

Abstract
Membrane-resealing agents such as poloxamer P188 improve the outcome in experimental brain injury paradigms; however, whether membrane resealing is a key mechanism for protection has not been shown in vivo. We previously reported that Kollidon VA64, a polymeric membrane-resealing agent, reduces cell membrane permeability and improves brain edema, brain tissue damage, and functional outcome after controlled cortical impact in mice, without rescuing resealed cells from death. To reconcile these disparate findings, we used a dual-pulse labeling protocol to determine membrane-resealing kinetics by VA64/P188 in vivo. Membrane resealing after controlled cortical impact in mice by intravenous or intracerebroventricular VA64 and poloxamer P188 was transient, with most cells becoming repermeabilized within 2 hours, even with multiple-dose paradigms that maintained high VA64 blood levels. Moreover, VA64 reduced cytotoxic brain edema in a water intoxication model devoid of plasmalemma permeability (P<0.05 versus P188, VA30, mannitol, and vehicle). We conclude that VA64 reduces cytotoxic and traumatic brain edema independent of membrane resealing. The results suggest that classic membrane-resealing agents such as poloxamer P188, and the newly discovered VA64, exert protective effects in central nervous system injury paradigms by mechanisms other than or in addition to maintaining permeable cell membranes sealed.
Keywords: brain injury, controlled cortical impact, Kollidon VA64, mice, models
Introduction
Plasmalemma damage is a marker of cellular injury and death in experimental models of traumatic brain injury, intracerebral hemorrhage, and stroke.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 We previously reported that controlled cortical impact (CCI) in mice induces plasmalemma permeability in injured cells, and that intravenous administration of Kollidon VA64, a vinylpyrrolidone–vinyl acetate copolymer, restores plasmalemma integrity and reduces brain edema, Evans blue leakage, brain tissue loss, and postinjury motor deficits.8, 11 However, VA64 administered as a single (1 hour) or multiple (6, 24, and 48 hours) intravenous dose regimens did not rescue injured (PI+) cells from ultimate death or removal from contused brain.11 These findings raised the possibility that resealing of injured cells by VA64 might be transient, and that prolonged dosing strategies might be needed to maintain membranes resealed and thereby rescue injured cells from death. Alternatively, the protective effects of VA64 in the CCI model might be independent of its membrane-resealing properties. Resolving these questions is important because VA64 is already FDA (Food and Drug Administration) approved for enteral administration, and could be fast tracked to clinical trials on traumatic brain injury if experimental studies prove its safety and efficacy by the intravenous route.
Here, we used multiple fluorescent probes and a pulse labeling protocol to interrogate plasma membrane integrity and assess the kinetics of membrane resealing by VA64 after CCI. We assessed the blood pharmacokinetics of intravenously administered VA64, and we used a water intoxication model that lacks plasmalemma damage to assess the effects of VA64 on cytotoxic brain edema independent of membrane resealing. With these methodologies we addressed the following questions: (1) How long does a single dose of VA64 reseal injured cell membranes? (2) Can prolonged dosing of VA64 (multiple intravenous doses, continuous intravenous infusion, or intravenous/intracerebroventricular dosing strategies) enhance membrane-resealing time beyond that of a single dose? (3) Are the protective effects of VA64 on brain edema11 necessarily related to membrane resealing, or could additional mechanism(s) be involved?
Materials and methods
Animals
A total of 199 mice were used for the studies. Studies were performed using 12- to 14-week-old adult male CD-1 mice (Charles River, Wilmington, MA, USA) weighing 33 to 37 g (water intoxication model), or C57Bl/6 mice weighing 25 to 30 g (all other experiments). Mice were housed in a pathogen-free environment with 12-hour day–night cycles. All procedures followed the protocols approved by the MGH Institution for Animal Care and Use Committee in accordance with the NIH Guide for Care and Use of Laboratory Animals. In all experiments, mice were randomized to treatment groups and data were obtained by investigators masked to the study group.
Mouse CCI Model
A CCI model was used as previously described.12 Mice were anesthetized with 4% isoflurane (Anaquest, Memphis, TN, USA) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, USA). The blow by anesthesia was maintained with 3.5% isoflurane for the duration of surgery. Side-stream ventilation of room air mixed with this amount of isoflurane produces unresponsiveness to tail and toe pinch and to surgical procedures, yet maintains blood pressure and blood gases within normal limits.13, 14 The head was positioned in a stereotaxic apparatus. Following a midline scalp incision, a 5-mm craniotomy was made with a portable drill and trephine over the left parieto-temporal cortex. The bone flap was removed and the dura was left intact. The impact was delivered using a 3-mm flat-tip pneumatic piston at a velocity of 6 m/s, duration of 100 ms, and depth of 0.6 mm. The bone flap was discarded and the scalp incision sutured closed. Mice were returned to their cages to recover from anesthesia.
Quantification of VA64 in Blood
The concentration of VA64 in mouse blood samples was determined photometrically using an adaptation of the iodine complexation protocol described by Muller.15 Briefly, 50 μL of 0.2 mol/L citric acid solution and 20 μL of freshly prepared 0.006 N iodine solution were mixed with 100 μL of plasma containing VA64 in a 96-well plate. The A420 was measured after 30 minutes using a SpectraMax M2 microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA). A standard curve was generated from serial dilutions of known VA64 plasma concentrations. Background values for the VA64 plasma standards were obtained by mixing plasma with phosphate-buffered saline (PBS) instead of VA64. A420 values of experimental samples were adjusted for background absorbance by subtracting the A420 of plasma samples obtained from mice administered PBS.
Preparation of Brain Tissue for Histochemistry
Mice were deeply anesthetized with isoflurane and brains removed and frozen in liquid nitrogen vapor and stored at −80 °C. Brains were sectioned (14 μm) in the coronal plane on a cryostat. Sections 250 μm apart from bregma −0.7 to +3.2 were collected on poly-L-lysine-coated glass slides and stored at −80 °C.
Experimental Protocols
Protocol 1
Membrane resealing by VA64 at 24 versus 48 hours after CCI: At 24 or 48 hours after CCI, mice were administered the green fluorescent membrane-impermeant dye YOYO-1 (Invitrogen, Grand Island, NY, USA; 1 μg/g mouse in 100 μL PBS, pH 7.4 (PBS)) intravenously, followed 5 to 10 minutes later by VA64 (BASF; Florham Park, NJ, USA; 1 mmol/L, 700 μL except as noted), poloxamer P188 (BASF; 700 μL, 5 mmol/L) or PBS (700 μL). At 5 to 10 minutes after VA64 or PBS, propidium iodide (PI, Sigma, St Louis, MO, USA; 1 μg/g in 100 μL PBS) was administered intravenously. Mice were euthanized at 10 minutes after PI administration and the brains removed and frozen in liquid nitrogen vapor. Cryostat brain sections were photographed with a Nikon Eclipse T300 fluorescence microscope (Tokyo, Japan) using the NIS Elements software (Nikon, Tokyo, Japan). Excitation and emission wavelengths for YOYO-1 were 490 and 520 nm, respectively, and 536 and 617 nm for PI. Injured cells were defined as YOYO-1+. Resealed cells were defined as YOYO-1+/PI−, and permeable cells as YOYO-1+/PI+. The percentage of resealed cells was defined as the number of resealed cells divided by the total number of injured, YOYO-1 permeable cells ((YOYO-1+/PI−)/(YOYO-1+) × 100). Cell counts were performed in 8 to 10 cortical × 200 fields randomly chosen from 10 brain sections separated by at least 250 μm, located within the center or periphery of the contusion. Cell count data were averaged per × 200 field for each mouse.11
Protocol 2
Membrane-resealing kinetics of single-dose VA64 or P188: At 1 to 2 or 24 hours post CCI, fluorophores and drugs (VA64 or P188) were administered as in protocol 1 (YOYO-1 followed by VA64 or P188 followed by PI). Mice were euthanized at various intervals after PI administration (30 to 45, 60 to 90, or 120 minutes) and brains examined for the presence or absence of resealed cells.
Protocol 3
Effect of multiple-dose or continuous infusion VA64 on membrane resealing: 1 hour after CCI, YOYO-1 was administered intravenously followed by VA64 and PI as in protocol 1. VA64 was administered every 30 minutes intravenously after the initial dose for up to 90 minutes for a total of four doses. Brains were obtained for analysis of membrane resealing 30 minutes after the final dose of VA64. The 30-minutes dosing interval was derived from data obtained in protocol 2 that indicated robust membrane resealing at 30 minutes after a single dose of VA64. In a subset of mice, VA64 was also administered intracerebroventricularly via a 1-mm burr hole made with a trephine and hand-held drill 1 mm lateral and 2 mm deep to bregma (10 μL VA64 into the left and right lateral ventricles) at the time of each intravenous dose. In another subset of mice, VA64 was bolused (700 μL intravenously) and then infused continuously at a rate of 700 μL/h for 2 hours. At the end of VA64 dosing mice were euthanized and brains examined for membrane resealing.
Protocol 4
Cytotoxic brain edema (water intoxication) model: Mice were administered free water 200 mL/kg intraperitoneally, followed immediately by PBS or VA64 and PI (but not YOYO-1) as described in protocol 1. At 55 minutes PI was administered (1 μg/g intraperitoneally) and mice were euthanized at 60 minutes for assessment of PI+ cells. PI+ cells were counted in randomly selected × 200 microscopic fields (1,100 × 1,100 μm2) in the cortex (n=10 fields per section, 5 sections per mouse=50 fields/mouse, n=5 mice) and hippocampus (6 fields/section, 5 sections per mouse=30 fields/mouse, n=5 mice). Cell count data for each mouse were the average of the total brain fields counted.
To determine blood–brain barrier (BBB) permeability to Evans blue in the free-water intoxication model, another set of mice was administered free water and Evans blue (2%, 5 mL/kg, intravenous) or no free water and Evans blue and brains were examined qualitatively by fluorescence microscopy at 1 hour for the presence or absence of Evans blue leakage.
To test the effect of various polymers on brain edema in the free-water intoxication model, mice were administered free water in the presence or absence of mannitol (20%, 20 mL/kg), polymers, or PBS (all 20 mL/kg) and brain edema was assessed at 1 hour. Polymer doses were based on near-maximum tolerable intravenous doses in naive mice and our previously published data,11 and mannitol was dosed based on clinically relevant doses to reduce intracranial hypertension: VA64 1 mmol/L; VA30, PEG 8000, P188 5 mmol/L; mannitol 20% all doses 20 mL/kg intravenous at the time of free-water administration.
Assessment of Brain Edema
Brain edema was assessed using the (wet–dry)/wet brain weight method. Brains were removed and bisected into left and right hemispheres. Each hemisphere was weighed (wet weight), then dried at 99 °C for 72 hours, and dry weights were obtained. The percentage of brain water content was expressed as (wet–dry weight)/wet weight × 100%.
Statistical Analyses
Data are mean±s.e.m. Sample sizes were estimated based on between-group differences of at least 25%, standard deviation 15–25%, alpha=0.05, and power 0.8. Data were analyzed using Graphpad PRISM V (Graphpad Software, La Jolla, CA, USA). Cell count and edema data were analyzed using t-test or analysis of variance followed by Bonferroni's test for multiple comparisons if more than two groups were analyzed. For all comparisons, P<0.05 was considered significant.
Results
Protocol 1: Temporal Course of Membrane Resealing by VA64
Figure 1 shows an overview of the experimental protocols. We began by assessing the spatial and temporal characteristics of membrane resealing by VA64 in the cortex and hippocampus after CCI. Previous studies by our group showed ∼60% resealing by VA64 in the injured cortex at 1 to 6 hours after CCI.11 To determine whether VA64 could induce membrane resealing at later times, mice were subjected to CCI and administered indicator dyes and VA64 at 24 or 48 hours. Figure 2 shows robust membrane resealing (58%) by VA64 at 24 hours, but only 12% resealing at 48 hours after injury. We next determined whether VA64 might preferentially reseal certain brain regions by assessing membrane resealing at 24 hours in contused cortex anterior versus posterior to bregma −2.30. Figure 2C shows that membrane resealing was approximately twofold greater in posterior brain regions compared with anterior.
Figure 1.

Experimental overview showing the different protocols used in the experiments. BBB, blood–brain barrier; CCI, controlled cortical impact; PI, propidium iodide.
Figure 2.

(A) Representative micrographs of cells in the injured cortex resealed by VA64 administered at 24 and 48 hours after controlled cortical impact (CCI). Cells were labeled with YOYO-1 (green) before and propidium iodide (PI, red) after administration of VA64. YOYO-1+/PI− cells indicate plasmalemma membrane resealing. Scale bar=50 μm. (B) Percentages of resealed cortical neurons in mice treated with VA64 at 24 (n=5) or 48 hours (n=7) after CCI versus phosphate-buffered saline (PBS) treated (n=4) mice (24 hours). *P<0.005 versus PBS; **P<0.0002 versus PBS and VA64 (48 hours). (C) Spatial distribution of injured cells resealed by VA64 at 1 to 2 hours after CCI. Cortical regions posterior to the midhippocampus had significantly greater resealing compared with anterior brain regions (*P<0.001, n=8/group).
Protocol 2: Pharmacokinetics of VA64 and Determination of Membrane-Resealing Time
We next determined pharmacokinetic data for blood VA64 after single and multiple VA64-dosing paradigms. Figure 3A shows that after intravenous administration (1 mmol/L, 20 mL/kg) blood VA64 levels peaked at 5 and 30 minutes and decreased by ∼50% at 1 to 6 hours. VA64 concentration further decreased between 6 and 24 hours. Repeated dosing (every 30 minutes × 4 total doses) of VA64 resulted in blood levels at 2 hours that were three- to fourfold higher than peak concentrations obtained from a single dose (Figure 3A). With these reference data we next determined the length of resealing time of a single intravenous dose of VA64 during the first 6 hours after CCI using protocol 2. By 30 to 60 minutes after VA64 administration, ∼35% of injured cells remained resealed, whereas <5% remained resealed at 120 minutes (Figure 3B). Membrane resealing by poloxamer P188, the most widely studied membrane-resealing agent, was significantly less than that of VA64 at 120 minutes (Figure 3B). When the experiment was performed at 24 hours after injury using VA64, resealing by VA64 was apparent at 120 minutes in only 2.1%±0.7% of YOYO-1-positive cells (n=4 mice).
Figure 3.

(A) Pharmacokinetic data of Kollidon VA64 assessed during the first 24 hours after controlled cortical impact (CCI). At 1 hour after CCI, VA64 was given as a single intravenous dose (1 mmol/L, 20 mL/kg; n=4 to 7/time point, total n=37), or as four doses (1 mmol/L, 20 mL/kg) every 30 minutes (2 hours MD, multiple-dose regimen over 2 hours; n=7). Blood VA64/P188 levels were obtained at the indicated times after drug administration or immediately after the 2-hour multiple-dose VA64 paradigm. Data are compared with the 30-minutes time point because this was chosen as the dosing interval based on single-dose blood pharmacokinetics and brain-resealing kinetics. *P<0.0001 versus 30 minutes; **P<0.002 versus 30 minutes; ***P<0.00005 versus 30 minutes. (B) Duration of resealing by Kollidon VA64 or poloxamer P188. Mice were subjected to CCI and administered YOYO-1 at 1 to 2 hours, followed by VA64, P188, or PBS and then PI every 5 to 10 minutes as in protocol 1. Mice were euthanized and brains removed at the times indicated after VA64 administration. Brain sections were evaluated for the presence or absence of resealed cells. By 120 minutes after a single dose of VA64 (n=6), <5% of cells remained resealed, whereas P188 maintained <1% of cells resealed by this time. *P<0.001 versus 30 to 45 minutes (n=8), *P<0.01 versus 60 to 90 minutes (n=11), and *P<0.001 versus P188 at 120 minutes (n=6).
Protocol 3: Multiple-Dose VA64 Paradigms to Maximize Resealing Time
To test the hypothesis that multiple-dose VA64 administration would reseal injured cells for more prolonged time periods, multiple intravenous doses, multiple-dose intravenous/intracerebroventricular, and continuous intravenous administration of VA64 was performed. Multiple intravenous doses of VA64 (four doses given every 30 minutes) maintained membrane resealing in <3% of permeabilized cells assessed at 120 minutes after the initial VA64 dose (Figures 4A–D). The multiple intravenous dose paradigm produced blood levels that should have been adequate for membrane resealing over the 2-hour period (Figure 3A). Similar to intravenous dosing alone, four intravenous/intracerebroventricular doses of VA64 spaced 30 minutes apart only slightly prolonged membrane resealing at 120 minutes compared with intravenous dosing alone (Figure 4D, intravenous/intracerebroventricular group). Of all dosing strategies examined, only continuous intravenous dosing (20 mL/kg bolus followed by 40 mL/kg over 2 hours) produced an increase, albeit modest, in resealed cells at 120 minutes (Figure 4D, intravenouscontinuous (IV Cont) group). Taken together, the data suggest that membrane resealing by VA64 is transient and cannot be maintained by VA64 blood levels even above those needed for resealing initially. These data suggest that additional mechanisms might contribute to the protective effects of single-dose VA64 observed previously in CCI.11
Figure 4.

Duration of resealing using multidose regimens of VA64 given intravenously (IV), intravenously plus intracerebroventricularly (IV/ICV), or as an intravenous bolus (20 mL/kg) followed by continuous infusion of 20 mL/kg/h × 2 hours (IV Cont). Mice were subjected to controlled cortical impact and administered YOYO-1 at 1 hour, followed 5 minutes later by the first dose of VA64 and 5 minutes later by PI. VA64 was administered every 30 minutes IV or IV+ICV after the initial dose for a total of four doses over 90 minutes. Mice were euthanized and brains obtained for analysis of membrane resealing at 120 minutes after the initial dose of IV or IV/ICV VA64. In continuous infusion groups, brains were obtained after the end of the 2-hour IV infusion of VA64. (A–C) Representative photomicrographs of YOYO-1+ and PI+ cells from the IV VA64 administration group. Note the lack of membrane resealing. (D) Quantitative analyses of membrane resealing by the three different multiple-dose paradigms. *P<0.03 IV Cont (n=5) versus IV MD (IV multiple dose, n=5) and IV/ICV (n=7) paradigms. Scale bar=100 μm.
Free-Water Intoxication Model to Examine Alternate Mechanisms of VA64
To dissociate membrane resealing from the protective effects of VA64 we used a free-water intoxication model that induces cytotoxic brain edema. No mice in this injury paradigm exhibited PI-positive cells (0±0 PI+ cells/ × 200 field in the cortex and hippocampus, n=5 mice) or leakage of Evans blue dye into the brain parenchyma assessed qualitatively compared with sham-treated (no water intoxication) controls (n=5 sham and n=5 water intoxicated; data not shown).
Compared with baseline, free-water administration produced a 2% increase in hemispheric brain water content by 60 minutes (Figure 5). Treatment with intravenous VA64 (coadministered with water) reduced brain edema in this model by ∼40%, an effect significantly greater than that of all other compounds tested (Figure 5). These data suggest that VA64 may reduce brain edema by mechanisms other than or in addition to membrane resealing.
Figure 5.
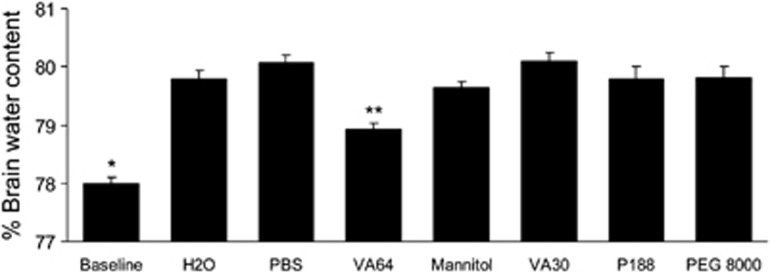
Effect of VA64 on cytotoxic brain edema after water intoxication. Mice were examined at baseline (n=9) or administered free water 200 mL/kg intraperitoneally (n=10) or free water plus VA64 (n=12) or other agents (mannitol, VA30, poloxamer P188, or PEG 8000; phosphate-buffered saline (PBS), n=5/group) intravenously (protocol 4). Brain water content was assessed at 1 hour. Compared with baseline values (normal brain), brain water content was significantly increased in all experimental conditions shown, including free water alone and PBS treatment (P<0.001, analysis of variance). VA64 significantly reduced brain water compared with PBS, mannitol, and all other treatments. *P<0.05 versus all other groups. **P<0.05 versus all other groups. PEG 8000, polyethylene glycol 8000.
Discussion
Kollidon VA64, a novel cell membrane-resealing agent, reduced brain edema, brain tissue damage, and postinjury motor deficits when administered intravenously 1 hour after CCI in mice.11 In that study, VA64 robustly resealed permeable cells but did not rescue them from ultimate demise. These observations raised two key questions addressed in the current report: (1) Is plasmalemma resealing by VA64 transient or permanent? (2) Could mechanisms other than membrane resealing account for the protective effects of VA64 in the CCI model? We found that regardless of dosing strategy (intravenous, intravenous+intracerebroventricular, multiple-dose, or continuous intravenous infusion), membrane resealing by VA64 is transient (<2 hours) despite favorable blood pharmacokinetics of VA64. Furthermore, VA64 reduced cytotoxic brain edema in a water intoxication model devoid of plasmalemma damage and BBB permeability to Evans blue. Taken together, these findings suggest that mechanisms in addition to membrane resealing may account for the protective effects of VA64 in traumatic brain injury.
We previously reported robust membrane resealing by VA64 early (<6 hours) after CCI;11 however, it was unknown whether cells that became permeable at later times could be efficiently resealed. We found that VA64 effectively resealed cells at 24 but not 48 hours after CCI. Assuming equivalent delivery of VA64 to injured brain regions at both time points, one explanation for these findings is that permeable cells at 48 hours have greater damage and hence are not efficiently resealed. Because permeable cells can remain in injured brain for up to 3 days after CCI,8 it is possible that some PI+ cells injured early after CCI are in advanced stages of neurodegeneration and less amenable to recovery of membrane integrity by 48 hours. This possibility is supported by our previous work showing that PI+ cells had morphologic, histochemical, and ultrastructural features of degeneration at early times after CCI.8 Alternatively, mode(s) of cell death may differ between 24 and 48 hours; however, necrotic death phenotypes (membrane permeability in the absence of or concomitant with DNA damage) predominate at both 24 and 48 hours in our CCI model.8 Another possibility is that VA64 diffusion into injured brain regions may be less at 48 hours. This could occur in the context of severe microvascular plugging and damage to cerebrovascular endothelium, or if the BBB reseals and is impermeable to VA64 at 48 hours after injury.
In our CCI model, BBB integrity is largely restored by 24 hours. Thus, we would not expect VA64 to penetrate the BBB and get into the brain when administered at later times after CCI. Moreover, because brain edema peaks by 24 to 48 hours, administration of VA64 at 48 hours would not be expected to reduce postinjury brain edema. The purpose of testing VA64 at 24 and 48 hours after CCI was to compare the efficiency of resealing at these times, not to infer the clinical utility of the polymer administered at late time points after injury.
Somewhat surprisingly, we found that membrane resealing by VA64 was nearly twice as effective in regions of the cortex posterior to the midhippocampus compared with anterior regions. The reasons for this observation are unknown. Cerebral blood flow in the contusion is sharply decreased within hours after CCI due in part to vasoconstriction and microvascular plugging by leukocytes and platelets,16 and perhaps by pericyte constriction of capillaries as occurs in focal stroke models.17 If cerebral blood flow were greater in posterior brain regions, more drug could be delivered, resulting in greater numbers of resealed cells. Alternatively, cellular injury and subsequent membrane damage in posterior brain regions may be less than that in anterior regions and therefore more amenable to plasmalemma resealing by VA64.
A major finding of the current study is that membrane resealing by VA64 is transient and lasts for less than 2 hours in most resealed cells. This conclusion is based on examining YOYO-1-positive (permeabilized) cells at multiple time points in the presence of PI and quantifying repermeabilized (YOYO-1+/PI+) cells at different time points. In so doing we determined the membrane-resealing kinetics for various VA64-dosing regimens. Although a fair degree of variability was noted among individual animals, by 2 hours after a single intravenous dose of VA64 <5% resealing was observed despite VA64-dosing strategies that maintained blood VA64 concentrations above those needed for initial membrane resealing (Figure 3). Similarly, <1% was observed using poloxamer P188, the prototype membrane-resealing agent. To address the possibility of inadequate brain delivery by the intravenous route alone, we administered VA64 using combined intravenous and intracerebroventricular administration. Again, we found no significant increase in membrane-resealing efficiency (Figure 4). A continuous intravenous infusion paradigm expected to maintain blood VA64 levels at least as high as those observed after a single intravenous dose only modestly increased the number of resealed cells remaining at 2 hours compared with intravenous or intravenous/intracerebroventricular administration of VA64 (Figure 4). Thus, membrane repermeabilization occurred over time despite what should have been more than adequate blood VA64 levels.
These findings support the notion that membrane resealing by VA64 (and poloxamer P188) is transient, and that mechanisms in addition to or other than membrane resealing may contribute to the beneficial effects of VA64 previously reported in the CCI model.11 Although the therapeutic window of VA64 to reduce edema and motor deficits after CCI (∼1 hour) corresponds to the peak of PI+ cells, cell death mechanisms per se may not fully account for the membrane effects of VA64 in the CCI model. Indeed, the data presented herein raise the question as to whether targeting membrane poration to reduce secondary injury after traumatic brain injury has therapeutic relevance at all, as even the best membrane-resealing agent thus far reported (VA64 compared with P188) did not effectively reverse membrane permeability (current study) or rescue permeabilized cells in our previous study.11 The transient nature of cell membrane resealing reported in the current study, and the failure of VA64 to rescue resealed cells from death reported in a prior study,11 raises significant doubt as to whether poloxamers, VA64, and other polymers reported to exert neuroprotective effects actually do so via membrane resealing.18, 19, 20
What is the basis for the transient nature of membrane resealing in the CCI model? One plausible explanation is that VA64 may be a pannexin channel blocker. Pannexin channels are multisubunit transmembrane proteins that when opened by mechanical or other stress release adenosine triphosphate into the extracellular space and activate inflammasome signaling, leading to brain edema, bystander cell death (pyroptosis), and exacerbation of brain tissue damage.21, 22, 23, 24, 25, 26 Open pannexin channels admit small molecules, such as fluorescent dyes including PI,26 and dye permeability is an accepted surrogate for detecting open pannexin channels in experimental models.23 The transient nature of membrane resealing by VA64 could be explained by the initial pannexin channel blockade that effectively prevents influx of PI, followed by cellular necrosis, which is not mitigated by VA64 because of widespread, irreparable membrane damage. In addition, the ability of VA64 to mitigate posttraumatic brain edema and tissue damage11 is consistent with the effects of pannexin channel blockers in other models of acute brain injury.27, 28, 29 We speculate that the uneven distribution of membrane resealing by VA64 in anterior versus posterior brain regions might be explained by the unequal distribution of pannexin channels. Finally, we found that carbenoxolone, a known pannexin channel blocker, administered intracerebroventricularly to mice 1 hour after CCI promoted membrane resealing similar to VA64 (unpublished data). Although beyond the scope of the current study, the possibility that VA64 is a novel pannexin channel blocker warrants further investigation.
Data from the current study suggest that ongoing cell death programs may overwhelm the ability of VA64 to maintain the plasmalemma resealed. This could explain why resealed cells are ultimately lost (not rescued from death11), and would suggest a rationale for combination therapy to concomitantly seal damaged membranes and inhibit cell death mechanisms. Such a strategy might allow for rescue of permeabilized cells, which so far has not been demonstrated in a brain injury model.8 Another explanation for the transient effects of VA64 might be rapid elimination of the polymer,30 but this explanation is not supported by the pharmacokinetic data presented herein.
VA64 reduced cytotoxic brain edema in a water intoxication model that is devoid of PI permeable cells and BBB permeability to Evans blue (Figure 5 and data not shown), thereby dissociating the effects on edema from its membrane-resealing properties. Although the mechanism(s) by which VA64 reduces edema is unknown, it is not likely to be related simply to its ability to absorb water, as VA30, which is three times more absorbent than VA64, failed to reduce edema in the water intoxication model (Figure 5). Another possible mechanism could involve interaction between VA64 and aquaporin-4 water channels that mediate cytotoxic brain edema,31, 32 as has recently been suggested for poloxamer P188.19
In conclusion, the current study calls into question the long-standing premise that membrane resealing accounts for the beneficial effects of poloxamers and other polymers, and more recently VA64.11 We believe that the current study is the first to directly test this central tenet in vivo. The ability of VA64 to reduce cytotoxic brain edema independent of plasmalemma damage further supports the idea that mechanisms other than membrane-resealing properties may explain its observed beneficial effects in acute central nervous system injury models.
The authors declare no conflict of interest.
Footnotes
This work was supported by RO1NS061255 (MJW).
References
- Geddes DM, LaPlaca MC, Cargill RS., 2nd Susceptibility of hippocampal neurons to mechanically induced injury. Exp Neurol. 2003;184:420–427. doi: 10.1016/s0014-4886(03)00254-1. [DOI] [PubMed] [Google Scholar]
- Kilinc D, Gallo G, Barbee K. Poloxamer 188 reduces axonal beading following mechanical trauma to cultured neurons. Conf Proc IEEE Eng Med Biol Soc. 2007;2007:5395–5398. doi: 10.1109/IEMBS.2007.4353562. [DOI] [PubMed] [Google Scholar]
- Marks JD, Pan CY, Bushell T, Cromie W, Lee RC. Amphiphilic, tri-block copolymers provide potent membrane-targeted neuroprotection. FASEB J. 2001;15:1107–1109. doi: 10.1096/fj.00-0547fje. [DOI] [PubMed] [Google Scholar]
- Prado GR, Ross JD, DeWeerth SP, LaPlaca MC. Mechanical trauma induces immediate changes in neuronal network activity. J Neural Eng. 2005;2:148–158. doi: 10.1088/1741-2560/2/4/011. [DOI] [PubMed] [Google Scholar]
- Singleton RH, Povlishock JT. Identification and characterization of heterogeneous neuronal injury and death in regions of diffuse brain injury: evidence for multiple independent injury phenotypes. J Neurosci. 2004;24:3543–3553. doi: 10.1523/JNEUROSCI.5048-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal Cevik I, Dalkara T. Intravenously administered propidium iodide labels necrotic cells in the intact mouse brain after injury. Cell Death Differ. 2003;10:928–929. doi: 10.1038/sj.cdd.4401250. [DOI] [PubMed] [Google Scholar]
- Unal-Cevik I, Kilinc M, Can A, Gursoy-Ozdemir Y, Dalkara T. Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke. 2004;35:2189–2194. doi: 10.1161/01.STR.0000136149.81831.c5. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Dalkara T, You Z, Qiu J, Bermpohl D, Mehta N, et al. Acute plasmalemma permeability and protracted clearance of injured cells after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:490–505. doi: 10.1038/sj.jcbfm.9600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Tao L, Tejima-Mandeville E, Qiu J, Park J, Garber K, et al. Plasmalemma permeability and necrotic cell death phenotypes after intracerebral hemorrhage in mice. Stroke. 2012;43:524–531. doi: 10.1161/STROKEAHA.111.635672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadichon SB, Le Hoang M, Wright DA, Curry DJ, Kang U, Frim DM. Neuroprotective effect of the surfactant poloxamer 188 in a model of intracranial hemorrhage in rats. J Neurosurg. 2007;106:36–40. doi: 10.3171/ped.2007.106.1.36. [DOI] [PubMed] [Google Scholar]
- Mbye LH, Keles E, Tao L, Zhang J, Chung J, Larvie M, et al. Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2012;32:515–524. doi: 10.1038/jcbfm.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager PH, You Z, Qin T, Kim HH, Takahashi K, Ezekowitz AB, et al. Mannose binding lectin gene deficiency increases susceptibility to traumatic brain injury in mice. J Cereb Blood Flow Metab. 2008;28:1030–1039. doi: 10.1038/sj.jcbfm.9600605. [DOI] [PubMed] [Google Scholar]
- Yang J, You Z, Kim HH, Hwang SK, Khuman J, Guo S, et al. Genetic analysis of the role of tumor necrosis factor receptors in functional outcome after traumatic brain injury in mice. J Neurotrauma. 2010;27:1037–1046. doi: 10.1089/neu.2009.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller K. [Identification and determination of polyvinylpyrrolidone(PVP) as well as determination of active substances in PVP-containing drug preparations] Pharm Acta Helv. 1968;43:107–122. [PubMed] [Google Scholar]
- Schwarzmaier SM, Kim SW, Trabold R, Plesnila N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J Neurotrauma. 2010;27:121–130. doi: 10.1089/neu.2009.1114. [DOI] [PubMed] [Google Scholar]
- Dalkara T, Gursoy-Ozdemir Y, Yemisci M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011;122:1–9. doi: 10.1007/s00401-011-0847-6. [DOI] [PubMed] [Google Scholar]
- Luo CL, Chen XP, Li LL, Li QQ, Li BX, Xue AM, et al. Poloxamer 188 attenuates in vitro traumatic brain injury-induced mitochondrial and lysosomal membrane permeabilization damage in cultured primary neurons. J Neurotrauma. 2013;30:597–607. doi: 10.1089/neu.2012.2425. [DOI] [PubMed] [Google Scholar]
- Bao HJ, Wang T, Zhang MY, Liu R, Dai DK, Wang YQ, et al. Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem Res. 2012;37:2856–2867. doi: 10.1007/s11064-012-0880-4. [DOI] [PubMed] [Google Scholar]
- Serbest G, Horwitz J, Jost M, Barbee K. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. 2006;20:308–310. doi: 10.1096/fj.05-4024fje. [DOI] [PubMed] [Google Scholar]
- Kimbler DE, Shields J, Yanasak N, Vender JR, Dhandapani KM. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PLoS ONE. 2012;7:e41229. doi: 10.1371/journal.pone.0041229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ, 3rd, Nonner D, Bullock M, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab. 2014;34:621–629. doi: 10.1038/jcbfm.2013.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl G, Keane RW. Pannexin: from discovery to bedside in 11+/−4 years. Brain Res. 2012;1487:150–159. doi: 10.1016/j.brainres.2012.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, et al. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem. 2009;284:18143–18151. doi: 10.1074/jbc.M109.004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1251–1261. doi: 10.1038/jcbfm.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Kocak E, Sen ZD, et al. Spreading depression triggers headache by activating neuronal Panx1 channels. Science. 2013;339:1092–1095. doi: 10.1126/science.1231897. [DOI] [PubMed] [Google Scholar]
- Xiong XX, Gu LJ, Shen J, Kang XH, Zheng YY, Yue SB, et al. Probenecid protects against transient focal cerebral ischemic injury by inhibiting HMGB1 release and attenuating AQP4 expression in mice. Neurochem Res. 2014;39:216–224. doi: 10.1007/s11064-013-1212-z. [DOI] [PubMed] [Google Scholar]
- Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, et al. Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci USA. 2011;108:20772–20777. doi: 10.1073/pnas.1018262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiotas P, Krenz A, Monyer H, Schwaninger M. Functional outcome of pannexin-deficient mice after cerebral ischemia. Channels. 2012;6:453–456. doi: 10.4161/chan.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blessing MH. Storage of vinylpyrrolidone-vinylacetate (VP-VA) in rats following endotracheal and subcutaneous injection. Virchows Arch A Pathol Anat Histol. 1974;362:115–128. doi: 10.1007/BF00432390. [DOI] [PubMed] [Google Scholar]
- Yang B, Zador Z, Verkman AS. Glial cell aquaporin-4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J Biol Chem. 2008;283:15280–15286. doi: 10.1074/jbc.M801425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 and brain edema. Pediatr Nephrol. 2007;22:778–784. doi: 10.1007/s00467-006-0411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]