

Abstract
The symptoms of Mn-induced neurotoxicity resemble those of Parkinson’s diseases. Since iron (Fe) appears to play a pivotal role in pathophysiology of Parkinson’s disease, we set out to test the hypothesis that alterations in Fe-requiring enzymes such as aconitase contribute to Mn-induced neurotoxicity. Mitochondrial fractions prepared from rat brain were preincubated with MnCl2 in vitro, followed by the enzyme assay. Mn treatment significantly inhibited mitochondrial aconitase activity (24% inhibition at 625 μM to 81% at 2.5 mM, p < 0.05). The inhibitory effect was reversible and Mn-concentration dependent, and was reversed by the addition of Fe (0.05–1 mM) to the reaction mixture. In an in vivo chronic Mn exposure model, rats received intraperitoneal injection of 6 mg/kg Mn as MnCl2 once daily for 30 consecutive days. Mn exposure led to a region-specific alteration in total aconitase (i.e., mitochondrial + cytoplasmic): 48.5% reduction of the enzyme activity in frontal cortex (p < 0.01), 33.7% in striatum (p < 0.0963), and 20.6% in substantia nigra (p < 0.139). Chronic Mn exposure increased Mn concentrations in serum, CSF, and brain tissues. The elevation of Mn in all selected brain regions (range between 3.1 and 3.9 fold) was similar in magnitude to that in CSF (3.1 fold) rather than serum (6.1 fold). The present results suggest that Mn alters brain aconitase activity, which may lead to the disruption of mitochondrial energy production and cellular Fe metabolism in the brain.
Keywords: Manganese, Aconitase, Mitochondria, Iron, Respiratory chain, Parkinson’s diseases
1. Introduction
Adverse health effects of manganese (Mn) have been associated with organic Mn compounds (pesticides) and inorganic Mn [8,30,40]. Recently, several countries including the United States have replaced tetraethyl lead (Pb) in gasoline with a Mn-containing antiknock compound methylcyclopentadienyl Mn tricarbonyl (MMT). The addition of MMT to gasoline supply has raised concerns about public health risks associated with the inevitable increase in the environmental levels of Mn. In fact, several studies have reported that Mn causes significant health hazard in heavily air-polluted areas [7,21,34].
A relationship between Mn intoxication and Parkinsonism has long been recognized [2,23,39]. Both human idiopathic Parkinson’s disease (IPD) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism in animals are associated with abnormal mitochondrial function, accumulation of iron (Fe) in the substantia nigra, and biochemical evidence of oxidative stress [15,20,42]. It has been postulated that cellular Fe overload in the substantia nigra may catalyze the generation of reactive oxygen species and enhance lipid peroxidation. Such an Fe-mediated oxidative stress may ultimately lead to the degeneration of nigrostriatal dopamine neurons in IPD patients [15,42].
The neurodegenerative damage in Mn intoxication differs from that of IPD. While the neurologic lesions of Mn intoxication occur in the globus pallidus and striatum of the basal ganglia, IPD is associated with damages mainly in dopaminergic neurons of the substantia nigra [26,27,41]. However, at the cellular and subcellular levels, Mn may directly interact with Fe since Mn shares numerous similarities with Fe: (a) both are transition elements adjacent to each other in the Periodic Table; (b) both carry similar valent charges (2 + and 3 +) in physiological conditions; (c) they have similar ionic radii; (d) both strongly bind transferrin (Tf) [1,38]; and (e) intracellularly, they are extensively transported into mitochondria [9,10,12]. Because of these similarities, we hypothesized that Mn may interfere with Fe-dependent cellular functions, particularly with mitochondrial and cytosolic [4Fe-4S]proteins, such as aconitase.
Both mitochondrial and cytosolic aconitases contain a unique [4Fe-4S] cubane cluster in their active catalytic sites, with one particularly labile Fe atom [16,29]. Mitochondrial aconitase (ACO2) catalyzes the interconversion of L-citrate and isocitrate in the tricarboxylic acid cycle. Cytoplasmic aconitase (ACO1), on the other hand, functions to regulate cellular Fe homeostasis. When the cellular Fe level is insufficient, cytoplasmic aconitase loses the fourth labile Fe and assumes a [3Fe-4S] configuration. In that state, the enzyme forfeits its enzymatic activity and binds with high affinity to mRNAs that contain an iron responsive element (IRE) stem-loop structure, i.e., to the mRNAs of the major proteins in Fe metabolism including ferritin, transferrin receptor, δ-aminolevulinic acid synthase, succinate dehydrogenase, and even ACO2 [3,16,17]. The net result of these interactions is a down-regulation of cellular Fe utilization and up-regulation of cellular Fe uptake. Since the coordination chemistry of Mn closely resembles that of Fe, we hypothesized that Mn may interact with Fe in both mitochondrial and cytoplasmic aconitases to alter cellular energy metabolism and Fe regulation.
A primary objective of this study, therefore, was to test the hypothesis that Mn exposure may alter the activity of mitochondrial aconitase in vitro. We chose mitochondrial aconitase as our initial enzyme target because Mn has been shown to accumulate exclusively in mitochondria [11]. Along the same line of investigation, we established a chronic Mn exposure model in rats to test the hypothesis that in vivo Mn exposure may also alter total aconitase activity (ACO1 + ACO2) in selected brain regions. In addition, we characterized the distribution of Mn in rat brain and cerebrospinal fluid (CSF) in this chronic Mn exposure model.
2. Materials and methods
2.1. Materials
Chemicals were obtained from the following sources: manganese chloride (MnCl2 · 4H2O), cis-aconitic acid, ferrous ammonium sulfate [Fe(NH4)2(SO4)2 · 6H2O], L-citric acid, isocitric acid, triethanolamine-HCl, Triton X-100, sucrose, β-nicotinamide adenine dinucleotide phosphate (β-NADP), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), phenazine methosulfate (PMS), isocitrate dehydrogenase, and purified porcine aconitase (30 units/g) from Sigma Chemical Co., St. Louis, MO; NH4H2PO4 from Aldrich Chemical Co., Milwaukee, WI; standard Mn for atomic absorption spectrophotometry from Alfa Products, Danvers, MA. All reagents were of analytical grade, HPLC grade or the best available pharmaceutical grade.
2.2. Preparation of brain mitochondrial fractions
Rat brains were removed from the skull, rinsed with 0.9% saline, and homogenized (1 g: 5 ml) in 0.25 M sucrose and 10 mM Tris, pH 7.4 on ice. The homogenates were centrifuged at 650 × g at 4°C for 10 min to remove the nuclei and membrane debris. The supernatants were further centrifuged at 25 000 × g at 4°C for 15 min. The pellet was resuspended in homogenizing buffer and centrifuged at 25 000 × g at 4°C for another 15 min. At the end of the centrifugation, the pellet containing mitochondrial fractions was resuspended in 0.1 M Tris, pH 8.0 and stored at −70°C [6].
Prior to the enzyme assay, the mitochondrial preparations were repeatedly thawed and frozen for at least three times, followed by sonication on ice using a Model-250 Sonifer at duty cycle 20 and output 3.5 for 10 pulses to break mitochondrial membranes. This procedure has proven effective in destroying mitochondrial membrane to yield a high enzyme activity. The preparations were then centrifuged at 13 600 × g for 5 min at 4°C. The supernatants were allowed to stand at room temperature for at least one hour prior to the enzyme assay. The prepared mitochondrial fractions were stored at −70°C and usually assayed within a week.
2.3. Aconitase activity assay
Aconitase catalyzes the interconversion of L-citrate and isocitrate, via cis-aconitate:
The activity of aconitase in our in vitro studies was assayed by determining the rate of formation of the intermediate product, cis-aconitate, from the substrate L-citrate. The mitochondrial fractions (10–30 μg proteins) were pretreated with various concentrations of Mn in a buffer consisting of 40 mM HEPES (pH 7.5) and 10 mM cysteine in a total volume of 0.1 ml at 22°C for 10 min. To achieve a measurable reaction velocity, the assay requires preactivation by Fe [13]. Thus, the preparations were incubated with 50 μM Fe2 + (as ferrous ammonium sulfate) in the same buffer at 22°C for another 10 min. The enzymatic reaction was initiated by adding 30 μl of the pretreated samples in a total of 1 ml assay buffer containing 20 mM triethanolamine-HCl (pH 7.5) and 1.0 mM L-citrate. The changes of absorbance at 240 nm were recorded for 5 min by a Perkin-Elmer Lambda-11 spectrophotometer and used for the calculation of the enzyme activity. Aconitase activity was expressed as the formation of nmol cis-aconitate/mg protein/min. All samples were run in duplicates. The method has a good reproducibility and is suitable for studying kinetic properties of aconitase.
Since our in vivo studies involved a large number of tissue samples from animals, there was a need to develop a rapid assay procedure. Thus, we subsequently established a method based on the conversion of cis-aconitate to isocitrate. The formed isocitrate was further catalyzed by isocitrate dehydrogenase, in presence of NADP, Fe2 +, MTT and PMS, to form a photochrome species [18]. The reaction was initiated by adding 10 μl of diluted tissue homogenates (20–30 μg protein) into a glass tube containing 1 ml of freshly made reaction solution, which consisted of 10 mM aconitate, 60 mM Mg2 +, 0.1 mg/ml β-NADP, 0.1 mg/ml MTT, 25 μg/ml PMS, 10 μg/ml Fe(NH4)2(SO4)2, and 0.04 unit/ml isocitrate dehydrogenase in 150 mM Tris, pH 8.0. Following thorough vortex mixing, the reaction was allowed to continue in a 37°C water bath for 30 min, and the absorbance at 575 nm was recorded. A standard curve of aconitase was constructed by adding known amounts (units) of purified porcine aconitase into the reaction solution and determining the absorbance under the same experimental conditions as described above. One unit of the enzyme activity was defined as the rate of conversion of 1 μmol aconitate to isocitrate/mg protein/min at 37°C. This method has proven to be sensitive and convenient for determination of tissue aconitase activity.
2.4. In vivo animal studies
Male Sprague–Dawley rats (Harlan, Indianapolis, IN), aged 20–22 days, weighing 30–50 g upon arrival, were assigned to two groups at 26–28 days of age such that the group mean body weights were comparable. The animals were housed in a temperature-controlled, 12/12 light/dark room, and allowed to have free access to pelleted rat chow (Teklad 4% Mouse-Rat Diet, Teklad, Madison, WI) and distilled, deionized water. At the age of 28–32 days, the animals began to receive the intraperitoneal injections of 6 mg/kg of Mn as MnCl2 once daily between 9:30am and 10:30am for 30 consecutive days. This dose regimen was chosen because it was known to be associated with a significant reduction of succinic dehydrogenase (another [4Fe-4S] enzyme) in rat brain [32,33,35]. The injection solution was prepared by dissolving MnCl2 in sterile saline (6 mg Mn/ml). For the control group, the animals received the daily injections of the equivalent volume of sterile saline.
Twenty-four hour after the last injection, rats were anesthetized with pentobarbital (50 mg/kg, i.p.). CSF samples were obtained through a 26-gauge needle inserted between the protruberance and the spine of the atlas, and were free of blood [44]. Blood samples were collected from the inferior vena cava. Rat brains were then removed from the skull and placed on a filter paper, saturated with saline, which rested on an ice-chilled glass plate. Striatum, substantia nigra, hippocampus, and frontal cortex were dissected out, the wet weights recorded, and frozen at −70°C for determination of enzyme activity and for analysis of Mn contents.
2.5. Atomic absorption spectrophotometry(AAS)analysis
Mn concentrations in the CSF, serum, and brain tissues were determined by a flameless graphite furnace AAS [24]. For Mn in CSF and serum, aliquots (50 μl) of CSF and serum were diluted with an appropriate volume of 8% Triton X-100 and 5% EDTA in distilled, deionized water prior to AAS. For Mn in tissues, the samples (30–50 mg) were combusted in acid-washed crucibles at 800°C for 4–6 h. The crucibles were then rinsed with 1.0 ml of washing solution consisting of 40 mM HCl and 8% Triton X-100 in distilled, deionized water. The samples were further diluted, if necessary, with the washing solution so as to keep the absorbance reading within the linear ranges of the measurement.
A Perkin-Elmer Model 3030 Zeeman atomic absorption spectrophotometer, equipped with an HGA-600 graphite furnace, was used for quantification. The detection limit for this method was 0.2 ng Mn/ml of assay solution.
2.6. Protein assay and statistics
All protein concentrations were determined by the method of Bradford [4] using a Bio-Rad Protein Assay Kit (Bio-Rad Lab, Richmond, CA). Bovine serum albumin was used as the standard.
Statistical analyses of the differences between groups were performed by using Student’s t test. The differences between two means were considered significant if p values were equal or less than 0.05.
3. Results
3.1. Mn inhibition of aconitase activities in vitro
When rat brain mitochondrial fractions were incubated with various concentrations of Mn, the aconitase activity was significantly decreased in a Mn concentration-dependent fashion (Fig. 1). The presence of Mn across a concentration range of 0.625 to 2.5 mM produced a linear decline in enzyme activities (r = 0.993, p < 0.05). At the concentration of 2.5 mM or higher, the inhibitory effect of Mn on the aconitase was maximal, about 19% of the control values. Further increase in Mn concentrations did not completely abolish the aconitase activity when the enzyme was preactivated by 50 μM of Fe using our standard in vitro assay procedure.
Fig. 1.

Inhibition by Mn of aconitase activity in mitochondrial fractions prepared from rat brain. Mitochondrial fractions (25 μg) were pretreated with various concentrations of Mn for 10 min followed by Fe (50 μM) activation for another 10 min. The rate of formation of aconitase from L-citrate (1.0 mM) was monitored at 240 nm. Values represent means ± S.D. (n = 3).
3.2. Reversal of Mn inhibitory effect by Fe
Our preliminary studies confirmed that aconitase requires the preactivation with Fe (see Section 2). As depicted in Fig. 2, in the absence of Mn treatment, the activity of aconitase increased as the concentrations of Fe in the reaction mixture was increased. The maximum velocity (Vmax) was achieved at an Fe concentration of 200 μM, with a Michaelis–Menten constant (Km) of 40.2 μM. In the presence of Mn, however, the reaction required Fe concentrations exceeding 1000 μM to restore the Vmax, which was about 93% of the control value. The data suggested that the increase in Fe concentrations in the reaction solution reversed the inhibitory effect of Mn on aconitase activity.
Fig. 2.

Reversal of the Mn-induced inhibition of aconitase by Fe. Mitochondrial fractions (25 μg) were pretreated with or without Mn (2.5 mM) for 10 min followed by Fe activation for 5 min. The rate of formation of aconitase from L-citrate (1.0 mM) was monitored at 240 nm. Values represent means ± S.D. (n = 4).
It was also noted that in a Mn-pretreated mitochondrial preparation, a much steeper Fe concentration–response curve was observed when Fe concentration exceeded 200 μM (Fig. 2), suggesting a competitive interaction between Fe and Mn at higher Fe-concentration range. However, in the lower Fe-concentration range (< 200 μM), the reversal effect of Fe on Mn appeared to arise slowly. This is likely due to an uncompetitive interaction (i.e. not acting on the same binding sites in the enzyme) between Fe and Mn on the enzyme. Thus, it appeared likely that in addition to acting directly on Fe binding, Mn influenced the substrate (citrate) binding as well.
3.3. Interaction with L-citrate
In order to substantiate the possibility that Mn may alter the binding of L-citrate to the enzyme, we studied the kinetics properties of Mn action using a purified aconitase (Sigma). Use of the purified aconitase, rather than mitochondrial fractions, allowed the study to examine substrate (L-citrate) binding, since the mitochondrial preparations usually contain substantial L-citrate. The results presented in Fig. 3 reveal that incubation of aconitase with Mn, followed by Fe activation at 50 μM, altered the Km of the reaction when L-citrate was used as the substrate. To reach the half-maximum velocity, the concentrations of L-citrate were raised from 0.50 mM in the controls to 1.07 mM in the Mn-treated groups. However, the Vmax (35.5 nmol/mg/min) of the enzymatic reaction could be reestablished when the excess amounts of the citrate were added to the Mn-treated samples. Thus, it appeared that Mn also interrupted L-citrate binding.
Fig. 3.

Reversal of the Mn-induced inhibition of aconitase by L-citrate. Purified enzyme (0.2 mg) was pretreated with 2.5 mM Mn followed by Fe (50 μM) activation for 10 min. The rate (V, nmol/mg/min) of formation of aconitase from L-citrate (S, mM) was monitored at 240 nm. Each point represents mean of three separate experiments.
3.4. Mn inhibition of aconitase activities in vivo
The effect of Mn on cellular aconitase activities was further evaluated in rats chronically exposed to Mn by i.p. injections of 6 mg Mn/kg for 30 days. Mn exposure led to a region-specific reduction in total aconitase (ACO1 + ACO2) activities in brain tissue (Fig. 4). In the striatum, reportedly a target for Mn neurotoxicity, Mn exposure caused a marked reduction (approximately 60% of controls) in aconitase activity, which approached, but did not attain statistical significance (p < 0.096). The same treatment caused a 48.5% reduction of the enzyme in frontal cortex (p < 0.01), 20.6% in substantia nigra (p < 0.139), and 19.3% in hippocampus (p < 0.323).
Fig. 4.

Effect of chronic exposure to Mn on total aconitase (ACO1 + ACO2) activities in selected brain regions. Rats received i.p. injections of 6 mg/kg Mn as MnCl2 once daily for 30 days. The whole tissue homogenates (20–30 μg proteins) were used for assay of aconitase activity at day 31. Data represent mean ± S.E. (n = 4–6). * *: p < 0.01, †: p = 0.096.
3.5. Brain distribution of Mn
Mn concentrations in normal control rats were detectable in serum, CSF, and in various brain regions. Following chronic Mn administration, the concentrations of Mn in serum and CSF were elevated by 6.1 and 3.1 fold, respectively, as compared to controls (Table 1). The concentrations of Mn in striatum, hippocampus, and frontal cortex were also significantly increased after long-term, repeated dosing of MnCl2 (Table 1). It is interesting to note that the magnitude of increase in Mn concentrations in all selected brain regions (range between 3.1 and 3.9 folds) was similar to that of CSF.
Table 1.
Concentrations of Mn in serum, CSF, and selected brain regions of Sprague–Dawley rats following chronic exposure to Mn
| Magnitude | Control | Mn-treated | Increase |
|---|---|---|---|
| Serum (ng/ml) | 5.90 ± 3.20 | 36.0 ± 17.0* | 6.1 |
| CSF (ng/ml) | 3.63 ± 1.85 | 11.2 ± 4.79** | 3.1 |
| Striatum (μg/g) | 0.52 ± 0.59 | 1.62 ± 0.82* | 3.1 |
| Hippocampus (μg/g) | 0.39 ± 0.20 | 1.54 ± 0.75* | 3.9 |
| Substantia nigra (μg/g) | 0.66 ± 0.30 | 2.21 ± 1.59 | 3.4 |
| Frontal cortex (μg/g) | 0.33 ± 0.14 | 1.18 ± 0.40** | 3.6 |
Rats received i.p. injection of 6 mg/kg Mn (as MnCl2) once daily for 30 consecutive days.
Mn concentrations were determined by flameless AAS.
Data represent means ± S.D., n = 3–4 for controls and n = 6 for Mn-treated group.
p < 0.05,
p < 0.01 as compared to the control.
4. Discussion
The present studies demonstrate that Mn exposure significantly inhibits mitochondrial aconitase activity. The inhibitory effect is Mn-concentration dependent and is reversible by subsequent Fe treatment. The results from our in vivo studies further substantiate that chronic exposure to Mn induces a marked reduction of total aconitase activities in the brain regions associated with Parkinsonism.
Aconitase requires Fe as a co-factor in its active center to catalyze reactions involved in mitochondrial energy production. The proposed interaction between Mn and Fe in biological systems is presumably due to similarities of both metals in their coordination chemistry. As a complete [4Fe-4S]cluster in aconitase is necessary for the enzyme to bind citrate, the replacement of Mn for the fourth labile Fe in the cubane structure would inhibit the enzyme’s catalytic function [3,17]. Our data corroborate that the rate of aconitase-catalyzed reaction varies as a function of the Fe concentrations of the reaction mixture. When excess Fe was added to Mn-pretreated mitochondrial fractions, Fe effectively revived the aconitase activities. Thus, it appears likely that the inhibitory effect of Mn on mitochondrial aconitase is due, at least in part, to the competition of Mn on Fe binding sites of the enzyme. The fourth, highly labile Fe binding site of the [4Fe-4S] cube is known to be involved in citrate binding as well. It is therefore not surprising that Mn exposure also altered citrate binding to purified pig aconitase enzyme. Collectively, we postulate that Mn suppresses the mitochondrial aconitase activity by altering the binding of Fe and/or L-citrate to the enzyme. The proposed interaction between Mn and Fe is shown below.
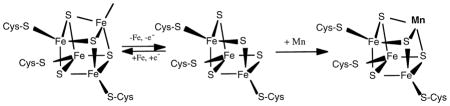
Our in vitro studies also show that the inhibition of mitochondrial aconitase requires a relatively high concentration of Mn in the reaction mixture. A possible explanation for this extraordinarily high Mn required for inhibition may relate to the active species of Mn in the reaction system. It is unclear which form of Mn is associated with toxicity, i.e., Mn2 + or Mn3 +. In our experiments, Mn was added to the buffers as the divalent salt, MnCl2. While it is reasonable to suggest that the effect on aconitase activities was associated with the divalent oxidation species of Mn2 +, our study does not preclude the possibility that a Mn species of higher oxidation state, such as Mn3 +, is required for the induction of these effects. From a chemical point of view, Mn3 +, whose radius is 65 ppm, possesses a similar ionic size to Fe3 + (65 ppm at the high spin state) in aconitasen [25,37]. We speculate that the higher oxidation state of Mn may be the preferred reactive species that fit into the geometric space of aconitase and alters enzyme activity. Under such a condition, the rate-limiting step of the toxic action may be the oxidation of Mn2 + to Mn3 +. This is a slow, oxygen-dependent process [1]. Hence, Mn-induced toxicity may become apparent only at high concentrations of MnCl2. Alternatively, the high amount of Mn could be due to the binding of Mn to cellular proteins, or due to the short course of exposure (10-min incubation) in these in vitro studies.
Inhibition of mitochondrial aconitase by Mn could be associated with the reduction of energy production in the targeted brain areas. In IPD, the bulk of evidence supports the view that oxidative stress and defects in energy metabolism are causally related to neuronal cell death [15,42]. For example, the failure of mitochondrial energy supplies has been linked to MPTP/MPP +-induced neurotoxicity [15,28]. Similar to MPP +, Mn preferentially accumulates in the mitochondria, apparently due to extremely slow efflux of Mn from mitochondria [9,12]. The mitochondrial ligands that bind to Mn remain uncertain; but it is clear that Mn accumulation in mitochondria decreases the rate of ADP-stimulated respiration, suggesting a direct effect of Mn on energy production [10]. Our results support this notion and further suggest that the alteration in mitochondrial energy production may also be related to an effect of Mn on Fe-requiring enzymes in respiratory chain.
Our in vivo Mn studies demonstrate a brain region-specific reduction of total aconitase activity, which represents the sum of mitochondrial and cytoplasmic aconitase. Since cytoplasmic aconitase is responsible for post-transcriptional regulation of the major proteins of Fe metabolism, it is possible that change in cytoplasmic aconitase activity may lead to altered intracellular Fe. Previous studies have indeed revealed that chronic exposure to Mn (20 mg MnCl2/ml in drinking water) produces a 95% increase of Fe levels in rat striatum [19]. High endogenous Fe has been suggested to potentiate Mn neurotoxicity [36]. The presence of Mn–Tf complex has also been reported to augment the cellular uptake of59 Fe when the comparison was made among various metal–transferrin (Tf) complexes [31]. It has also been found that dietary Mn supplements in rats bring about an increased Fe uptake by the brain, liver, and kidney in a synergistic rather than competitive manner [5]. Furthermore, a recent study by Olanow et al. [26] has demonstrated that Mn intoxication in monkeys causes an elevated Fe deposition in globus pallidus and substantia nigra pars reticulata. Therefore, it appears plausible that Mn exposure, in addition to its direct action on respiratory enzymes, may lead to altered cellular Fe regulation in the basal ganglia, bringing about cellular Fe overload via an effect on cytoplasmic aconitase. The latter hypothesis is currently under investigation in this laboratory.
Following chronic Mn administration, Mn concentrations in selected brain regions were relatively evenly increased (3.1 to 3.9 fold compared to the controls). However, while Mn exposure led to a marked (but not statistically significant) reduction in total aconitase activities in striatum, the same treatment caused a significant reduction of the enzyme in frontal cortex. The limited numbers of animals in each study group may have attenuated the statistical power to reveal the significance in other brain regions. At present, we cannot explain the decrease in aconitase activity in the frontal cortex. It is known, however, that the frontal cortex is actively involved in the motor regulation [22].
In addition, we have noticed that the CSF concentrations of Mn were significantly increased in Mn-treated animals from our chronic studies. Studies by Aposhian’s group [14] illustrate that acute exposure to Mn in rats produces a low Mn concentration in CSF and brain tissues in comparison to many fold increases of Mn in blood and choroid plexus, suggesting a protective function of the choroid plexus in Mn intoxication. The role of the choroid plexus in metal toxicity including Mn has been reviewed elsewhere [43]. It is interesting to note that the increase in brain Mn in the current chronic study was similar in magnitude to that of CSF Mn, but not to serum Mn. This observation supports the view that CSF Mn may serve as the source for Mn deposition in brain tissues.
In conclusion, Mn exposure in vitro significantly inhibits mitochondrial aconitase activity. The inhibitory effect is reversible, Mn-concentration dependent, and possibly due to the competitions of Mn with Fe and/or citrate binding. Chronic Mn exposure produces a region-specific alteration of total aconitase in rat brain. The present results suggest that the alteration of aconitase activity by Mn may lead to the disruption of mitochondrial energy production and cellular Fe metabolism, which may underlie Mn-induced neurotoxicity.
Acknowledgments
The authors gratefully acknowledge the technical assistance of Dr. Qiuqu Zhao. This research was supported in part by grants RO1-ES07042 and P20-ES06831.
Footnotes
Published on the World Wide Web on 3 June 1998.
References
- 1.Aschner M, Aschner JL. Manganese transport across the blood–brain barrier: relationship to iron homeostasis. Brain Res Bull. 1996;24:857–860. doi: 10.1016/0361-9230(90)90152-p. [DOI] [PubMed] [Google Scholar]
- 2.Barbeau A, Inoué N, Cloutier T. Role of manganese in dystonia. Adv Neurol. 1976;14:339–352. [PubMed] [Google Scholar]
- 3.Beinert H, Kennedy MC. Aconitase, a two-faced protein: enzyme and iron regulatory factor. FASEB J. 1993;7:1442–1449. doi: 10.1096/fasebj.7.15.8262329. [DOI] [PubMed] [Google Scholar]
- 4.Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 5.Chua AC, Morgan EH. Effects of iron deficiency and iron overload on manganese uptake and deposition in the brain and other organs of the rat. Biol Trace Elem Res. 1996;55:39–54. doi: 10.1007/BF02784167. [DOI] [PubMed] [Google Scholar]
- 6.Clark JB, Nicklas WJ. The metabolism of rat brain mitochondria. J Biol Chem. 1970;245:4724–4731. [PubMed] [Google Scholar]
- 7.Cooper WC. The health implications of increased manganese in the environment resulting from the combustion of fuel additives: a review of the literature. J Toxicol Environ Health. 1984;14:23–46. doi: 10.1080/15287398409530561. [DOI] [PubMed] [Google Scholar]
- 8.Ferraz HB, Bertolucci PHF, Pereira JS, Lima JGC, Andrade LAF. Chronic exposure to the fungicide maneb may produce symptoms and signs of CNS manganese intoxication. Neurology. 1988;38:550–553. doi: 10.1212/wnl.38.4.550. [DOI] [PubMed] [Google Scholar]
- 9.Gavin CE, Gunter KK, Gunter TE. Manganese and calcium efflux kinetics in brain mitochondria. Relevance to manganese toxicity. Biochem J. 1990;266:329–334. doi: 10.1042/bj2660329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavin CE, Gunter KK, Gunter TE. Mn2 + sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol Appl Pharmacol. 1992;115:1–5. doi: 10.1016/0041-008x(92)90360-5. [DOI] [PubMed] [Google Scholar]
- 11.Gavin CE, Gunter TE. Manganese dynamics in brain mitochondria. In: Yasui M, Strong MJ, Ota K, Verity MA, editors. Mineral and Metal Neurotoxicology. CRC Press; New York: 1996. pp. 305–310. [Google Scholar]
- 12.Grafstein B, Forman DS. Intracellular transport in neurons. Physiol Rev. 1980;60:1167–1283. doi: 10.1152/physrev.1980.60.4.1167. [DOI] [PubMed] [Google Scholar]
- 13.Henson CP, Cleland WW. Purification and kinetic studies of beef liver cytoplasmic aconitase. J Biol Chem. 1967;17:3833–3838. [PubMed] [Google Scholar]
- 14.Ingersoll RT, Montgomery EB, Aposhian HV. Central nervous system toxicity of manganese. I. Inhibition of spontaneous motor activity in rats after intrathecal administration of manganese chloride. Fundam Appl Toxicol. 1995;27:106–113. doi: 10.1006/faat.1995.1113. [DOI] [PubMed] [Google Scholar]
- 15.Jenner P, Schapira AH, Marsden CD. New insights into the cause of Parkinson’s disease. Neurology. 1992;42:2241–2250. doi: 10.1212/wnl.42.12.2241. [DOI] [PubMed] [Google Scholar]
- 16.Kennedy MC, Emptage MH, Dreyer JL, Beinert H. The role of iron in the activation–inactivation of aconitase. J Biol Chem. 1983;258:11098–11105. [PubMed] [Google Scholar]
- 17.Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: The control of cellular iron metabolism. Cell. 1993;72:19–28. doi: 10.1016/0092-8674(93)90046-s. [DOI] [PubMed] [Google Scholar]
- 18.Koen AL, Goodman M. Aconitase hydratase isozymes: subcellular location, tissue distribution and possible subunit structure. Biochim Biophys Acta. 1969;191:698–701. doi: 10.1016/0005-2744(69)90363-5. [DOI] [PubMed] [Google Scholar]
- 19.Lai JC, Chan AW, Leung TK, Minski MJ, Lim L. Neurochemical changes in rats chronically treated with a high concentration of manganese chloride. Neurochem Res. 1992;17:841–847. doi: 10.1007/BF00993259. [DOI] [PubMed] [Google Scholar]
- 20.Loeffler DA, Connor JR, Juneau PL, Snyder BS, Kanaley L, DeMaggio AJ, Nguyen H, Brickman CM, LeWitt PA. Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J Neurochem. 1995;65:710–724. doi: 10.1046/j.1471-4159.1995.65020710.x. [DOI] [PubMed] [Google Scholar]
- 21.Loranger S, Zayed J. Environmental and occupational exposure to manganese: a multimedia assessment. Int Arch Occup Environ Health. 1995;67:101–110. doi: 10.1007/BF00572233. [DOI] [PubMed] [Google Scholar]
- 22.Martin JH, editor. Neuroanatomy. Appleton and Lange; Stamford: 1989. pp. 323–350. [Google Scholar]
- 23.Mena I, Court J, Fuenzalida S, Papavasiliou PS, Cotzias GC. Modification of chronic manganese poisoning. Treatment with L-dopa or 5-OH tryptophane. New Engl J Med. 1970;282:5–10. doi: 10.1056/NEJM197001012820102. [DOI] [PubMed] [Google Scholar]
- 24.Neve J, Leclercq N. Factors affecting determinations of manganese in serum by atomic absorption spectrometry. Clin Chem. 1991;37:723–728. [PubMed] [Google Scholar]
- 25.Nieboer E, Fletcher GG. Determinants of reactivity in metal toxicology. In: Chang LW, editor. Toxicology of Metals. CRC Press; New York: 1996. pp. 113–132. [Google Scholar]
- 26.Olanow CW, Good PF, Shinotoh H, Hewitt KA, Vingerhoets F, Snow BJ, Beal MF, Calne DB, Perl DP. Manganese intoxication in the rhesus monkey: a clinical, imaging, pathologic, and biochemical study. Neurology. 1996;46:492–498. doi: 10.1212/wnl.46.2.492. [DOI] [PubMed] [Google Scholar]
- 27.Pentschew A, Ebner FF, Kovatch RM. Experimental manganese encephalopathy in monkeys. J Neuropathol Exp Neurol. 1963;22:488–499. doi: 10.1097/00005072-196307000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Ramsay RR, Singer TP. Energy-dependent uptake of N-methyl-4-phenylpyridinium, the neurotoxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, by mitochondria. J Biol Chem. 1986;261:7585–7587. [PubMed] [Google Scholar]
- 29.Robbins AH, Stout CD. Iron–sulfur cluster in aconitase, crystallographic evidence for a three-iron center. J Biol Chem. 1985;260:2328–2333. [PubMed] [Google Scholar]
- 30.Roels H, Lauwerys R, Buchet JP, Genet P, Sarhan MJ, Hanotiau I, deFays M, Bernard A, Stanescu D. Epidemiological survey among workers exposed to manganese: effects on lung, central nervous system, and some biological indices. Am J Ind Med. 1987;11:307–327. doi: 10.1002/ajim.4700110308. [DOI] [PubMed] [Google Scholar]
- 31.Seligman PA, Chitambar C, Vostrejs M, Moran PL. The effects of various transferrins on iron utilization by proliferating cells. Ann NY Acad Sci. 1988;526:136–140. doi: 10.1111/j.1749-6632.1988.tb55499.x. [DOI] [PubMed] [Google Scholar]
- 32.Seth PK, Husain R, Mushtaq M, Chandra SV. Effect of manganese on neonatal rat: Manganese concentration and enzymatic alterations in brain. Acta Pharmacol Toxicol. 1977;40:553–560. [PubMed] [Google Scholar]
- 33.Seth PK, Hong JS, Kilts CD, Bondy SC. Alteration of cerebral neurotransmitter receptor function by exposure of rats to manganese. Toxicol Lett. 1981;9:247–254. doi: 10.1016/0378-4274(81)90157-0. [DOI] [PubMed] [Google Scholar]
- 34.Sierra P, Loranger S, Kennedy G, Zayed J. Occupational and environmental exposure of automobile mechanics and nonautomotive workers to airborne manganese arising from the combustion of methylcyclopentadienyl manganese tricarbonyl (MMT) Am Ind Hyg Assoc J. 1995;56:713–716. doi: 10.1080/15428119591016746. [DOI] [PubMed] [Google Scholar]
- 35.Singh J, Husain R, Tandon SK, Seth PR, Chandra SV. Biochemical and histopathological alterations in early manganese toxicity in rats. Environ Physiol Biochem. 1974;4:16–23. [PubMed] [Google Scholar]
- 36.Sloot WN, Sluijs-Gelling AJ, Gramsbergen JBP. Selective lesions by manganese and extensive damage by iron after injection into rat striatum or hippocampus. J Neurochem. 1994;62:205–216. doi: 10.1046/j.1471-4159.1994.62010205.x. [DOI] [PubMed] [Google Scholar]
- 37.Sneed NC, Maynard JL, Brasted RC. Comprehensive Inorganic Chemistry. Nostrand; New York: 1953. p. 68. [Google Scholar]
- 38.Suarez N, Eriksson H. Receptor-mediated endocytosis of a manganese complex of transferrin into neuroblastoma (SHSY5Y) cells in culture. J Neurochem. 1993;61:127–131. doi: 10.1111/j.1471-4159.1993.tb03546.x. [DOI] [PubMed] [Google Scholar]
- 39.Tepper LB. Hazards to health: Manganese. New Engl J Med. 1961;264:347–348. doi: 10.1056/NEJM196102162640710. [DOI] [PubMed] [Google Scholar]
- 40.Wang DX, Zhou WM, Wang SZ, Zheng W. Occupational exposure to manganese in welders and associated neurodegenerative diseases in China. Toxicol Sci. 1998;42:24. [Google Scholar]
- 41.Yamada M, Ohno S, Okayasu I, Hatakeyama S, Watanabe H, Ushio K, Tsukagoshi H. Chronic manganese poisoning: a neuropathological study with determination of manganese distribution in the brain. Acta Neuropathol. 1986;70:273–278. doi: 10.1007/BF00686083. [DOI] [PubMed] [Google Scholar]
- 42.Youdim MBH, Ben-Shachar D, Riederer P. The possible role of iron in the etiopathology of Parkinson’s disease. Mov Disord. 1993;8:1–12. doi: 10.1002/mds.870080102. [DOI] [PubMed] [Google Scholar]
- 43.Zheng W. The choroid plexus and metal toxicities. In: Chang LW, editor. Toxicology of Metals. C.R.C. Press; New York, NY: 1996. pp. 605–622. [Google Scholar]
- 44.Zheng W, Perry DF, Nelson DL, Aposhian HV. Protection of cerebrospinal fluid against toxic metals by the choroid plexus. FASEB J. 1991;5:2188–2193. doi: 10.1096/fasebj.5.8.1850706. [DOI] [PubMed] [Google Scholar]