activity by sulindac sulfide. MZF1 directly bound to the putative MZF1-binding site of the DR5 promoter and the binding was increased by sulindac sulfide. The expression of MZF1 was also increased by sulindac sulfide, and MZF1 siRNA attenuated the upregulation of DR5 by sulindac sulfide. These results indicate that sulindac sulfide induces the expression of DR5 by up-regulating MZF1.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a cytokine that belongs to the TNF family1,2 and plays an important role in immunesurveillance for cancer3,4. Consistent with the function of TRAIL, a TRAIL deficiency in mice was shown to accelerate carcinogenesis5. Previous studies have demonstrated that TRAIL selectively induced apoptosis in cancer cells both in vitro and in vivo, with little or no toxicity in normal cells6,7,8. These unique properties of TRAIL have promoted clinical trials on recombinant TRAIL and agonistic antibodies for TRAIL receptors for various malignant tumors including colorectal, renal, and ovarian cancers and melanoma9,10. Therefore, TRAIL is one of the most promising new candidates for cancer therapeutics. However, some tumor types have been shown to exhibit resistance to TRAIL11. Thus, overcoming this resistance is of importance.
TRAIL induces apoptosis through its specific receptors, death receptor 5 (DR5, also called TRAIL-R2, Apo2, TRICK2, or KILLER) and death receptor 4 (DR4), which are expressed on the cell surface12,13,14,15,16. DR5 and DR4 mediate TRAIL-induced apoptosis through interactions with adapter proteins, such as FADD, and the activation of caspases17. DR5 and DR4 expression levels are a limiting factor for the TRAIL sensitivity of malignant tumor cells and we and other groups have reported various types of agents that can induce the expression of DR5 and sensitize TRAIL-induced apoptosis in malignant tumor cells18,19. To overcome the resistance of malignant tumors to TRAIL therapy, we previously proposed a strategy combining TRAIL and a DR5 inducer19, because the amount of DR5 is known to regulate TRAIL sensitivity in cancer cells.
Sulindac sulfide is one of the nonsteroidal anti-inflammatory drugs (NSAIDs). Although NSAIDs have generally been used to treat pain, inflammation, and fever, the continuous usage of NSAIDs has been shown to reduce the risk of various cancers, especially colon cancer20,21,22, and metastasis in patients who subsequently develop cancer23.
A single treatment with sulindac sulfide inhibited the growth of cancer cells by inducing apoptosis and necrosis through gadd45a upregulation24 and phosphodiesterase 5 inhibition25. Sulindac sulfide has also been shown to sensitize cancer cells to TRAIL-induced apoptosis via the upregulation of DR526,27, which indicates that sulindac sulfide is a promising candidate for overcoming TRAIL resistance in cancer cells. However, the precise mechanism by which sulindac sulfide up-regulates DR5 remains unclear.
In the present study, we demonstrated for the first time that sulindac sulfide up-regulated DR5 through the transcription factor myeloid zinc finger 1 (MZF1) and overcame TRAIL-induced apoptosis in cancer cells.
Results
Sulindac sulfide enhanced TRAIL-induced apoptosis in SW480 colon cancer cells
We examined the effect of sulindac sulfide on TRAIL-induced apoptosis by measuring the sub-G1 population, which reflected hypodiploid cells. Sulindac sulfide or TRAIL alone slightly induced apoptosis in SW480 colon cancer cells; however, the combined treatment with sulindac sulfide and TRAIL markedly induced apoptosis (Fig. 1a). As a result of DAPI staining, the combined treatment with sulindac sulfide and TRAIL induced condensed nuclei (Fig. 1b). The combined effect was blocked by the pan-caspase inhibitor zVAD-fmk, which indicated that apoptosis was caspase-dependent (Fig. 1c). Moreover, the combination cleaved caspase-8, caspase-3 and PARP (Fig. 1d). These results indicate that sulindac sulfide enhanced the efficacy of TRAIL to induce apoptosis and overcame TRAIL resistance in SW480 colon cancer cells.
Figure 1. Sulindac sulfide enhanced TRAIL-induced apoptosis in SW480 cells.

(a) SW480 cells were treated with 200 μM sulindac sulfide and/or the indicated concentrations of TRAIL for 24 h. Cells were analyzed for DNA content by PI staining (FL2-H) using a flow cytometer. The percentages of sub-G1 are shown as a bar graph. (b) DAPI staining of SW480 cells. SW480 cells were treated with 200 μM sulindac sulfide and/or 10 ng/ml TRAIL for 24 h. Nuclear morphology was visualized using DAPI staining under a fluorescence microscope. (c) SW480 cells were treated with 200 μM sulindac sulfide and/or 10 ng/ml TRAIL with or without 20 μM zVAD-fmk for 24 h. The effects were analyzed as described in (a). Data represent the means +/− S.D. of three determinations. *: p < 0.05 (d) Western blotting for caspase-8, caspase-3 or PARP. SW480 cells were treated with 200 μM sulindac sulfide and/or 10 ng/ml TRAIL for 24 h. β-actin was used as a loading control.
Sulindac sulfide induced DR5 expression in SW480 cells
We next examined whether sulindac sulfide affected gene expression related to cell death using the RNase protection assay. As shown in Figure 2a, sulindac sulfide increased DR5, DR4, and TRAIL mRNA levels. Among these, the upregulation of DR5 by sulindac sulfide was the most prominent. Therefore, we confirmed that sulindac sulfide increased DR5 mRNA level in a dose-dependent manner by Northern blotting (Fig. 2b). Sulindac sulfide also increased DR5 protein expression in a dose- and time-dependent manner (Fig. 2c and d). Using luciferase reporter plasmids carrying the DR5 promoter, we examined the mechanism underlying how sulindac sulfide up-regulated the expression of DR5 (Fig. 3). Previous studies reported that the transcription factors p53 and NF-κB increased DR5 promoter activity through these consensus elements on intron 133,34,35,36. With or without p53- and NF-κB- binding sites, DR5 promoter activity was enhanced by the sulindac sulfide treatment. These results demonstrated that sulindac sulfide up-regulated DR5 expression at a transcriptional level and the responsive element against sulindac sulfide was on the upstream promoter region of the DR5 gene.
Figure 2. Sulindac sulfide induced DR5 expression in SW480 cells.

(a) RNase protection assay. SW480 cells were treated with or without 200 μM sulindac sulfide for 24 h. Total RNA from SW480 cells was hybridized with probes, and then digested with RNase as described in the Materials and Methods. The housekeeping genes GAPDH and ribosomal protein L32 are shown as controls. (b) Northern blot analysis. SW480 cells were treated with various concentrations of sulindac sulfide for 24 h. Total RNA was probed with human DR5 cDNA. Ethidium bromide staining of 28S and 18S rRNA are shown as loading controls. (c) Western blotting for DR5. SW480 cells were treated with the indicated concentrations of sulindac sulfide for 24 h. β-actin was used as a loading control. (d) Western blotting for DR5. Cells were treated with or without 200 μM sulindac sulfide for the period indicated. β-actin was used as a loading control. −, treated with solvent DMSO.
Figure 3. Sulindac sulfide induced DR5 promoter activity in SW480 cells.

SW480 cells were transiently transfected with reporter plasmids containing various sizes of DR5 promoters and the luciferase gene. Twenty-four hours after the transfection, cells were treated with or without 200 μM sulindac sulfide for 24 h, and cell lysates were the harvested for the luciferase assay, as described in the Materials and Methods. Relative luciferase activity is shown as raw light units (RLU) standardized with the protein concentrations. Data represent the means of triplicate experiments (bars, S.D.). −: treated with solvent DMSO. *: p < 0.05.
Identification of sulindac sulfide-responsive elements in the DR5 promoter
We investigated transcription factors contributing to the upregulation of DR5 by sulindac sulfide. Using a series of deletion mutants in the DR5 promoter flanking the luciferase reporter gene, we performed luciferase assays with or without sulindac sulfide (Fig. 4). The DR5 promoter-luciferase construct pDR5/−448 as well as pDR5PF increased promoter activity by sulindac sulfide more than 2-fold, whereas the promoter activity of pDR5/−198 was not increased (Fig. 4a). This result indicates that sulindac sulfide-responsive elements are located between −448 and −199. Thus, we generated additional reporter plasmids between −448 and −199 and performed luciferase assays (Fig. 4b). Sulindac sulfide enhanced the promoter activity of pDR5/−301 more than 2-fold. On the other hand, pDR5/−252 did not respond to the sulindac sulfide treatment, which indicated that the sulindac sulfide-responsive elements of the DR5 promoter are located within a 50-bp region between −301 and −253 relative to the first base of the translation initiation codon.
Figure 4. Analysis of sulindac sulfide-responsive elements in the DR5 promoter.

(a)(b) The luciferase assay was performed as described in Figure 3 with the indicated reporter plasmids. Data represent the means of triplicate experiments (bars, S.D.). +: treated with 200 μM sulindac sulfide for 24 h, −: treated with solvent DMSO. *: p < 0.05.
We examined the predicted transcription factor-binding sites using TFSEARCH. This sequence represents the possible binding region for the transcription factor MZF1 (Fig. 5a). We introduced a site-directed mutation to the putative MZF1-binding site on the DR5 promoter and generated pDR5/mtMZF1 (Fig. 5a). As shown in Figure 5b, sulindac sulfide did not enhance the promoter activity of pDR5/mtMZF1.
Figure 5. Mutation in the MZF1-binding site attenuated activation of the DR5 promoter due to sulindac sulfide.
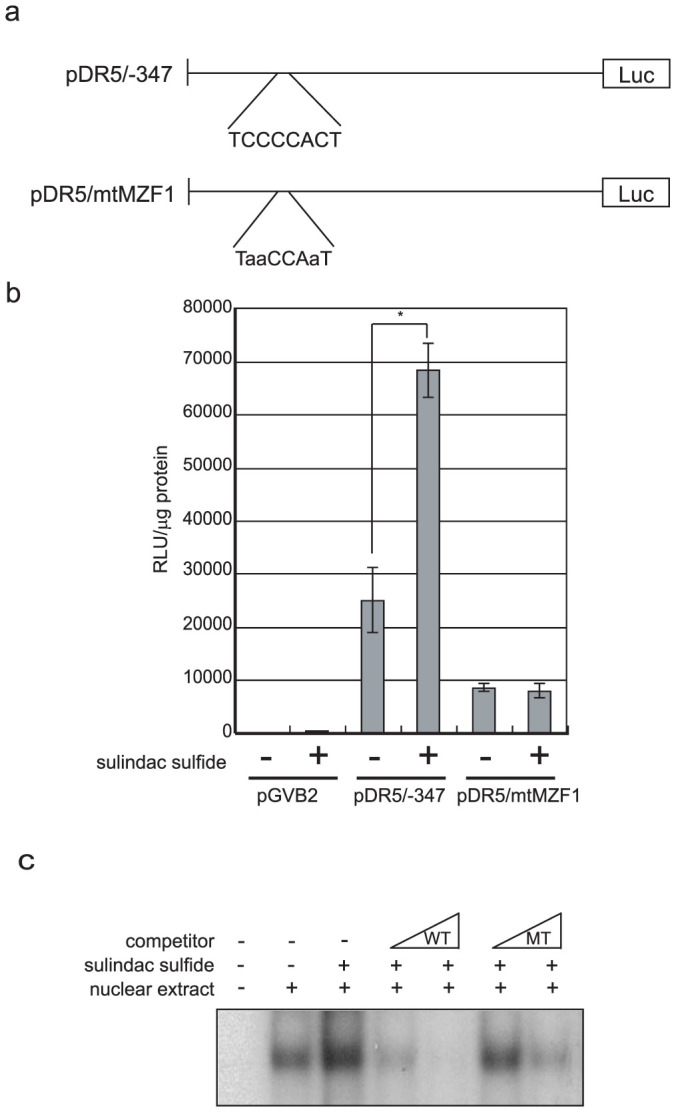
(a) The sulindac sulfide-responsive region in the DR5 promoter and MZF1-binding sequences are shown. The predicted transcription factor-binding sites are shown using TFSEARCH 1.3. Mutation sequences are described in small letters. (b) The luciferase assay was performed as described in Figure 3 with the indicated reporter plasmids. Data represent the means of triplicate experiments (bars, S.D.). +: treated with 200 μM sulindac sulfide for 24 h, −: treated with solvent DMSO. *: p < 0.05 (c) Nuclear extract from cells treated with sulindac sulfide or DMSO were analyzed by gel shift assay, as described in Materials and methods. Increased amounts of the unlabeled oligonucleotides (×2 or ×8) were used as competitors. WT: wild-type MZF1-binding site of DR5 promoter. MT: mutant MZF1-binding site of DR5 promoter.
Next, we examined the direct protein binding to the putative MZF1-binding site of the DR5 promoter at the condition with or without sulindac sulfide by gel shift assay. We detected the protein-DNA complex on the site and the complex was increased by sulindac sulfide treatment (Fig. 5c). Competitor oligonucleotides with an MZF1-binding sequence but not a mutant sequence strongly competed out the protein-DNA complex, indicating that the protein specifically bound to the site depending on the MZF1-binding sequence. These results indicated that sulindac sulfide up-regulated DR5 transcription via the MZF1-binding site of the DR5 promoter.
Sulindac sulfide also induced MZF1 expression and knockdown of MZF1 blocked the upregulation of DR5 by sulindac sulfide
As described above, MZF1 was suggested to contribute to the enhancement of the DR5 promoter by sulindac sulfide. We investigated the behavior of MZF1 when treated with sulindac sulfide. The expression of MZF1 was increased by the sulindac sulfide treatment (Fig. 6a). Thus, we examined whether MZF1 induction was related to the expression of DR5. In another human colon cancer HCT116 cells, the expressions of DR5 and MZF1 were similarly induced by sulindac sulfide (Supplemental Figure). The overexpression of MZF1 increased DR5 promoter activity, whereas the DR5 promoter with a mutation in the MZF1-binding site was not activated by the overexpression of MZF1 (Fig. 6b). We also examined the MZF1 knockdown effect on the upregulation of DR5 by sulindac sulfide. MZF1 siRNA reduced the upregulation of DR5 by sulindac sulfide (Fig. 6c), which indicated that MZF1 at least partially mediated the upregulation of DR5 induced by sulindac sulfide.
Figure 6. Sulindac sulfide induced MZF1 expression, and MZF1 siRNA reduced the enhancement in DR5 expression by sulindac sulfide.
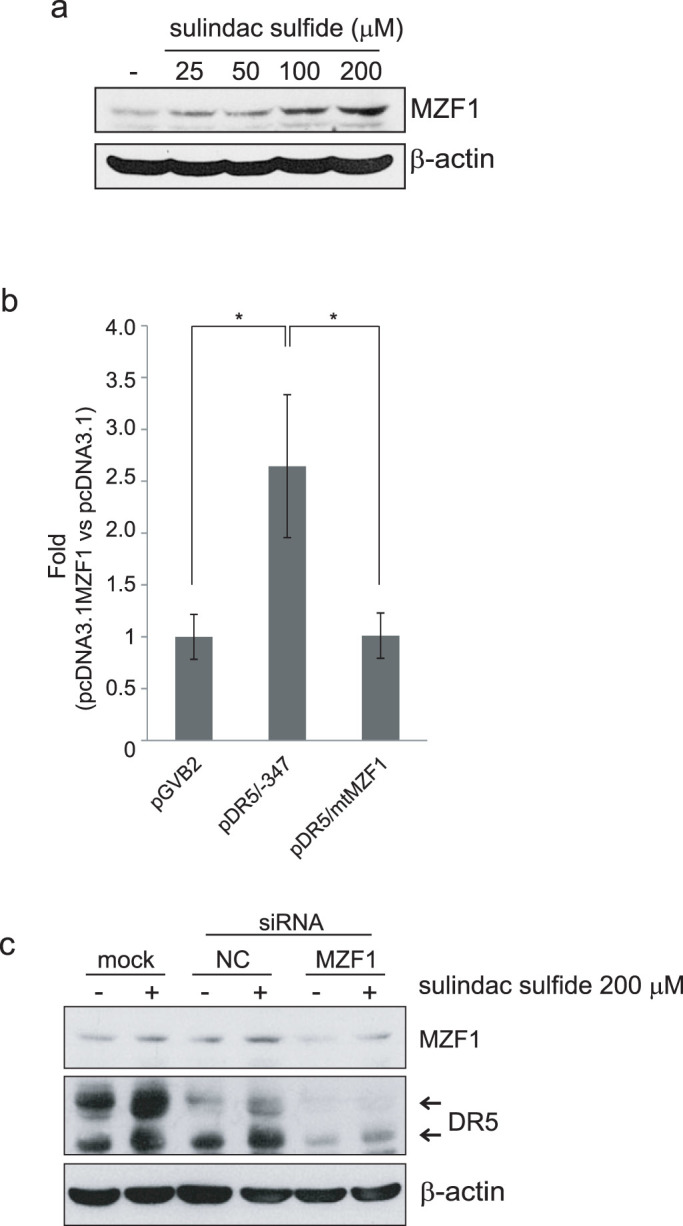
(a) Western blotting for MZF1. SW480 cells were treated with the indicated concentrations of sulindac sulfide for 24 h. β-actin was used as a loading control. (b) The luciferase assay was performed as described in Figure 5 with the indicated reporter plasmids. The MZF1 overexpression plasmid pcDNA3.1MZF1 was co-transfected with luciferase reporter plasmids as described in the Materials and Methods. The fold induction in promoter activity (pcDNA3.1MZF1 vs control vector pcDNA3.1) was shown as a bar graph. Data represent the means +/− S.D. of three determinations. *: p < 0.05 (c) Western blotting for DR5 and MZF1. SW480 cells were transfected with MZF1 siRNA or negative control siRNA at 80 nM. Twenty-four hours after the transfection, cells were treated with or without 200 μM sulindac sulfide for 24 h. β-actin was used as a loading control. −: treated with DMSO, mock: transfection was performed without any siRNA.
Discussion
Sulindac sulfide was previously suggested to be a candidate for a TRAIL sensitizer due to the upregulation of DR527. However, the mechanism underlying how sulindac sulfide up-regulates DR5 has not yet been elucidated. In the present study, we demonstrated for the first time that MZF1, a transcription factor containing the Zinc-finger domain, mediated the sulindac sulfide-induced upregulation of DR5. Sulindac sulfide increased DR5 protein and mRNA levels as well as upstream promoter activity (Fig. 2 and 3). We identified a responsive element regulated by sulindac sulfide using a series of mutant plasmids in the DR5 promoter, and the element contained a consensus sequence of the MZF1-binding site with 100% identity (Fig. 4 and 5a, b). Next, we confirmed the direct MZF1 binding to the putative MZF1-binding site of the DR5 promoter (Fig. 5c). Furthermore, we showed the expression of MZF1 was increased by sulindac sulfide (Fig. 6a). The overexpression of MZF1 increased DR5 promoter activity through the element and knockdown of MZF1 abrogated the upregulation of DR5 by sulindac sulfide (Fig. 6b, c). These results indicate that MZF1 is a transcriptional regulator of DR5 expression and a mediator for the induction of DR5 by sulindac sulfide (Fig. 7).
Figure 7. The scheme of mechanisms enhancing DR5 expression by sulindac sulfide.
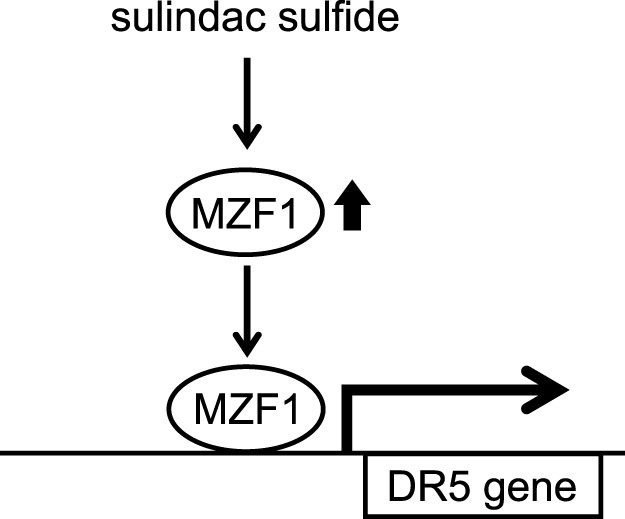
Sulindac sulfide increases MZF1. The transcription factor MZF1 then upregulates DR5 promoter activity through its binding site.
Some direct transcription factors for DR5 expression have so far been reported and include p5333,34, NF-κB35,36, c-Myc37, CHOP38,39,40, and YY141. Oncogenic stresses, such as NF-κB and c-Myc, have been shown to enhance the expression of DR5. In the present study, we also showed that MZF1, which is known to possess an oncogenic function, is a novel transcriptional regulator of DR5. The aberrant expression of MZF1 facilitates cancer cell proliferation and invasion42,43. In this study, we showed that sulindac sulfide increased the expression of MZF1 (Fig. 6); however, sulindac sulfide did not induce proliferation in human colon cancer SW480 cells (data not shown). The mechanism underlying the induction of MZF1 by sulindac sulfide remains unclear; however, a non-steroidal anti-inflammatory drug activated gene (NAG-1) was shown to be induced by sulindac sulfide and correlated to the induction of DR544. Therefore, we postulated that NAG-1 may be a candidate regulator for the induction of MZF1.
Sulindac sulfide induces DR5 in a tumor-suppressor p53-independent manner, and DR5 is a downstream gene of the tumor-suppressor p5333,34. We showed that the intronic p53-resposive site was not responsible for the activation of the promoter by sulindac sulfide (Fig. 3). Moreover, human colon cancer SW480 cells, which were mainly used in the present study, carried a mutated p53. These findings support the clinical benefit of combining sulindac sulfide and TRAIL, because more than half of malignant tumors possess inactivating mutations in the p53 gene45,46. Our results suggest that the combined treatment with sulindac sulfide and TRAIL is a promising strategy against tumors carrying a p53 mutation.
We proposed that sulindac sulfide can enhance TRAIL sensitivity against cancer cells. However, our results also suggested that TRAIL may enhance the chemopreventive efficacy of NSAIDs such as sulindac sulfide. Although, NSAIDs are frequently prescribed worldwide, adverse effects such as gastrointestinal bleeding are considered problematic. Based on our present results, TRAIL inducers may be candidates that can reduce the effective dose of NSAIDs. We previously reported that lactobacilli and Clostridium butyricum MIYAIRI 588 facilitated the production of TRAIL47,48. These results suggest that the combination with NSAIDs and probiotics as a TRAIL inducer may be a useful strategy for cancer chemoprevention through the TRAIL-DR5 pathway.
Methods
Reagents
Sulindac sulfide (Calbiochem, La Jolla, CA) was dissolved in DMSO. Soluble recombinant human TRAIL/Apo2L was purchased from PeproTech (London, UK). zVAD-fmk, a pan-caspase inhibitor, was purchased from R&D Systems (Minneapolis, MN).
Cell culture
Human colon cancer SW480 cells and HCT116 cells were purchased from the American Type Culture Collection (ATCC) and propagated and maintained according to protocols supplied by ATCC. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 4 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C, with humidity and 5% CO2. Cell line authentication was not performed by the authors within the last 6 months.
Detection of apoptosis
DNA fragmentation was quantified by the percentage of hypodiploid DNA (sub-G1). Cells were treated with the agent and harvested from culture dishes. After being washed with PBS, cells were suspended in 0.1% Triton-X100/PBS solution. Cells were then treated with RNase A (Sigma, St Louis, MO) and the nuclei were stained with propidium iodide (Sigma). The DNA content was measured using FACS Calibur (Becton Dickinson, Franklin Lakes, NJ). A total of 10,000 events were collected for each experiment. Cell Quest software (Becton Dickinson) was used to analyze the data.
DAPI staining was performed out as previously described28.
RNA analysis
Total RNA was extracted from SW480 cells using Sepasol-RNA I (Nacalai Tesque, Kyoto, Japan) according to the manufacturer's instructions. The RNase protection assay was performed using MAXI script (Ambion, Austin, TX), RPA III kit (Ambion), and hAPO3d (death receptor and death ligand) template sets (BD Biosciences PharMingen, San Jose, CA), as described previously28.
Total RNA (10 μg) was separated with electrophoresises on a 1% agarose gel and transferred to a Biodyne B nylon membrane (Pall, Pensacola, FL). Full-length DR5 cDNA was used as a probe for Northern blot analysis. Hybridization was performed with a 32P-labelled probe in PerfectHyb PLUS Hybridization buffer (Toyobo, Osaka, Japan) at 68°C for 16 h, and the membrane was washed at 68°C in 2 × SSC containing 0.1% SDS. The blot was exposed to X-ray films (Kodak, Chalon-sur-Saone, France).
Western blot analysis
Whole cell lysates were prepared as described previously29. The cell lysate was separated on 10% SDS-polyacrylamide gel for electrophoresis, and blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). Rabbit polyclonal antibodies for DR5 (Prosci, Poway, CA), caspase-3 (Cell Signaling Technology, Beverly, MA), and MZF1 (Santa Cruz Biotechnology, Santa Cruz, CA), and mouse monoclonal antibodies for caspase-8 (MBL, Nagoya, Japan), poly(ADP-ribose) polymerase (PARP) and β-actin (Sigma) were used as the primary antibodies. The blots were incubated with the appropriate HRP-conjugated secondary antibody (GE Healthcare, Piscataway, NJ), and signals were detected with Chemilumi-One (Nacalai Tesque) and an ECL Western blot analysis system (GE Healthcare).
DNA transfection and luciferase assay
As previously described30,31,32, a digested SacI-NcoI fragment from the DR5 promoter region of human genomic DNA was subcloned into the SacI-NcoI site of the pGVB2 luciferase assay vector (Toyo Ink, Tokyo, Japan) to produce pDR5PF. 5′-deletion mutants were generated with a Mungbean-Exonuclease III system from the Kilo-sequence Deletion Kit (Takara, Tokyo, Japan). pDR5/mtMZF1 was generated with a site-directed mutagenesis kit (Stratagene, La Jolla, CA) using synthesized oligonucleotides (5′-cgcttgcggaggaggtagttgacgagac, and 5′-agagtctcgtcaactacctcctccgc). SW480 cells (1 × 105 cells) were seeded on 6-well plates, and 1.0 μg of plasmids were transfected with the DEAE-dextran method using a CellPhect transfection kit (GE Healthcare). After 24 h incubation, cells were treated with sulindac sulfide or solvent DMSO. Twenty-four hours after the treatment with the agent, cells were collected for the luciferase assay. The luciferase activities of cell lysates were measured with a PicaGene luciferase assay system (Toyo Ink) and normalized for the amount of protein in the cell lysate, which was measured with the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA).
To overexpress MZF1, MZF1 cDNA was subcloned into a pcDNA3.1 mammalian expression vector (Invitrogen, Carlsbad, CA) and termed as pcDNA3.1 MZF1. In brief, 1 day prior to transfection, SW480 cells were seeded in a 6-cm dish without antibiotics at a density of 80%. Plasmid DNA (8 μg) was transfected into cells using Lipofectamine2000 reagent (Invitrogen) according to the manufacturer's instructions.
siRNA
Knockdown of MZF1 was achieved by transfection with small interfering RNA (siRNA) (Ambion) using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions.
Gel shift assay
Nuclear extracts were prepared from SW480 cells treated with DMSO or 200 μM sulindac sulfide for 12 h. The cells were lysed in buffer containing 10 mM HEPES-KOH (pH7.8), 10 mM KCl, 0.1 mM EDTA, 0.1% NP-40, 1 mM DTT, 0.5 mM PMSF, 2 μg/ml aprotinin and 2 μg/ml leupeptin, and the lysate was precipitated. The pellets were re-suspended with 50 mM HEPES-KOH (pH7.8), 420 mM KCl, 0.1 mM EDTA, 5 mM MgCl2, 20% glycerol, 1 mM DTT, 0.5 mM PMSF, 2 μg/ml aprotinin and 2 μg/ml leupeptin, rotated for 30 min at 4°C and precipitated. The supernatant was used as a nuclear extract after dialysis in reaction buffer as described below. Double-stranded DNA probes containing synthetic oligonucleotides (5′-cgcgtctccccacttggaa, and 5′-gatcttccaagtggggaga) were labeled with [α-32P] dCTP and Klenow. The reaction mixture contained 10 mM Tris-HCl (pH8.0), 50 mM NaCl, 2 mM MgCl2, 0.5 mM EDTA, 4% glycerol, 25 μg/ml poly dI-dC, 300 μg/ml bovine serum albumin, 10 μg nuclear extracts and 32P-labeled probe (50,000 cpm). The reaction mixture was incubated on ice for 20 min and resolved on 6% polyacrylamide gel in 0.25 × TBE buffer in a cold room. After electrophoresis, the gels were dried and autoradiographed at −80°C. Cold double-stranded nucleotides (wild-type: 5′-cgcgtctccccacttggaa, and 5′-gatcttccaagtggggaga, mutant: 5′-cgcgtctaaccaattggaa, and 5′-gatcttccaattggttaga) were pre-incubated with the nuclear extracts for 10 min on ice before the addition of the radiolabeled probes.
Statistical analysis
Measurements were repeated three times and statistically analyzed using a two-tailed, paired Student's t-test. A significant difference was determined at p < 0.05.
Equipment and settings
For image acquisition, all data were scanned on a Fuji Xerox DocuCentre IV C3370, multifunction printer. Scanned pictures were rotated slightly using Adobe Photoshop software.
Author Contributions
T.S., M.H. and T.Y. designed the research and wrote the manuscript. M.H. and T.Y. performed the majority of the experiments. M.T., S.Y. and Y.S. supported several experiments. All authors reviewed the manuscript.
Supplementary Material
Supplementary Figure
Acknowledgments
This work was partly supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Wiley S. R. et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682 (1995). [DOI] [PubMed] [Google Scholar]
- Pitti R. M. et al. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 271, 12687–12690 (1996). [DOI] [PubMed] [Google Scholar]
- Srivastava R. K. TRAIL/Apo-2L: Mechanisms and clinical applications in cancer. Neoplasia 3, 535–546 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K. et al. Critical role for tumor necrosis facter-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 195, 161–169 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerafa N. et al. Cutting edge: TRAIL deficiency accelerates hematological malignancies. J. Immunol. 175, 5586–5590 (2005). [DOI] [PubMed] [Google Scholar]
- Walczak H. et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 5, 157–163 (1999). [DOI] [PubMed] [Google Scholar]
- Ashkenazi A. et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 104, 155–162 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence D. et al. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat. Med. 7, 383–385 (2001). [DOI] [PubMed] [Google Scholar]
- Ashkenazi A., Holland P. & Eckhardt S. G. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J. Clin. Oncol. 26, 3621–3630 (2008). [DOI] [PubMed] [Google Scholar]
- Johnstone R. W., Frew A. J. & Smyth M. J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 8, 782–798 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang L. & Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 12, 228–237 (2005). [DOI] [PubMed] [Google Scholar]
- MacFarlane M. et al. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J. Biol. Chem. 272, 25417–25420 (1997). [DOI] [PubMed] [Google Scholar]
- Pan G. et al. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277, 815–818 (1997). [DOI] [PubMed] [Google Scholar]
- Sheridan J. P. et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277, 818–821 (1997). [DOI] [PubMed] [Google Scholar]
- Screaton G. R. et al. TRICK2, a new alternatively spliced receptor that transduces the cytotoxic signal from TRAIL. Curr. Biol. 7, 693–696 (1997). [DOI] [PubMed] [Google Scholar]
- Walczak H. et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 16, 5386–5397 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc H. N. & Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 10, 66–75 (2003). [DOI] [PubMed] [Google Scholar]
- Horinaka M. et al. Aclarubicin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis through death receptor 5 upregulation. Cancer Sci. 103, 282–287 (2012). [DOI] [PubMed] [Google Scholar]
- Yoshida T., Horinaka M. & Sakai T. Combination-oriented molecular-targeting prevention" of cancer: a model involving the combination of TRAIL and a DR5 inducer. Environ. Health Prev. Med. 15, 203–210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huls G., Koornstra J. J. & Kleibeuker J. H. Non-steroidal anti-inflammatory drugs and molecular carcinogenesis of colorectal carcinomas. Lancet 362, 230–232 (2003). [DOI] [PubMed] [Google Scholar]
- Chan A. T., Ogino S. & Fuchs C. S. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med. 356, 2131–2142 (2007). [DOI] [PubMed] [Google Scholar]
- Rothwell P. M. et al. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 377, 31–41 (2011). [DOI] [PubMed] [Google Scholar]
- Rothwell P. M. et al. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 379, 1591–1601 (2012). [DOI] [PubMed] [Google Scholar]
- Chiou S. K. & Hoa N. Up-regulation of GADD45α expression by NSAIDs leads to apoptotic and necrotic colon cancer cell deaths. Apoptosis 14, 1341–1351 (2009). [DOI] [PubMed] [Google Scholar]
- Tinsley H. N. et al. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol. Cancer Ther. 8, 3331–3340 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q., Luo X., Huang Y. & Sheikh M. S. Apo2L/TRAIL differentially modulates the apoptotic effects of sulindac and a COX-2 selective non-steroidal anti-inflammatory agent in Bax-deficient cells. Oncogene 21, 6032–6040 (2002). [DOI] [PubMed] [Google Scholar]
- Huang Y., He Q., Hillman M. J., Rong R. & Sheikh M. S. Sulindac sulfide-induced apoptosis involves death receptor 5 and the caspase 8-dependent pathway in human colon and prostate cancer cells. Cancer Res. 61, 6918–6924 (2001). [PubMed] [Google Scholar]
- Horinaka M. et al. The dietary flavonoid apigenin sensitizes malignant tumor cells to tumor necrosis factor-related apoptosis-inducing ligand. Mol. Cancer Ther. 5, 945–951 (2006). [DOI] [PubMed] [Google Scholar]
- Nakata S. et al. Histone deacetylase inhibitors up-regulate death receptor 5/TRAIL-R2, and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 23, 6261–6271 (2004). [DOI] [PubMed] [Google Scholar]
- Yoshida T., Maeda A., Tani N. & Sakai T. Promoter structure and transcription initiation sites of the human death receptor 5/TRAIL-R2 gene. FEBS Lett. 507, 381–385 (2001). [DOI] [PubMed] [Google Scholar]
- Yoshida T. & Sakai T. Promoter of TRAIL-R2 gene. Vitam. Horm. 67, 35–49 (2004). [DOI] [PubMed] [Google Scholar]
- Shiraishi T. et al. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 65, 6364–6370 (2005). [DOI] [PubMed] [Google Scholar]
- Wu G. S. et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 17, 141–143 (1997). [DOI] [PubMed] [Google Scholar]
- Takimoto R. & El-Deiry W. S. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 19, 1735–1743 (2000). [DOI] [PubMed] [Google Scholar]
- Shetty S. et al. Transcription factor NF-κB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol. Cell Biol. 25, 5404–5416 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi R. et al. Regulation of death receptor expression and TRAIL/Apo2L-induced apoptosis by NF-κB. Nat. Cell Biol. 3, 409–416 (2001). [DOI] [PubMed] [Google Scholar]
- Wang Y. et al. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 5, 501–512 (2004). [DOI] [PubMed] [Google Scholar]
- Yamaguchi H. & Wang H. G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279, 45495–45502 (2004). [DOI] [PubMed] [Google Scholar]
- Yoshida T. et al. Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res. 65, 5662–5667 (2005). [DOI] [PubMed] [Google Scholar]
- Taniguchi H. et al. Baicalein overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance via two different cell-specific pathways in cancer cells but not in normal cells. Cancer Res. 68, 8918–8927 (2008). [DOI] [PubMed] [Google Scholar]
- Baritaki S. et al. Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J. Immunol. 179, 5441–5453 (2007). [DOI] [PubMed] [Google Scholar]
- Hromas R. et al. Aberrant expression of the myeloid zinc finger gene, MZF-1, is oncogenic. Cancer Res. 55, 3610–3614 (1995). [PubMed] [Google Scholar]
- Gaboli M. et al. Mzf1 controls cell proliferation and tumorigenesis. Genes Dev. 15, 1625–1630 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang T. J., Kang H. J., Kim J. R. & Yang C. H. Non-steroidal anti-inflammatory drug activated gene (NAG-1) expression is closely related to death receptor-4 and -5 induction, which may explain sulindac sulfide induced gastric cancer cell apoptosis. Carcinogenesis 25, 1853–1858 (2004). [DOI] [PubMed] [Google Scholar]
- Greenblatt M. S., Bennett W. P., Hollstein M. & Harris C. C. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 54, 4855–4878 (1994). [PubMed] [Google Scholar]
- Levine A. J. p53, the cellular gatekeeper for growth and division. Cell 88, 323–331 (1997). [DOI] [PubMed] [Google Scholar]
- Horinaka M. et al. Lactobacillus strains induce TRAIL production and facilitate natural killer activity against cancer cells. FEBS Lett. 584, 577–582 (2010). [DOI] [PubMed] [Google Scholar]
- Shinnoh M. et al. Clostridium butyricum MIYAIRI588 shows antitumor effects by enhancing the release of TRAIL from neutrophils through MMP-8. Int. J. Oncol. 42, 903–911 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure