

Abstract
Cyclooxygenase-2 (COX-2), a source of inflammatory mediators and a multifunctional neuronal modulator, is rapidly induced in select populations of cortical neurons after status epilepticus. The consequences of rapid activity-triggered induction of COX-2 in neurons have been the subject of much study and speculation. To address this issue directly, we created a mouse in which COX-2 is conditionally ablated in selected forebrain neurons. Results following pilocarpine-induced status epilepticus indicate that neuronal COX-2 promotes early neuroprotection and then delayed neurodegeneration of CA1 pyramidal neurons, promotes neurodegeneration of nearby somatostatin interneurons in the CA1 stratum oriens and dentate hilus (which themselves do not express COX-2), intensifies a broad inflammatory reaction involving numerous cytokines and other inflammatory mediators in the hippocampus, and is essential for development of a leaky blood–brain barrier after seizures. These findings point to a profound role of seizure-induced neuronal COX-2 expression in neuropathologies that accompany epileptogenesis.
Introduction
In adult rodent models of status epilepticus (SE), systemically administered pilocarpine induces prolonged SE resulting in neuronal damage in the hippocampus and other brain regions (Sloviter et al., 1991; Mello et al., 1993; Obenaus et al., 1993; Gorter et al., 2001; Borges et al., 2006), accompanied by an inflammatory reaction (Borges et al., 2003, 2004; Vezzani and Granata, 2005; Binder and Steinhauser, 2006; Ravizza et al., 2008; Marcon et al., 2009). During seizures, cyclooxygenase-2 (COX-2) is rapidly induced in principal forebrain neurons including hippocampal pyramidal cells and dentate granule cells (Yamagata et al., 1993; Marcheselli and Bazan, 1996; Sandhya et al., 1998; Ciceri et al., 2002; Takemiya et al., 2006). Previous studies correlate these phenomena and additionally reveal a role for COX-2 in synaptic plasticity (Murray and O'Connor, 2003; Chen and Bazan, 2005; Slanina and Schweitzer, 2005; Yang and Chen, 2008). However, the role of COX-2 expressed by neurons has remained elusive because COX-2 is expressed in many nonneuronal cells, and the only tools available have been pharmacologic inhibitors and global knock-outs. Inhibition of COX-2 can either exacerbate (Baik et al., 1999; Kim et al., 2008; Toscano et al., 2008) or attenuate (Baran et al., 1994; Kunz and Oliw, 2001; Jung et al., 2006; Takemiya et al., 2006; Zhang et al., 2008) the neurodegeneration observed after SE depending on the strategies used to delete or inhibit COX-2. The global COX-2 knock-out mouse is more susceptible to SE (Toscano et al., 2008), preventing conclusions on the role of postseizure COX-2 induction. Moreover, global knock-outs or systemic administration of COX-2 inhibitors cannot uncover the cell autonomous functions of COX-2, and these approaches cannot distinguish between peripheral and central involvement of COX-2.
Neuroinflammatory consequences of SE include leukocyte infiltration, robust astrogliosis, and microglial activation (Borges et al., 2004; Binder and Steinhauser, 2006; Ravizza et al., 2008). One broad class of seizure-induced inflammatory mediators includes cytokines and their receptors. Cytokines, specifically chemokines, are released by several cell types after injury and promote leukocyte infiltration to the CNS (Boztug et al., 2002; Babcock et al., 2003; Cardona et al., 2008). Moreover, cytokines such as interleukin 1 β (IL-1β), chemokine (C-X-C motif) ligand 10 (CXCL10), and tumor necrosis factor α (TNF-α) are well recognized to be involved in related functions such as gliosis induction and damage to the blood–brain barrier, as well as increased neuronal excitability and seizure intensity, which may secondarily contribute to neuronal cell loss (Jankowsky and Patterson, 2001; Vlkolinsky et al., 2004; Balosso et al., 2005; Vezzani and Granata, 2005; Oby and Janigro, 2006; Vezzani and Baram, 2007; Cho et al., 2009). These considerations led us to determine whether the principal neurons of the hippocampus are the source of COX-2 that triggers hippocampal neurodegeneration and neuroinflammation after SE.
The results point to a broad, deleterious role for neuronal COX-2 after SE, including the demonstration for the first time of a direct effect of neuron derived COX-2 on inflammation.
Materials and Methods
Creation of neuron-specific conditional COX-2 knock-outs.
Mice were bred for six to eight generations from the C57BL/6 (Jackson ImmunoResearch) strain into the C57BL/6 (Charles River Laboratories) strain and were housed under a 12 h light/dark cycle with food and water ad libidum. A transgenic mouse line was created in which 34 bp loxP elements were inserted into introns 5 and 8 of the COX-2 gene (J. Wang et al., 2009). We bred these mice with female mice expressing cre recombinase under control of the synapsin 1 promoter (Hoesche et al., 1993; He et al., 2004) to generate females expressing Cre and heterozygous for floxed COX-2. These females were further bred with heterozygous floxed COX-2 males to generate wild-type (WT) and neuron-specific conditional COX-2 knock-out (nCOX-2 cKO) offspring. DNA isolated from the tail of each mouse was genotyped for the presence of Syn Cre, loxP elements, and COX-2. By restricting COX-2 ablation to selected forebrain neurons (Hoesche et al., 1993; Zhu et al., 2001), the known developmental effects of global COX-2 ablation are avoided, which include severe kidney and reproductive deficits (Morham et al., 1995; Lim et al., 1997; Reese et al., 2001), and the opportunity for confounding developmental or compensatory adjustments after ablation is reduced.
Pilocarpine treatment.
Pilocarpine injections were performed in nCOX-2 cKO mice and littermates (or cre+ but unfloxed mice of the same breeding generation) as described previously (Borges et al., 2003, 2004). Mice were injected with methylscopolamine and terbutaline (2.5–3 mg/kg each, i.p., in 0.9% NaCl) 15–30 min before intraperitoneal pilocarpine. Mice initially received an intraperitoneal pilocarpine injection of 280 mg/kg; if they did not enter SE within 30 min, they received another dose of 140 mg/kg. Adult male Sprague Dawley rats were injected with methylscopolamine and terbutaline (2.5 mg/kg, s.c., in 0.9% NaCl) 30 min before pilocarpine administered subcutaneously (380–400 mg/kg in 0.9% NaCl). Rats that did not enter SE within 30 min received another injection 30–50% of the initial dose. The total pilocarpine doses received were 364 ± 33 mg/kg for wild-type control mice, 357 ± 89 mg/kg for nCOX-2 cKO mice, and 688 ± 37 mg/kg (s.c.) for rats. The number of Fluoro-Jade-stained profiles in the CA1 region of the hippocampus was not correlated with the total dose of pilocarpine received (1 d after SE in mice, R2 = −0.13; 4 d after SE in mice, R2 = 0.00; rats, R2 = −0.04).
Mice injected with pilocarpine were allowed to experience SE for 1 h as defined by continuous behavioral seizure activity consisting of mainly whole body clonic seizures (for a more detailed description of seizure behavior including a modified Racine scale, see Borges et al. 2003). Pentobarbital (30 mg/kg, i.p.) was then administered to terminate SE, and lactated dextrose Ringer's was administered subcutaneously as needed to maintain hydration. Mice showing behavioral seizure activity for <1 h were not included in our analysis. Control animals received terbutaline, methylscopolamine, pentobarbital, and lactated dextrose Ringer's, but no pilocarpine. Animals were killed under isoflurane anesthesia 24 h, 4 d, or 4 months after SE. Animals used for double-label COX-2 and somatostatin immunohistochemistry were killed 5 h after SE, by which time COX-2 induction was robust but somatostatin neuron death had not yet occurred. Mice were decapitated, and their brains were removed. Each brain hemisphere was divided; one half was used for RNA isolation and the other half for immunohistochemistry, Nissl, Fluoro-Jade, and TUNEL staining.
EEG surgeries and recordings.
Two- to 3-month-old mice were placed under deep isoflurane anesthesia and surgically implanted subdurally with four sterile 0–80 × 3/32 screw electrodes for EEG analysis. Two electrodes were placed on the right hemisphere above the frontal and parietal cortex (2 mm anterior to bregma and 1.2 mm lateral to the midline, and 1.5 mm posterior to bregma and 1.2 mm lateral to the midline, respectively). Contralaterally, the other two electrodes were placed on the left hemisphere above the parietal and visual cortex (0.5 mm posterior to bregma and 2.2 mm lateral to the midline, and 3.5 mm posterior to bregma and 2.2 mm lateral to the midline, respectively). Fine-wire electrodes were inserted into the left and right neck muscles for EMG acquisition. The animals were allowed to recover from surgery for 7 d.
All experiments were approved by the Institutional Animal Care and Use Committee of Emory University and conducted in accordance with its guidelines. Every effort was made to minimize animal suffering.
EEG analysis.
Amplified EEG and EMG signals and real-time video signals were collected and processed by the Stellate Harmonie headbox, amplifier, and software (version 6.1; Stellate). Filters for the EEG were 0.3 and 35 Hz, and signals were sampled at 200 Hz. The EMG filter was 65 Hz. We measured and reported the latency to the first EEG seizure and persistent continuous seizures (SE). In addition, to measure SE intensity, we used a modification of the coastline burst index (Korn et al. 1987), in which we measured the total linear length of the EEG tracing and expressed it in each mouse as a ratio to the baseline EEG signal before SE onset. To validate this new metric, we measured both total power of the EEG signal and the EEG coastline index in 15 8 min recording samples across three mice. The total power of the EEG signal showed good correlation with the EEG coastline index (Pearson's correlation index, 0.904; p < 0.0001), so this simplified method for estimating seizure intensity appears to be a feasible alternative to conventional power analysis.
Immunohistochemistry, Fluoro-Jade, and Nissl staining.
Brain hemispheres were fixed overnight in 4% paraformaldehyde at 4°C. Brains taken 4 d or 4 months after SE were then paraffin embedded. For the paraffin-embedded brains, 8 μm coronal sections were cut; for the fresh-fixed brains, 30 μm coronal sections were cut on a sliding microtome and sections through the hippocampus were collected. Every fifth section was labeled by the original Fluoro-Jade method as described by Schmued et al. (1997). The In Situ Cell Death Detection kit, TMR red (Roche) was used for a Fluoro-Jade/TUNEL double-label protocol. The other sections were used for immunohistochemistry. Endogenous peroxidase activity was reduced by 10 min incubation in 1% hydrogen peroxide. Sections were washed in 1% Tris-buffered saline (TBS) and incubated in 10% normal horse serum diluted in 0.15% Triton X-100 TBS buffer for 60 min to minimize nonspecific staining. This procedure was followed by 24 h incubation at 4° in goat anti-COX-2 (1:1000; Santa Cruz Biotechnology), rabbit anti-somatostatin (1:500; Peninsula), mouse anti-Neu N (1:2000; Millipore Bioscience Research Reagents), rabbit anti-glial fibrillary acidic protein (GFAP; 1:2000; Dako), or rabbit anti-ionized calcium-binding adaptor molecule 1 (IBA1; 1:1000; Wako Chemicals), followed by a 12 h incubation with fluorescent secondary antibodies, Alexa Fluor 488 or 546 (Goat anti mouse or goat anti rabbit). Hoechst 33258 dye was used (1 μm) in some occasion to visualize cell nuclei. All antisera dilutions were done in TBS containing 1% normal horse serum and 0.15% Triton X-100. Paraffin-embedded sections were used for Nissl staining using a 0.1% cresyl violet solution.
A Zeiss Axio Observer A1/D1 fluorescent microscope equipped with an AxioCam MRc 5 (D) camera was used to take images. AxioVision Rel. 4.7 was used to overlay double-labeled sections and adjust image intensity and contrast; any adjustments made were the same for all images that were compared.
GFAP and IBA1 protein staining intensity in each section were quantified using Image J software. RGB pixels were converted to brightness values using the formula V = (R + G + B)/3. The mean brightness was calculated from all pixels located within 20× fields of each section. Measurements from three fields, centered over the CA1, CA3, and hilar regions, were averaged from each of three to five sections from five to eight mice in each group. To minimize technical variation, all sections were stained the same day in the same Fluoro-Jade solutions, and all images were photographed the same day using the same lamp intensity. Images were not manipulated further to avoid differences in background intensity.
Protein extraction and Western blot for albumin.
Four days following SE, animals were perfused with PBS, and each cerebrum was flash frozen in liquid nitrogen. Tissue was homogenized in RIPA buffer (Pierce) and centrifuged for 10 min at 14,000 rpm (4°C). Proteins were separated by SDS-PAGE and transferred to a polyvinylidene fluoride membrane. Rabbit anti mouse albumin (1:1000) was incubated overnight, as well as the loading control mouse anti Actin (1:5000). Secondary antibodies (goat anti-rabbit IRDye 800, 1:5000; goat anti mouse Alexa Fluor 680, 91:15,000) were incubated for 1 h. An Odyssey infrared imaging system was used to visualize the Western blot.
Butaprost injections.
Adult male Sprague Dawley rats with an intralateral ventricular cannula (IVC) were purchased from Charles River Laboratories at 220 g and used at 250–450 g body weight. Rats experienced 90 min of pilocarpine-induced SE, which was terminated by pentobarbital. Immediately after, rats received an injection of 2 μl of 1 μm butaprost (a specific EP2 agonist) or vehicle though the IVC. This concentration of butaprost was selected based on its EC50 for activating EP2 receptors (21 nm) (Jiang et al., 2010), its >20-fold selectivity over EP3 (Abramovitz et al., 2000), and the expected dilution of 10- to 20-fold after injection. One day later, the brains were bisected and fixed overnight in 4% paraformaldehyde at 4°C, then processed for immunohistochemistry and Fluoro-Jade staining. For this, rat brain hemispheres were mounted onto a tissue-cutting block and frozen in optimum cutting temperature compound (VWR). Coronal sections (40 μm) through the hippocampus were cut using a cryostat CM 1850 (Leica) and placed in 0.05 m TBS (free floating). Fluoro-Jade staining was then performed as for mice.
Cell counting.
In each animal, Fluoro-Jade-stained neuron profiles in the hippocampal CA1 cell layer were counted by an examiner blinded to experimental conditions. All stained profiles with near-complete cell bodies were counted from 7–15 sections in each mouse between bregma −1.22 and −2.92 (Paxinos and Franklin, 2001). Profile counts were corrected for section thickness by a modified Abercrombie factor (Abercrombie, 1946), in which the cell diameter was estimated by averaging its long and short axes (Franklin and Craig, 1978); typically, 50–55 stained profiles from each mouse were used for this average. The average correction factor was very similar for the two genotypes (e.g., for CA1 1 d after SE, 0.858 for WT, 0.862 for cKO), suggesting that differential swelling of the tissue did not occur. The number of injured neurons in each mouse was expressed as the average corrected number of Fluoro-Jade-stained cells per section. Similarly, interneurons staining for somatostatin in CA1 stratum oriens and dentate hilus were counted at 20× by an examiner blind to experimental conditions and recorded as number of cells per section.
RNA isolation and inflammatory PCR array.
RNA was extracted from each mouse cerebral hemisphere using Trizol reagent, and after purification the SABiosciences RT2 First Strand kit was used to generate first-strand DNA. The mouse Inflammatory Cytokines and Receptors PCR Array (SABiosciences) was used to detect changes in expression of 84 inflammatory cytokine transcripts. RNA expression of each cytokine was normalized using the average of five housekeeping genes included in the array [β-glucuronidase; hypoxanthine guanine phosphoribosyl transferase 1; heat shock protein 90 kDa α (cytosolic), class B member 1; glyceraldehyde-3-phosphate dehydrogenase; β-actin, cytoplasmic]. Additional primers were design to detect COX-2, GFAP, IBA1, and L-selectin expression in the brain. To normalize their RNA expression, β-actin was used as housekeeping gene. The Bio-Rad IQ5 real-time PCR optical detection system was used.
Statistical analysis.
For parametric values, we calculated mean and SEM. Group means were compared by ANOVA with post hoc Bonferroni tests or t tests as appropriate. For comparisons of the number of injured neurons in the CA1 region (see Figs. 4A,B, 7C), the data were not normally distributed across animals, so a nonparametric test (Mann–Whitney) was used. GraphPad InStat was used for all statistical comparisons, except for the cytokine PCR array, which was analyzed in Excel by ANOVA with a Holm–Bonferroni post-test of the four groups (WT and cKO with and without pilocarpine), followed by a t test of individual cytokine inductions with Benjamini and Hochberg (1995) correction for significance of each of the 70 cytokines tested.
Figure 4.

Quantitation of neuronal injury after SE. A, B, Each symbol represents the average number of Fluoro-Jade-positive neurons per section counted in the CA1 region from individual mice, with an Abercrombie correction for profile size. N = 6 mice for 1 d WT; n = 7 mice for 1 d cKO; n = 4 mice each for 4 d WT and cKO. The bars indicate the median of each group. The Mann–Whitney test was used to compare WT and cKO groups. C, Abercrombie-corrected Nissl counts (mean and SEM) in CA1 for WT and cKO mice 4 months after SE, with the number of mice tested shown in parentheses for each column. The WT mice suffered 21.3% cell loss (p < 0.001, one-way ANOVA with Bonferroni post hoc tests), whereas the COX-2 cKOs lost 8.9% of neurons [not significant (ns)]. Error bars indicate SEM.
Figure 7.
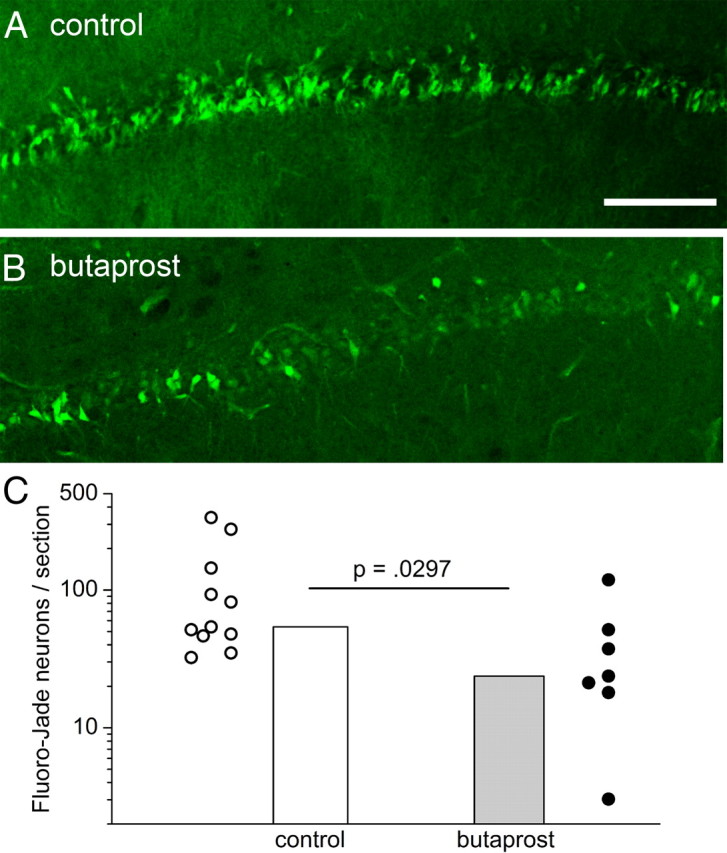
EP2 receptor activation in vivo protects neurons from injury. A, B, Fluoro-Jade staining in CA1 1 d after SE in rats with intraventricular injection of butaprost (B) or vehicle (A). C, The Abercrombie-corrected number of Fluoro-Jade-positive neurons was counted in 40 μm hippocampal sections from rats that had experienced SE 1 d before. Each symbol is from a different rat. Control, n = 11; butaprost, n = 7; p = 0.0297 by Mann–Whitney test. Scale bar, 20 μm.
Results
Creation of transgenic mice with neuronal COX-2 ablation
A mouse was recently created in which 34 bp loxP elements were inserted into introns 5 and 8 of the COX-2 gene (Fig. 1A) such that excision of the sequence between the loxP elements by cre recombinase disrupted the COX-2 coding sequence (Vardeh et al., 2009; D. Wang et al., 2009; J. Wang et al., 2009). To create neuron-specific conditional COX-2 knock-outs (nCOX-2 cKOs) in the mouse brain, we crossed the floxed COX-2 mouse with mice that express cre recombinase driven from the synapsin 1 promoter. This transgenic line expresses active cre recombinase postnatally mainly in principal neurons of the hippocampus (CA1 and CA3 pyramidal cells and dentate granule cells), as well as piriform and neocortex (Layer IV), amygdala, and thalamus (Hoesche et al., 1993; Rempe et al., 2006). Constitutive expression of COX-2 protein in CA3 pyramidal neurons (Fig. 1B1) and CA1 pyramidal and dentate granule cells (data not shown), which is normally observed in WT animals, was absent in nCOX-2 cKO animals (Fig. 1B2), but inspection of Figure 1, B1 and B2, shows that nonneuronal immunostaining due to COX-2 expression in astrocytes, microglia, and/or blood vessels was maintained. COX-2 immunostaining from animals expressing just Synapsin 1 cre or just loxP elements was similar to that observed in WT animals. Constitutive COX-2 RNA expression in the cerebrum was not significantly different between WT and nCOX-2 cKO animals (p > 0.05 by ANOVA with Bonferroni post-test; Fig. 1C), suggesting that constitutive COX-2 is much more abundant in nonneuronal cells than in principal neurons. Isolation of DNA from mouse tails was used to confirm that the COX-2 sequence was not globally deleted; indeed the COX-2 gene was intact in the tail of both WTs and nCOX-2 cKOs (Fig. 1D).
Figure 1.

Development and characterization of nCOX-2 cKOs. A, Diagram shows the mating scheme used to generate progeny with COX-2 coding sequence disrupted in all principal neurons of the hippocampus. Synapsin 1-driven cre recombinase mice were crossed with mice that had 34 bp loxP elements inserted into introns 5 and 8 of the COX-2 gene. B1–B4, Immunostaining for COX-2 demonstrates that COX-2 is expressed constitutively in CA3 pyramidal neurons in WT mice (B1), but not in the cKOs (B2). One day after pilocarpine-induced SE, COX-2 induction is observed in WT pyramidal neurons (B3), but not in cKO (B4). In all four panels, a pattern of nonneuronal COX-2 staining is preserved in the nCOX-2 cKO. C, Real time PCR demonstrates that only COX-2 expression from WT mice is enhanced 1 d after SE [Saline WT, n = 5; Pilocarpine (Pilo) WT, n = 6; Saline cKO, n = 5; Pilo cKO, n = 8; **p < 0.001, one-way ANOVA with Bonferroni post-tests]. Saline-treated WTs and nCOX-2 cKOs were not different (p > 0.05), but pilocarpine-treated WTs and nCOX-2 cKOs were significantly different (p < 0.001). D, PCR from DNA extracted from tail demonstrates that WT and cKO mice retain the 2 kb band reflecting the COX-2 gene in the periphery (primer locations shown in A). Scale bar, 10 μm. Error bars indicate SEM.
Unaltered seizure intensity in nCOX-2 cKO
COX-2 is induced in hippocampal neurons within a few hours after SE (Kunz and Oliw, 2001), declining to near basal levels within several days. We allowed animals to experience 1 h of intense continuous seizure. One day after SE, COX-2 protein was highly induced in principal hippocampal neurons of WT mice (Fig 1B3) as expected, but induction was not observed in the nCOX-2 cKO mice (Fig. 1B4), although nonneuronal staining was intact. Normal neuronal COX-2 induction was observed in animals expressing just Synapsin 1 cre or just loxP elements. In addition, 1 d after SE, COX-2 RNA expression in cerebrum was highly induced in WT animals but not in nCOX-2 cKOs (Fig. 1C), demonstrating that most if not all COX-2 induction occurred in the principal neurons. These data together confirm that COX-2 induction after seizures is overwhelmingly neuronal in origin (Yamagata et al., 1993) and demonstrate further that the nCOX-2 cKO mouse lacks constitutive and inducible COX-2 in hippocampal pyramidal neurons and dentate granule cells.
To interpret the phenotypic consequences of SE in the nCOX-2 cKO animals, we must first determine whether seizure intensity itself was altered. To do this, we measured mortality and behavioral and electrographic measures of seizure intensity. The following measures were similar between nCOX-2 cKO and wild-type littermates: mortality during the 60 min period of SE (Fig. 2A), latency to the first behavioral or electrographic seizure (Fig. 2B,E), and the temporal evolution of behavioral seizures following pilocarpine injection (Fig. 2C). Furthermore, the intensity of electrographic seizures during SE [evaluated by the EEG coastline index, modified from Korn et al. (1987)] was not significantly different in the two genotypes (Fig. 2D,F). Thus, ablation of COX-2 limited to principal forebrain neurons did not reduce seizure threshold, in contrast to the effect of global COX-2 ablation, which did (Toscano et al., 2008). The nCOX-2 cKO mouse can thus be used to explore the consequences of SE-triggered neuronal COX-2 induction.
Figure 2.

Wild-type and nCOX-2 cKO mice experienced similar intensity of SE. A, The mortality observed during SE was similar in both genotypes (n = 14 of 31 WT; n = 13 of 27 nCOX-2 KO). B, C, The latency to reach behavioral SE (B) and the temporal evolution of SE (C) as judged by the behavioral seizure score (modified Racine scale) (Borges et al., 2003) were also similar in WT and nCOX-2 cKO mice (WT, n = 14; nCOX-2 KO, n = 13). D–F, EEG recordings (D) also demonstrated similar latency to reach electrographic SE (E; p = 0.44) as well as similar seizure intensity judged by the EEG coastline index (F; p = 0.44). Fisher's exact test was used in A; t tests were used in B, E, and F, with n = 4 in each group. Error bars indicate SEM.
Pyramidal neuron degeneration after SE
Status epilepticus produces a pattern of pyramidal neuron loss in adult rodents that can be quite variable from animal to animal (Buckmaster and Dudek, 1997; Buckmaster and Jongen-Relo, 1999). We used Fluoro-Jade staining to determine whether neuronal COX-2 induction serves an autocrine role in seizure-induced neuron injury of pyramidal cells. After SE, there was intense but patchy Fluoro-Jade staining in the CA1 region. One day after SE, the number of Fluoro-Jade-positive neurons in CA1 was not significantly different in the nCOX-2 cKO mice compared to WT mice (Figs. 3A,B, 4A; p > 0.05), although a strong trend existed for about triple the number of injured neurons in the cKO compared to WT genotype. However, 4 d after SE, nCOX-2 cKO mice exhibited significantly fewer Fluoro-Jade-stained neurons in the CA1 region than WT mice (Figs. 3C,D, 4B; p = 0.0286 by Mann–Whitney test). Most Fluoro-Jade-positive neurons 4 d after SE were colabeled by TUNEL staining (Fig. 3E1–E3), consistent with a predominantly apoptotic mechanism for delayed neurodegeneration. These results suggest an early autocrine neuroprotective role for neuronal COX-2 in the CA1 region of the hippocampus that appears to convert to an injury-producing role within 4 d after SE.
Figure 3.
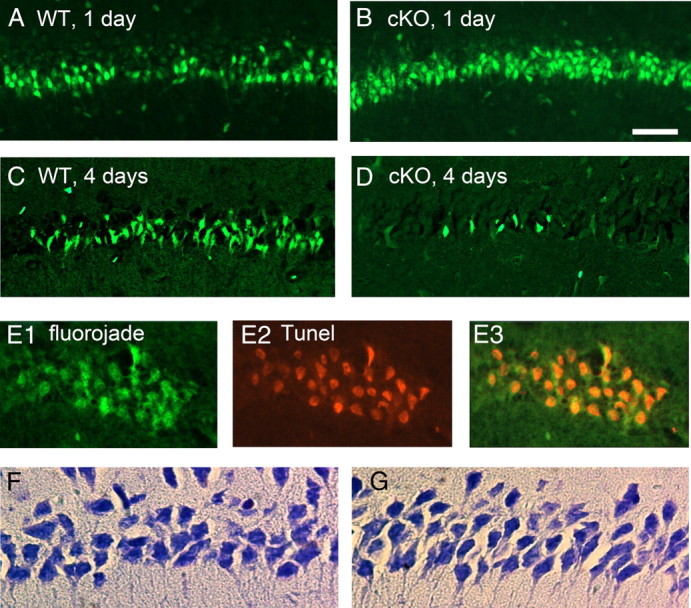
Effect of neuronal COX-2 on neurodegeneration after pilocarpine. A–D, Fluoro-Jade staining in CA1 shows more than twice as many injured neurons in the nCOX-2 cKO 1 d after SE (compare A, B), but far fewer injured neurons 4 d after SE (compare C, D). E, Four days after SE in the WT, Fluoro-Jade and TUNEL staining are colocalized in pyramidal neurons, suggestive of a delayed apoptotic component of neuronal injury. F, G, Nissl stains of CA1 4 months after SE indicate less neuronal loss in the nCOX-2 cKO (G) than WT mice (F). Scale bar, 10 μm.
Inspection of Figure 4, A and B, suggests that between days 1 and 4, the number of Fluoro-Jade-positive neurons in CA1 increases approximately twofold in WT mice, but decreases 25-fold in the cKO. There are several possible explanations for this intriguing observation. First, pyramidal neurons might die early in the COX-2 cKOs and then become quickly phagocytized, such that eventually neurodegeneration is similar in COX-2 cKO and WT. Second, some Fluoro-Jade-positive neurons in the COX-2 cKO might recover by 4 d. Finally, the proteins recognized by Fluoro-Jade have not been identified, and it is possible that the COX-2 cKO downregulates one or more of these proteins after pilocarpine, in which case Fluoro-Jade would not be an appropriate indicator of neuron injury in this genotype. To select among these hypotheses, we performed Nissl stains on brains from mice that experienced 1 h of SE 4 months before being killed, by which time all acute injury would have resolved. Nissl staining demonstrated a significant neurodegeneration of CA1 hippocampal pyramidal neurons in the WT mice but less cell loss in the nCOX-2 cKOs (Figs. 3F,G, 4C; p < 0.001, one-way ANOVA with Bonferroni post hoc tests), demonstrating a clear net neuroprotective effect of the COX-2 cKO in CA1.
These findings suggest that, following rapid neuronal COX-2 induction by SE, an early degree of neuroprotection is overcome by the later appearance of subsequent COX-2-dependent processes that lead to delayed neurodegeneration of CA1 pyramidal cells. Later we explore the role of COX-2 in the inflammatory response following SE, and of EP2 receptor activation in the early neuroprotection.
Degeneration of somatostatin interneurons after SE
In human epileptic hippocampus, or in the days and weeks following SE in rodent epilepsy models produced by pilocarpine or electrical stimulation, GABAergic inhibition onto dentate granule cells is reduced despite the appearance of more inhibitory synapses onto these neurons (Sloviter, 1987; Kapur and Macdonald, 1997; Shumate et al., 1998; Williamson et al., 1999; Doherty and Dingledine, 2001; Cohen et al., 2003; Kobayashi and Buckmaster, 2003; Sun et al., 2007). A large fraction of the somatostatin-expressing interneurons succumb after prolonged seizures (Sloviter, 1987; Buckmaster and Dudek, 1997; Buckmaster and Jongen-Relo, 1999), and loss of this GABAergic neuron population could contribute to dysregulation of synaptic inhibition after SE. We counted neurons in the CA1 stratum oriens and dentate hilus that express somatostatin (Fig. 5B,D, green) 1 d after SE in WT and nCOX-2 cKO mice to investigate COX-2 involvement in seizure-induced death of these highly vulnerable interneurons. We observed the expected significant (p < 0.001) loss of somatostatin-labeled neurons in the CA1 stratum oriens and dentate hilus in WT mice after SE (Fig. 5A,C). However, in the nCOX-2 cKO mice, somatostatin neurons appeared to be nearly completely protected in the stratum oriens after pilocarpine (Fig. 5A), and partially protected in the hilus (Fig. 5B). Interestingly, COX-2 immunoreactivity is undetectable in this population of interneurons (Fig. 6). These data indicate that COX-2 induction in principal neurons after SE triggers degeneration of nearby somatostatin neurons, which themselves do not express COX-2.
Figure 5.

Somatostatin interneurons in the CA1 stratum oriens and dentate hilus are protected 1 d after SE in the nCOX-2 cKO. A, The Abercrombie-corrected number of somatostatin-expressing interneurons per section in the CA1 stratum oriens was significantly reduced 1 d after SE in WT mice, whereas nCOX-2 cKOs had less [nonsignificant (n.s.)] neuron injury. B, Double-label immunohistochemistry shows somatostatin-expressing interneurons (green) in the CA1 stratum oriens; for comparison, neurons are stained with NeuN (red). The separate NeuN and somatostatin stains are shown for three interneurons in the inset. C, Hilar somatostatin-expressing interneurons were partially protected in nCOX-2 cKOs. D, Overlayed somatostatin (green) and NeuN (red) staining in dentate hilus. Scale bars: 10 μm. WT saline, n = 4; WT pilocarpine (pilo), n = 6; cKO saline, n = 5; cKO pilocarpine, n = 5. One-way ANOVA with Bonferroni post hoc tests was used. Error bars indicate SEM.
Figure 6.
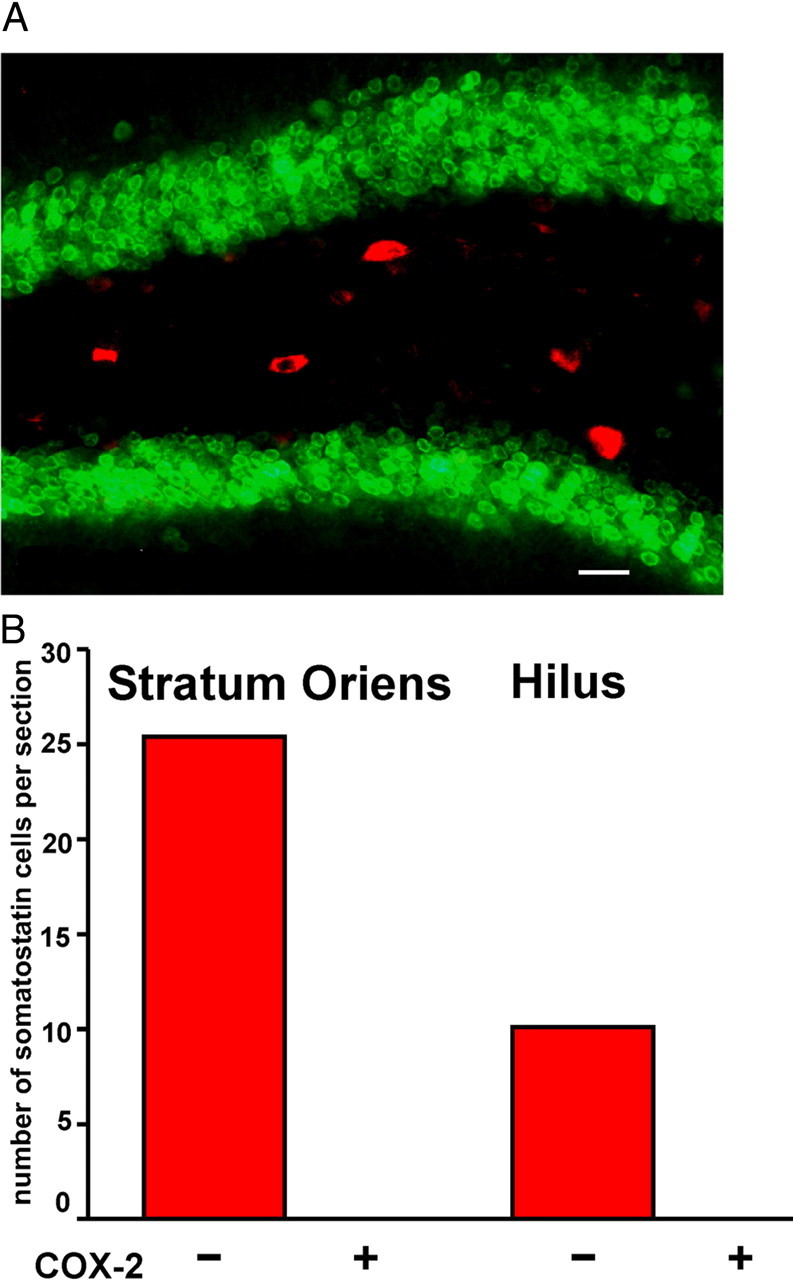
COX2 is not expressed by somatostatin interneurons in WT mice. A, Five hours after SE, double staining for COX-2 (green) and somatostatin (red) in the hilus shows no colocalization. B, Quantification of somatostatin-expressing interneurons that lacked or expressed COX-2 staining in the stratum oriens and hilus 5 h after SE demonstrates that these proteins do not colocalize (n = 4 mice). Scale bar, 10 μm.
Neuroprotection by the EP2 receptor agonist butaprost
A prominent COX-2 product in the brain is PGE2 (Yue et al., 2007), which activates four receptors, EP1, EP2, EP3, and EP4. EP2 receptor activation can be neuroprotective in models of focal ischemia (McCullough et al., 2004), and EP2 receptors are expressed by hippocampal principal neurons (Zhang and Rivest, 1999). Thus, we determined whether the early autocrine neuroprotective role for neuronal COX-2 in the CA1 region of the hippocampus could be mediated through EP2 receptor activation. Within a few minutes of SE termination, rats received 2 μl of vehicle or 1 μm butaprost, a selective EP2 agonist (Abramovitz et al., 2000), directly into the lateral ventricle. As expected, 1 d later, robust neuronal injury was detected with Fluoro-Jade histochemistry in the CA1 region of rats that experienced SE (Fig. 7A), as observed in mice. However, in rats that received the EP2 receptor agonist butaprost, the number of Fluoro-Jade-positive neurons was reduced by 50% (Fig. 7B). No neurodegeneration was observed in sham controls or in rats injected with butaprost without SE. These data identify a neuroprotective role for EP2 receptor activation in CA1 pyramidal cells and support the notion that the mechanism by which neuronal COX-2 induction might cause early neuroprotection could involve EP2 receptor activation by PGE2.
Inflammatory cytokines and cytokine receptors after SE
SE unleashes a florid inflammatory reaction in the brain marked by astrogliosis, microglial activation, leukocyte infiltration, and the attendant production of dozens of cytokines (Borges et al., 2003; Vezzani and Granata, 2005; Ravizza et al., 2008). We used qRT-PCR to measure the abundance of RNA encoding cytokines and cytokine receptors in the cerebrum to determine the breadth of inflammatory events triggered by neuronal COX-2 induction. Variability of expression levels for individual transcripts from animals of the same genotype and treatment was very low, with average coefficients of variation of ΔCT ranging from 3.2 to 1.8%. Four groups of mice were studied: WTs and nCOX-2 cKOs treated with either saline or pilocarpine. Out of 84 transcripts measured, 70 were expressed above background in at least one of the four groups. The relative levels of these cytokines and receptors in WTs and nCOX-2 cKOs were virtually identical in mice that had not experienced SE (Fig. 8C). However, 42 of the 70 were differentially expressed 1 d after SE in the WT genotype [ANOVA comparing the four groups with a Holm–Bonferroni post hoc test (p < 0.05), followed by a Benjamini and Hochberg (1995) correction for multiple comparisons within all 70 genes (p < 0.05)]. Most of these inflammatory mediators (38 of 42) were upregulated after SE in the WT mouse [e.g., a 155-fold increase for CXCL10, a 106-fold increase for chemokine (C-C motif) ligand 4 (CCL4), and 79-fold increase for CCL2]. In the nCOX-2 cKO mouse, upregulation was blunted in 34 of the 38 inflammatory mediators 1 d after SE (Fig. 8A; paired t test, p < 0.001), with seizure-induced upregulation of 11 genes being reduced two- to threefold (i.e., by 50–70%) in the COX2 cKO, including CXCL10, CCL2, CCL3, CCL4, interleukin 11 (IL-11), and CXC chemokine receptor 2 (CXCR2), as depicted in Figure 8B. This experiment demonstrates that neuron-derived COX2 induction contributes significantly to the inflammatory reaction observed after SE.
Figure 8.

Induction of inflammatory cytokines 1 d after SE is less robust in nCOX-2 cKO mice. A, Induction of inflammatory mediators after SE. Most cytokines and other mediators underwent higher induction in WT than in nCOX-2 cKO mice. Transcripts (42) with statistically significant expression changes in WT mice after SE are represented by solid symbols, whereas transcripts that were not differentially expressed after pilocarpine-induced SE are represented by open circles. B, Mean induction of five selected cytokines and the CXCR2 receptor is shown on linear scale. C, Basal expression of inflammatory cytokines was similar in WTs and nCOX-2 cKOs. D, Similar RNA induction of the astrocyte protein, GFAP, was observed 1 d after SE in WTs and nCOX-2 cKOs (p < 0.001, one-way ANOVA with Bonferroni post hoc tests). E, The microglial protein IBA1 was not induced in either group 1 d after SE. WT saline, n = 5; WT pilocarpine (pilo), n = 6; cKO saline, n = 5; cKO pilo, n = 8. Error bars indicate SEM. n.s., Nonsignificant.
We took two approaches to investigate whether blunted cytokine induction in the nCOX-2 cKO was correlated with features of glial activation related to altered morphology. We first measured the RNA levels of astrocytic GFAP and microglia protein IBA1. One day after SE, GFAP expression was highly induced in both genotypes (p < 0.001), with no significant difference between WTs and nCOX-2 cKOs (Fig. 8D). No difference was observed in IBA1 mRNA expression 1 d after SE in either genotype (Fig. 8E), consistent with the later appearance of microgliosis after pilocarpine (Borges et al., 2003). Next, we measured GFAP and IBA1 protein expression 4 d after SE and found that nCOX-2 cKO mice had significantly less gliosis as measured by GFAP and IBA1 protein expression (Fig. 9A–F), particularly in the cell layers. These data together indicate that neuron-derived COX-2 induction exacerbates inflammation and later frank gliosis after brain injury caused by SE.
Figure 9.

Blunted inflammatory reaction 4 d after SE in nCOX-2 cKO mice. A–D, GFAP (green) and IBA1 (red) were used as astrocyte and microglial markers, respectively. Hoechst staining (blue) was used to visualize cell nuclei and distinguish the hippocampal CA1 pyramidal layer. E, F, Four days after SE, there was more astrocyte (E) and microglia (F) activation in WT animals than in nCOX-2 cKOs, as judged by measuring the overall stain intensities across sections (n = 4 each group). G, RNA encoding the leukocyte-specific adhesion molecule, L-selectin, was significantly induced in the cerebrum in WT mice 1 d after SE, but not in nCOX-2 cKOs (n = 12). H, The levels of albumin in the forebrains of saline-perfused mice were measured by Western blot 4 d after SE. Serum albumin was significantly higher in the brains of WT mice but not nCOX-2 cKOs. WT saline (sal), n = 3; WT pilocarpine (pilo), n = 5; cKO sal, n = 3; cKO pilo, n = 4. One-way ANOVA with Bonferroni post hoc tests was used. Error bars indicate SEM.
Blood–brain barrier integrity after SE
The blood–brain barrier is opened after pilocarpine-induced SE (Marchi et al., 2009), and the ensuing penetration of serum albumin into the parenchymal brain tissue has been suggested to trigger or exacerbate the process of epileptogenesis (Ivens et al., 2007). Based on our finding of reduced gliosis after SE in the nCOX-2 cKO, we explored the role of neuronal COX-2 in leukocyte infiltration and albumin leakage into the brain after SE. We first measured by qRT-PCR the mRNA level in cerebrum of a cell adhesion protein expressed selectively by leukocytes, L-selectin (Springer, 1994). Most interesting was the finding that L-selectin expression in cerebrum is increased fivefold in WT mice (p < 0.05) 1 d after SE, but not at all in the nCOX-2 cKO (Fig. 9G), consistent with our finding of blunted expression of the chemokines CCL2, CCL3, and CCL4 at that time (Fig. 8B). Second, 4 d after SE, we perfused mice with saline to wash the blood out of the brain and performed Western blots for albumin in the cerebrum. As expected, the albumin protein level was increased about fourfold in WT mice; however, in the nCOX-2 cKOs, albumin did not penetrate the brain (Fig. 9H). These findings strongly suggest that neuronal COX-2 signaling initiates the breakdown of the blood–brain barrier after pilocarpine-induced SE, but the mechanism of this effect and whether it is a direct action on the neurovascular unit or indirectly mediated by COX-2-dependent neuronal injury or inflammation remain to be determined.
Discussion
We now realize that the brain is immunologically active, communicating directly with the peripheral immune system as well as performing its own surveillance and repair activities. COX-2 mediates inflammation in the periphery and, in the brain, regulates signaling pathways involved in synaptic plasticity. Although COX-2 is highly inducible in select populations of forebrain neurons, this enzyme is also expressed by microglia, capillary endothelial cells, and leukocytes that infiltrate the injured brain (Parfenova et al., 1997; Laflamme et al., 1999; Yamagata et al., 2001; Campuzano et al., 2008; Li et al., 2009), so it is important to identify the cells that mediate each COX-2 effect. We created a conditional knock-out mouse that lacks COX-2 in selected forebrain neurons beginning postnatally and used it to uncover the role of neuron-derived COX-2 in seizure-induced pathology. Although seizure intensity is increased in the global COX-2 cKO or when COX-2 is pharmacologically inhibited before pilocarpine administration (Baik et al., 1999; Toscano et al., 2008), the conditional ablation of COX-2 limited to selected forebrain neurons had no noticeable effect on mortality during SE or seizure onset or severity. Thus, meaningful interpretation of the effects of the conditional ablation on postseizure events is possible.
SE in man and rodents can trigger a series of molecular and cellular events that eventually culminate in the appearance of spontaneous seizures, i.e., epilepsy. These events include a multidimensional local inflammatory reaction primarily involving activated microglia and infiltrating leukocytes, but also reactive astrocytes and neurons themselves, as well as selective neuronal degeneration, selective axonal sprouting, new synapse formation, neurogenesis, and a myriad of changes in synaptic efficacy in the hippocampus. One day after SE, there was a trend toward increased numbers of Fluoro-Jade-stained neurons in the CA1 pyramidal region in the nCOX-2 cKO, implying a potential early neuroprotective role of COX-2 signaling pathways for these neurons. However, 4 d after SE, the delayed neuronal injury in CA1 was drastically reduced in nCOX-2 cKO mice. Furthermore, when we analyzed the population of CA1 pyramidal neurons 4 months after SE, there was significant (p < 0.001) loss of neurons in WT mice, but not in nCOX-2 cKO mice. We conclude that COX-2 signaling results in early neuroprotection of CA1 pyramidal neurons, which then transforms later into degeneration and more profound neurotoxicity that persists. Early nCOX-2-dependent neuroprotection might be mediated by autocrine EP2 activation by PGE2, because intraventricular injection in rats of the selective EP2 agonist, butaprost, 1 d after SE was neuroprotective by approximately the same degree as observed in nCOX-2 cKOs (compare Figs. 4A, 7C).
We found striking neuroprotection of somatostatin interneurons of the CA1 stratum oriens and dentate hilus after SE in the nCOX-2 cKO mouse (Fig. 5), which is interesting because these neurons do not themselves express COX-2 (Fig. 6). Therefore, COX-2 induction in nearby pyramidal and granule cells during or after SE appears to precipitate neurodegeneration in this highly susceptible GABAergic interneuron population. Two possibilities for COX-2 involvement in their neurodegeneration can be raised. First, COX-2-derived prostaglandins produced and released by nearby pyramidal or granule neurons could act in a paracrine fashion to activate receptors on these interneurons. For example, EP1 receptor activation by PGE2, leading to disruption of intracellular Ca2+ buffering, has been shown to mediate neurodegeneration in mice after NMDA-induced injury and cerebral ischemia (Kawano et al., 2006). Alternatively, enhanced membrane excitability and synaptic transmission of pyramidal and granule cells produced by PGE2 (Chen and Bazan, 2005; Sang et al., 2005; Yang and Chen, 2008) could have resulted in increased excitatory input to the interneurons, resulting in excitotoxic death. Conditional ablation of PGE2 receptors in somatostatin interneurons is needed to pursue these two alternatives.
COX-2 inhibition after an insult can be both proinflammatory (Blais et al., 2005; Wang et al., 2007; Aid et al., 2008, 2009) and anti-inflammatory (Kyrkanides et al., 2002; Candelario-Jalil et al., 2007; Funakoshi-Tago et al., 2008), but the myriad of cell types expressing COX-2 has limited our ability to reconcile these findings. One day after SE, cytokine induction was broadly but moderately (two- to threefold) reduced in nCOX-2 cKO compared with WT mice (Fig. 8A), implying that neuron-derived COX-2 contributes to a proinflammatory response after seizures. By contrast, systemic COX-2 inhibition or global COX-2 ablation increases the lipopolysaccharide-induced expression of IL-1β, TNF-α, and other inflammatory mediators in the brain (Aid et al., 2008), just the opposite of the effect of ablating neuronal COX-2. The substantial upregulation of chemokines that attract macrophages (CCL12, CCL2, CCL3, CCL7, CXCL10) and of cytokines (35-fold for IL-11, 17-fold for TNF-α) is blunted in the nCOX-2 cKO. Many of these cytokines are produced largely in astrocytes or especially microglia (Johnstone et al., 1999; Hanisch, 2002; Vezzani and Granata, 2005; Vezzani and Baram, 2007), highlighting the value of examining the effect of cell-specific COX-2 ablations and pointing to a critical cooperation between neurons and glia in the neuroinflammation that follows SE. The observation that microglial-derived cytokines are induced before evidence of IBA1 induction (compare Figs. 8A,E, 9F) is consistent with the notion that all manifestations of neuroinflammation need not develop at a uniform rate.
One day after SE, mRNA encoding the leukocyte cell adhesion protein, L-selectin, was observed in the cerebrum of WT mice but was absent in the nCOX-2 cKO. Four days after SE, there was significantly less gliosis in the nCOX-2 cKO and no excess albumin in the brain parenchyma. These data together suggest that neuronal COX-2 signaling also triggers processes that mediate breakdown of the blood–brain barrier after SE.
Whether nCOX-2-mediated inflammation and breakdown of the blood–brain barrier are causes or consequences of neurodegeneration is not clear, nor is the source of the COX-2-dependent cytokine release. One day after SE in the COX-2 cKO mouse, we observed (1) blunted cytokine release but no molecular evidence for altered microglial morphology or differential astrogliosis (Fig. 8), and (2) protection of somatostatin interneurons (Fig. 5) but a trend toward more injury of CA1 pyramidal neurons (Figs. 3, 4), suggesting that these early effects might be independent consequences of COX-2 ablation. On the fourth day after SE, by contrast, there is clear evidence for less pyramidal cell injury (Fig. 3, 4) and less gliosis in the COX-2 cKO (Fig. 9); net neuroprotection in the COX-2 cKO was confirmed by Nissl stains performed 4 months after SE. Considering these temporal patterns, we suggest that COX-2-dependent inflammatory reactions arising 1 d after SE in response to acute excitotoxic neuronal injury can nudge injured pyramidal neurons to die during the ensuing days, resulting in further glial activation. This hypothesis, if supported by further experiments, would reinforce the benefits of blunting inflammation early after SE.
We cannot compare these results directly with SE induction in the global COX-2 cKO because global ablation increases the severity of seizures (Toscano et al., 2008), which itself would be expected to intensify the inflammatory response. However, global ablation of COX-2 aggravated the neuroinflammation induced by lipopolysaccharide (Aid et al., 2008; Choi et al., 2009). These findings together suggest that in the brain neuronal COX-2 might exert a net proinflammatory effect, whereas nonneuronal COX-2 (microglia, endothelial cells, and/or lymphocytes) can be net anti-inflammatory.
In summary, by identifying multiple specific roles for neuronal COX-2 on neuronal survival, neuroinflammation, and blood–brain barrier integrity, we have initiated a deeper understanding of the consequences of COX-2 induction in principal forebrain neurons after SE. The dual role of cyclooxygenase—combating early excitotoxicity but promoting delayed inflammation-mediated secondary neurotoxicity—has also been reported in ischemia models (Andreasson, 2010). Recognition of the time sequence of processes mediated by neuronal COX-2 is essential to developing rational therapies targeted to COX-2 signaling pathways. Extension of the conditional knock-out approach to astrocytes, endothelial cells, and other cell types, and investigation of the mechanism by which COX-2 signaling jeopardizes blood–brain barrier integrity, will provide a more systematic picture of how this multifunctional signaling enzyme regulates higher circuit function in the hippocampus.
Footnotes
This work was supported by the CounterACT Program, National Institutes of Health Office of the Director, and NINDS Grant U01NS058158 (R.D.), and by NIH Grants F32-NS064695 (G.E.S.), T32-DA015040 (A.R.), and P01 HL062250 (G.F.).
References
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Abramovitz M, Adam M, Boie Y, Carrière M, Denis D, Godbout C, Lamontagne S, Rochette C, Sawyer N, Tremblay NM, Belley M, Gallant M, Dufresne C, Gareau Y, Ruel R, Juteau H, Labelle M, Ouimet N, Metters KM. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- Aid S, Langenbach R, Bosetti F. Neuroinflammatory response to lipopolysaccharide is exacerbated in mice genetically deficient in cyclooxygenase-2. J Neuroinflammation. 2008;5:17. doi: 10.1186/1742-2094-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aid S, Silva AC, Candelario-Jalil E, Choi SH, Rosenberg GA, Bosetti F. Cyclooxygenase-1 and -2 differentially modulate lipopolysaccharide-induced blood-brain barrier disruption through matrix metalloproteinase activity. J Cereb Blood Flow Metab. 2009;30:370–380. doi: 10.1038/jcbfm.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson K. Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediat. 2010;91:104–112. doi: 10.1016/j.prostaglandins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik EJ, Kim EJ, Lee SH, Moon C. Cyclooxygenase-2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res. 1999;843:118–129. doi: 10.1016/s0006-8993(99)01797-7. [DOI] [PubMed] [Google Scholar]
- Balosso S, Ravizza T, Perego C, Peschon J, Campbell IL, De Simoni MG, Vezzani A. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann Neurol. 2005;57:804–812. doi: 10.1002/ana.20480. [DOI] [PubMed] [Google Scholar]
- Baran H, Vass K, Lassmann H, Hornykiewicz O. The cyclooxygenase and lipoxygenase inhibitor BW755C protects rats against kainic acid-induced seizures and neurotoxicity. Brain Res. 1994;646:201–206. doi: 10.1016/0006-8993(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hoechberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Series B. 1995;57:289–300. [Google Scholar]
- Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54:358–368. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- Blais V, Turrin NP, Rivest S. Cyclooxygenase 2 (COX-2) inhibition increases the inflammatory response in the brain during systemic immune stimuli. J Neurochem. 2005;95:1563–1574. doi: 10.1111/j.1471-4159.2005.03480.x. [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Borges K, McDermott DL, Dingledine R. Reciprocal changes of CD44 and GAP-43 expression in the dentate gyrus inner molecular layer after status epilepticus in mice. Exp Neurol. 2004;188:1–10. doi: 10.1016/j.expneurol.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Borges K, McDermott D, Irier H, Smith Y, Dingledine R. Degeneration and proliferation of astrocytes in the mouse dentate gyrus after pilocarpine-induced status epilepticus. Exp Neurol. 2006;201:416–427. doi: 10.1016/j.expneurol.2006.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boztug K, Carson MJ, Pham-Mitchell N, Asensio VC, DeMartino J, Campbell IL. Leukocyte infiltration, but not neurodegeneration, in the CNS of transgenic mice with astrocyte production of the CXC chemokine ligand 10. J Immunol. 2002;169:1505–1515. doi: 10.4049/jimmunol.169.3.1505. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate-treated rats. J Comp Neurol. 1997;385:385–404. [PubMed] [Google Scholar]
- Buckmaster PS, Jongen-Relo AL. Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J Neurosci. 1999;19:9519–9529. doi: 10.1523/JNEUROSCI.19-21-09519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano O, Castillo-Ruiz MM, Acarin L, Castellano B, Gonzalez B. Distinct pattern of microglial response, cyclooxygenase-2, and inducible nitric oxide synthase expression in the aged rat brain after excitotoxic damage. J Neurosci Res. 2008;86:3170–3183. doi: 10.1002/jnr.21751. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Leon OS, Fiebich BL. Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood-brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J Neurochem. 2007;100:1108–1120. doi: 10.1111/j.1471-4159.2006.04280.x. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Li M, Liu L, Savarin C, Ransohoff RM. Chemokines in and out of the central nervous system: much more than chemotaxis and inflammation. J Leukoc Biol. 2008;84:587–594. doi: 10.1189/jlb.1107763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Bazan N. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;93:929–941. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- Cho J, Nelson TE, Bajova H, Gruol DL. Chronic CXCL10 alters neuronal properties in rat hippocampal culture. J Neuroimmunol. 2009;207:92–100. doi: 10.1016/j.jneuroim.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Aid S, Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol Sci. 2009;30:174–181. doi: 10.1016/j.tips.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciceri P, Zhang Y, Shaffer AF, Leahy KM, Woerner MB, Smith WG, Seibert K, Isakson PC. Pharmacology of celecoxib in rat brain after kainate administration. J Pharmacol Exp Ther. 2002;302:846–852. doi: 10.1124/jpet.302.3.846. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Lin DD, Quirk GL, Coulter DA. Dentate granule cell GABA(A) receptors in epileptic hippocampus: enhanced synaptic efficacy and altered pharmacology. Eur J Neurosci. 2003;17:1607–1616. doi: 10.1046/j.1460-9568.2003.02597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Dingledine R. Reduced excitatory drive onto interneurons in the dentate gyrus after status epilepticus. J Neurosci. 2001;21:2048–2057. doi: 10.1523/JNEUROSCI.21-06-02048.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin CD, Craig GT. Stereological quantification of histological parameters in normal hamster cheek pouch epithelium. Arch Oral Biol. 1978;23:337–345. doi: 10.1016/0003-9969(78)90090-0. [DOI] [PubMed] [Google Scholar]
- Funakoshi-Tago M, Shimizu T, Tago K, Nakamura M, Itoh H, Sonoda Y, Kasahara T. Celecoxib potently inhibits TNFalpha-induced nuclear translocation and activation of NF-kappaB. Biochem Pharmacol. 2008;76:662–671. doi: 10.1016/j.bcp.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Aronica E, Lopes da Silva FH. Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. Eur J Neurosci. 2001;13:657–669. doi: 10.1046/j.1460-9568.2001.01428.x. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Hoesche C, Sauerwald A, Veh RW, Krippl B, Kilimann MW. The 5′-flanking region of the rat synapsin I gene directs neuron-specific and developmentally regulated reporter gene expression in transgenic mice. J Biol Chem. 1993;268:26494–26502. [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. The role of cytokines and growth factors in seizures and their sequelae. Prog Neurobiol. 2001;63:125–149. doi: 10.1016/s0301-0082(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Jiang J, Ganesh T, Du Y, Thepchatri P, Rojas A, Lewis I, Kurtkaya S, Li L, Qui M, Serrano G, Shaw R, Sun A, Dingledine R. Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor. Proc Natl Acad Sci U S A. 2010;107:2307–2312. doi: 10.1073/pnas.0909310107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone M, Gearing AJ, Miller KM. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol. 1999;93:182–193. doi: 10.1016/s0165-5728(98)00226-4. [DOI] [PubMed] [Google Scholar]
- Jung KH, Chu K, Lee ST, Kim J, Sinn DI, Kim JM, Park DK, Lee JJ, Kim SU, Kim M, Lee SK, Roh JK. Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol Dis. 2006;23:237–246. doi: 10.1016/j.nbd.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Chung JI, Lee SH, Jung YS, Moon CH, Baik EJ. Involvement of endogenous prostaglandin F2alpha on kainic acid-induced seizure activity through FP receptor: the mechanism of proconvulsant effects of COX-2 inhibitors. Brain Res. 2008;1193:153–161. doi: 10.1016/j.brainres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz T, Oliw EH. The selective cyclooxygenase-2 inhibitor rofecoxib reduces kainate-induced cell death in the rat hippocampus. Eur J Neurosci. 2001;13:569–575. doi: 10.1046/j.1460-9568.2001.01420.x. [DOI] [PubMed] [Google Scholar]
- Kyrkanides S, Moore AH, Olschowka JA, Daeschner JC, Williams JP, Hansen JT, Kerry O'Banion M. Cyclooxygenase-2 modulates brain inflammation-related gene expression in central nervous system radiation injury. Brain Res Mol Brain Res. 2002;104:159–169. doi: 10.1016/s0169-328x(02)00353-4. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-kappaB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Kaur C, Lu J, Sivakumar V, Dheen ST, Ling EA. Expression of cyclooxygenase-2 and microsomal prostaglandin-E synthase in amoeboid microglial cells in the developing brain and effects of cyclooxygenase-2 neutralization on BV-2 microglial cells. J Neurosci Res. 2009;88:1577–1594. doi: 10.1002/jnr.22319. [DOI] [PubMed] [Google Scholar]
- Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197–208. doi: 10.1016/s0092-8674(00)80402-x. [DOI] [PubMed] [Google Scholar]
- Marcheselli VL, Bazan NG. Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus. Inhibition by a platelet-activating factor antagonist. J Biol Chem. 1996;271:24794–24799. doi: 10.1074/jbc.271.40.24794. [DOI] [PubMed] [Google Scholar]
- Marchi N, Fan Q, Ghosh C, Fazio V, Bertolini F, Betto G, Batra A, Carlton E, Najm I, Granata T, Janigro D. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcon J, Gagliardi B, Balosso S, Maroso M, Noe F, Morin M, Lerner-Natoli M, Vezzani A, Ravizza T. Age-dependent vascular changes induced by status epilepticus in rat forebrain: implications for epileptogenesis. Neurobiol Dis. 2009;34:121–132. doi: 10.1016/j.nbd.2008.12.018. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, Smithies O. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Murray HJ, O'Connor JJ. A role for COX-2 and p38 mitogen activated protein kinase in long-term depression in the rat dentate gyrus in vitro. Neuropharmacology. 2003;44:374–380. doi: 10.1016/s0028-3908(02)00375-1. [DOI] [PubMed] [Google Scholar]
- Obenaus A, Esclapez M, Houser CR. Loss of glutamate decarboxylase mRNA-containing neurons in the rat dentate gyrus following pilocarpine-induced seizures. J Neurosci. 1993;13:4470–4485. doi: 10.1523/JNEUROSCI.13-10-04470.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. 2006;47:1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- Parfenova H, Eidson TH, Leffler CW. Upregulation of COX-2 in cerebral microvascular endothelial cells by smooth muscle cell signals. Am J Physiol. 1997;273:C277–C288. doi: 10.1152/ajpcell.1997.273.1.C277. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. New York: Academic; 2001. [Google Scholar]
- Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–160. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Reese J, Zhao X, Ma WG, Brown N, Maziasz TJ, Dey SK. Comparative analysis of pharmacologic and/or genetic disruption of cyclooxygenase-1 and cyclooxygenase-2 function in female reproduction in mice. Endocrinology. 2001;142:3198–3206. doi: 10.1210/endo.142.7.8307. [DOI] [PubMed] [Google Scholar]
- Rempe D, Vangeison G, Hamilton J, Li Y, Jepson M, Federoff HJ. Synapsin I Cre transgene expression in male mice produces germline recombination in progeny. Genesis. 2006;44:44–49. doi: 10.1002/gene.20183. [DOI] [PubMed] [Google Scholar]
- Sandhya TL, Ong WY, Horrocks LA, Farooqui AA. A light and electron microscopic study of cytoplasmic phospholipase A2 and cyclooxygenase-2 in the hippocampus after kainate lesions. Brain Res. 1998;788:223–231. doi: 10.1016/s0006-8993(97)01552-7. [DOI] [PubMed] [Google Scholar]
- Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–9870. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Shumate MD, Lin DD, Gibs JW, III, Holloway LI, Coulter DA. GABA(A) receptor function in epileptic human dentate granule cells: comparison to epileptic and control rat. Epilepsy Res. 1998;32:114–128. doi: 10.1016/s0920-1211(98)00045-x. [DOI] [PubMed] [Google Scholar]
- Slanina K, Schweitzer P. Inhibition of cyclooxygenase-2 elicits a CB1-mediated decrease of excitatory transmission in rat CA1 hippocampus. Neuropharmacology. 2005;49:653–659. doi: 10.1016/j.neuropharm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Sollas AL, Barbaro NM, Laxer KD. Calcium-binding protein (calbindin-D28K) and parvalbumin immunocytochemistry in the normal and epileptic human hippocampus. J Comp Neurol. 1991;308:381–396. doi: 10.1002/cne.903080306. [DOI] [PubMed] [Google Scholar]
- Springer. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Sun C, Mtchedlishvili Z, Bertram EH, Erisir A, Kapur J. Selective loss of dentate hilar interneurons contributes to reduced synaptic inhibition of granule cells in an electrical stimulation-based animal model of temporal lobe epilepsy. J Comp Neurol. 2007;500:876–893. doi: 10.1002/cne.21207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemiya T, Maehara M, Matsumura K, Yasuda S, Sugiura H, Yamagata K. Prostaglandin E2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neurosci Res. 2006;56:103–110. doi: 10.1016/j.neures.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Toscano CD, Kingsley PJ, Marnett LJ, Bosetti F. NMDA-induced seizure intensity is enhanced in COX-2 deficient mice. Neurotoxicology. 2008;29:1114–1120. doi: 10.1016/j.neuro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardeh D, Wang D, Costigan M, Lazarus M, Saper CB, Woolf CJ, Fitzgerald GA, Samad TA. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J Clin Invest. 2009;119:287–294. doi: 10.1172/JCI37098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Baram TZ. New roles for interleukin-1 beta in the mechanisms of epilepsy. Epilepsy Curr. 2007;7:45–50. doi: 10.1111/j.1535-7511.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- Vlkolinsky R, Siggins GR, Campbell IL, Krucker T. Acute exposure to CXC chemokine ligand 10, but not its chronic astroglial production, alters synaptic plasticity in mouse hippocampal slices. J Neuroimmunol. 2004;150:37–47. doi: 10.1016/j.jneuroim.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J, Zhang H, Hui Y, Cheng Y, Petrenko N, Yu Y, FitzGerald GA. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci U S A. 2009;106:7548–7552. doi: 10.1073/pnas.0805806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cao MJ, Pan GT, Lu YL, Rong TZ. RNA editing of mitochondrial functional genes atp6 and cox2 in maize (Zea mays L.) Mitochondrion. 2009b;9:364–369. doi: 10.1016/j.mito.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Wang XM, Wu TX, Hamza M, Ramsay ES, Wahl SM, Dionne RA. Rofecoxib modulates multiple gene expression pathways in a clinical model of acute inflammatory pain. Pain. 2007;128:136–147. doi: 10.1016/j.pain.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Spencer DD. Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol. 1999;45:92–99. doi: 10.1002/1531-8249(199901)45:1<92::aid-art15>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J Neurosci. 2001;21:2669–2677. doi: 10.1523/JNEUROSCI.21-08-02669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Chen C. Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des. 2008;14:1443–1451. doi: 10.2174/138161208784480144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue H, Jansen SA, Strauss KI, Borenstein MR, Barbe MF, Rossi LJ, Murphy E. A liquid chromatography/mass spectrometric method for simultaneous analysis of arachidonic acid and its endogenous eicosanoid metabolites prostaglandins, dihydroxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and epoxyeicosatrienoic acids in rat brain tissue. J Pharm Biomed Anal. 2007;43:1122–1134. doi: 10.1016/j.jpba.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HJ, Sun RP, Lei GF, Yang L, Liu CX. Cyclooxygenase-2 inhibitor inhibits hippocampal synaptic reorganization in pilocarpine-induced status epilepticus rats. J Zhejiang Univ Sci B. 2008;9:903–915. doi: 10.1631/jzus.B0820018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Rivest S. Distribution, regulation and colocalization of the genes encoding the EP2- and EP4-PGE2 receptors in the rat brain and neuronal responses to systemic inflammation. Eur J Neurosci. 1999;11:2651–2668. doi: 10.1046/j.1460-9568.1999.00682.x. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]