

Abstract
ALKBH1 is a mammalian AlkB homolog that possesses abasic site (AP) lyase activity. The AP lyase reaction is catalyzed by imine formation with an active site Lys, and a covalent intermediate can be trapped in the presence of NaBH4. Surprisingly, ALKBH1 also forms a stable protein-DNA adduct in the absence of a reducing agent. Experiments with different substrates demonstrated that the protein covalently binds to the 5′ DNA product; i.e., the fragment containing an α,β-unsaturated aldehyde. The amino terminal domain of ALKBH1 was identified as the main site of linkage with DNA. By contrast, mutagenesis studies suggest that the primary catalytic residue forming the imine linkage is Lys133, with Lys154 and other Lys residues in this region serving in opportunistic roles. These findings confirm the classification of ALKBH1 as an AP lyase, identify the primary and a secondary Lys involved in the lyase reaction, and demonstrate the protein forms a covalent adduct with the 5′ DNA product. We propose two plausible chemical mechanisms to account for the covalent attachment.
Keywords: Abasic site, Lyase, Schiff base, Covalent Protein-DNA Adduct, Demethylase
INTRODUCTION
Genomic DNA is constantly exposed to endogenous and exogenous damaging agents that cause a variety of lesions, resulting in more than 10,000 apyrimidinic/apurinic (AP or abasic) sites produced per human cell each day [1–3]. AP sites, which leave the deoxyribose of the DNA backbone intact, can arise spontaneously by hydrolytic cleavage of the N-glycosidic bond or they can form as intermediates during DNA repair. For example, monofunctional glycosylases of the base excision repair (BER) pathway remove alkylated and oxidized nucleic acid bases thereby generating AP sites; in mammals, these sites are mainly recognized by the AP endonuclease Ape1 leading to single strand breaks. Alternatively, bifunctional glycosylases remove the damaged base and cleave the DNA strand to directly produce single strand breaks. These breaks are processed and then religated by the concerted action of polymerase β and ligase III [4–6].
Enzymes that cleave at AP sites are divided into two distinct categories, as illustrated in Figure 1 [2, 7]. AP hydrolases such as Ape1 contain an active site Mg2+ and cleave the DNA on the 5′ side of the AP site, producing a new 3′-OH group and 5′-deoxyribophosphate. In contrast, AP/deoxyribophosphate (dRP) lyases incise the DNA on the 3′ side of the lesion either by catalyzing a β-elimination leading to a 3′-α,β-unsaturated aldehyde and a 5′-phosphate (as shown in Figure 1) or by a β,δ-elimination producing 5′-phosphate and 3′-phosphate products, along with a 4-oxo-2-pentenal fragment [5]. Both types of AP/dRP lyase enzymes form an imine intermediate between an amino acid nucleophile of the protein and the C1′ atom of the deoxyribose. Members of the Nth group, such as Escherichia coli endonuclease III (EndoIII), catalyze β-elimination and use an internal active site Lys as the nucleophile [8, 9]. In contrast, members of the formamidopyrimidine DNA glycosylase (Fpg)/Nei family exhibit β,δ-elimination activity and form the intermediate using an N-terminal Pro or Val residue, as has been reported for E. coli Fpg [10] and the mouse ortholog NEIL3 [11]. For either lyase mechanism, the Schiff base intermediate can be trapped with a strong reducing agent such as NaBH4 and the resulting stable DNA-protein species can be visualized by SDS-PAGE.
Figure 1. Comparison of AP hydrolase and AP lyase reactions.

AP sites can be cleaved by an AP hydrolase reaction (top) or by an AP lyase reaction acting on the open-ring aldehyde form of the deoxyribose (bottom).
Recently, several mammalian proteins other than glycosylases have been shown to possess AP/dRP lyase activities, expanding the role of this reaction beyond the BER pathway. Ku, for example, recognizes DNA double strand breaks (DSBs) in the non-homologous end joining pathway and nicks DNA at AP sites near the DNA breaks [12]. In addition, poly(ADP-ribose) polymerase-1 [13], the human ribosomal protein S3 [14], and histone H4 [15] exhibit AP lyase activity. Finally, relevant to the present investigation, human AlkB homolog 1 (ALKBH1) is an AP lyase [16].
ALKBH1 is an ortholog of E. coli AlkB [17], an Fe2+/2-oxoglutarate dependent dioxygenase that catalyzes the oxidative demethylation of 1-methyl adenine and 3-methyl cytosine in single stranded DNA with the concomitant decarboxylation of 2-oxoglutarate to succinate [18, 19]. The DNA demethylase specificity of the human protein is more limited, since it only acts on single stranded 3-methyl cytosine [20]. In addition, this protein is reported to be a histone H2A demethylase [21]. ALKBH1’s AP lyase activity does not require Fe2+ or 2-oxoglutarate and is unaffected by mutation of the putative metal-binding residues [16]. Of use for a facile assay, ALKBH1 can introduce DSBs into oligonucleotides containing AP sites in close proximity on opposing strands [16]. AP lyase activity also was seen in Alkbh1 of Saccharomyces pombe [22].
In this study, we demonstrate that ALKBH1 covalently binds to the 5′ product of its lyase activity as an irreversible protein-DNA adduct. The site of DNA linkage was localized to the amino terminal region of the protein and shown to be separate from the key lyase active site residue. Trapping experiments previously had shown that the lyase reaction proceeds through a Schiff base intermediate, suggesting an active site Lys. By mutational studies we identify the primary Lys responsible for this chemistry and demonstrate that alternative Lys residues are able to substitute for catalyzing the lyase reaction.
EXPERIMENTAL
Construction of plasmids and mutants
Plasmid pBAR67 encodes a His6-tagged version of human ALKBH1 [23]. C-terminal His6-tagged ALKBH1 was created by PCR amplifying the alkbh1 gene from pBAR67 with primers 5′-gcgcggcagccccatggggaagatg-3′ and 5′-ctgtagatcctctcgaggctgtgaggg-3′ (introducing NcoI and XhoI restriction sites at the start and at the end, respectively), while also removing the stop codon. The product was cloned into pET28b using the newly introduced restriction sites and sequence verified (Davis Sequencing). Mutants were created by using the QuickChange Site-Directed mutagenesis kit (Qiagen) with pBAR67 as a template. Primers were purchased from Integrated DNA Technologies and all sequences were confirmed (Davis Sequencing).
Overexpression and Purification
N- and C-terminal His6-tagged ALKBH1 and their variants were overproduced and purified as previously described [16]. For selected studies, the N-terminal His6-tag was removed by incubation with thrombin (0.1 U per 5 – 10 μg ALKBH1; Sigma-Aldrich) for 20 min at room temperature, resulting in protein containing three extra residues derived from the linker compared to the native enzyme. The cleavage was verified by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) analysis. To physically remove the His6-tag, the mixture was loaded onto a Ni-nitrilotriacetic acid Sepharose column (GE Healthcare Bio-Sciences) and the untagged protein was eluted with 50 mM imidazole in 50 mM phosphate buffer containing 300 mM NaCl (pH 8) and immediately subjected to buffer exchange using a disposable desalting column (GE Healthcare Bio-Sciences). When necessary, His6-tagged and untagged ALKBH1-containing samples were concentrated by using an Amicon Ultra-4 centrifugal filter device (Millipore).
Oligonucleotide Substrates
The oligonucleotides (Integrated DNA Technologies) used in the assays are listed in Table 1. Where indicated, the single stranded substrates were radioactively labeled at the 5′-end by incubating with T4 polynucleotide kinase (PNK, New England Biolabs) and [γ-32P]-ATP (Perkin-Elmer) for 1 h at 37 °C. To make double stranded substrates, equimolar amounts of the reverse complement oligonucleotides were annealed by incubating at 95 °C and slowly cooling to room temperature. To create the abasic sites, the uracil-containing substrates (10 or 50 μM) were treated with uracil DNA glycosylase (UDG, 15 U per 50 μl assay, New England Biolabs) for 30 min at 37 °C.
Table 1.
Oligonucleotide substrates used in these studies
| Name | Oligonucleotide | Comment |
|---|---|---|
| Oligo1 | 5′-AGT AGA CAG CTA CCA TGC CTG CAC GAA G(dU)T AGC AAT TCG TAA TCA TGG TCA TAG CTA GTA-3′ | 60-mer containing dU |
| Oligo2 | 5′-TAC TAG CTA TGA CCA TGA TTA CGA ATT GCT AG(dU) TTC GTG CAG GCA TGG TAG CTG TCT ACT-3′ | Reverse complement of oligo1, with dU opposite dG in each strand |
| Oligo3 | 5′-AGT AGA CAA G(dU)T ACC ATG CCT GCA CGA AGT T-3′ | 30-mer containing dU |
| Oligo4 | 5′-AAC TTC GTG CAG GCA TGG TAG (dU)TT GTC TAC T-3′ | Reverse complement of oligo3, with dU opposite dG in each strand |
| Oligo5 | 5′-AAC TTC GTG CAG GCA TGG TAG CTT GTC TAC T-3′ | Reverse complement of oligo3, with dC instead of dU |
AP Lyase Activity and Protein-DNA Adduct Formation Assays
Standard AP lyase activity assays were carried out as described previously [16]. Briefly, 5 μM His6-tagged or untagged ALKBH1 was incubated with 1 μM substrate for 1 h at 37 °C. For analysis of protein-DNA adduct formation, the samples were directly loaded onto an 18% native gel, a denaturing urea (7 M) gel, or an SDS-PAGE gel. For product analysis, the samples (10 μl) were treated with 0.4 U fungal proteinase K (Life Technologies) in the presence of 0.5% SDS for 30 min at 65 °C. The substrate and products were separated by either native or denaturing PAGE. Gels containing 32P-labeled substrates were dried and exposed to a Typhoon 9410 phosphorimager (GE Healthcare) to visualize the DNA. Gels using unlabeled DNA were stained with ethidium bromide.
Endopeptidase Digests
ALKBH1 (5 μM) was reacted with the 32P-labeled oligonucleotides (1 μM) for 1 h at 37 °C and then denatured by heating at 95 °C for 2 min. The protein-DNA adducts were incubated with LysC (0.1 μg/2.3 μg protein, Promega) overnight at 37 °C. The reactions were stopped by addition of SDS-PAGE loading dye and heating the samples to 95 °C for 5 min. All samples were analyzed by 15% SDS-PAGE, stained with Coomassie dye, dried overnight, and exposed to a phosphorimager screen.
BNPS-Skatole Cleavage and N-Terminal Sequencing
ALKBH1 (5 μM) was incubated with 7.5 μM unlabeled substrate in a total volume of 100 μl for 1 h at 37 °C. The samples were adjusted to 70% acetic acid containing 0.1% phenol plus 1 mM BNPS-skatole (Sigma-Aldrich) and treated for 72 h at room temperature in the dark. β-Mercaptoethanol was added to a final concentration of 5 mM and the samples were further incubated overnight at room temperature in the dark. All samples were concentrated in a speed vacuum concentrator to about 2 μl, then resuspended in H2O and reduced again (repeated twice). The pellet was resuspended in 10 μl 1x SDS-PAGE buffer and loaded onto a 10–16% Tris-Tricine gel [24]. To sequence the N-termini of peptides of interest, the gel bands were transferred to a 0.2 μm polyvinylidene fluoride membrane (Millipore) for 30 min at 0.35 A, the membrane was stained and destained according to the instructions on the UC Davis website (http://msf.ucdavis.edu/protein_gel_protocols.html), and the appropriate spots were provided to the Edman sequencing facility of UC Davis.
Examination of the 5′-end of the 3′-product
ALKBH1 (5 μM, N-terminal His6-tagged and untagged), EndoIII (10 U, New England Biolabs), and Ape1 (10 U, New England Biolabs) were incubated with unlabeled oligo4 and, where indicated, subjected to proteinase K digestion as described above. All samples were then split in two; one aliquot was treated with Antarctic alkaline phosphatase (5 U) for 1 h at 37 °C followed by inactivation at 65 °C for 30 min, whereas the other aliquot was left untreated and kept on ice. All samples were incubated with PNK and [γ-32P]-ATP for 1 h at 37 °C and analyzed by denaturing urea PAGE (20%). The signals were detected by exposing the dried gel to a phosphorimager.
RESULTS
Protein-DNA Adduct Formation in the Absence of Reducing Agent
A previous study reported that human ALKBH1 possesses AP lyase activity, but the number of AP sites cleaved was, at best, equimolar with the amount of added protein [16]. This near stoichiometric reactivity could arise from a highly stabilized interaction between the protein and its imine-linked product or from covalent attachment of a lyase product to the enzyme, thus resulting in a single turnover reaction. Proteinase K digestion of denatured sample was required in order to visualize product DNA, consistent with an irreversible linkage between the components. Further experiments were carried out to investigate this covalent interaction between ALKBH1 and AP-containing DNA.
As with other Lys-dependent AP lyases, ALKBH1 can be linked to its substrate DNA by NaBH4 reduction of the imine intermediate [16]; unexpectedly however, a protein-DNA signal of greater intensity and greater mobility was observed in ALKBH1 samples incubated with 32P-labeled AP oligonucleotides in the absence of NaBH4 reductant (Fig. 2A). Formation of this distinct species required enzyme, shown by the lack of this band when enzyme was absent, and an AP site in the DNA. Notably, a corresponding band was not generated when using the control protein EndoIII. The absence of NaBH4 ensured that the substrate aldehyde was not partially reduced, thus likely accounting for the increased band intensity compared to the reduced ALKBH1 sample. The greater mobility of the new NaBH4-free species versus that obtained when using NaBH4 suggested that it may contain product DNA, rather than substrate DNA. The faster migrating species also was observed when samples were treated with the less potent reductant NaCNBH3 (data not shown), consistent with less trapping of the intact substrate with the lyase Schiff base intermediate and accompanying formation of the enzyme-DNA product adduct. In addition to the major bands, both ALKBH1 samples also contained more slowly migrating species that may represent two protein molecules binding per double strand oligonucleotide with its dual AP sites. The molecular masses of these protein-DNA adducts cannot be accurately measured by comparison to the protein molecular weight markers, but the relative sizes can be compared. These results were consistent with ALKBH1 forming an adduct with the AP lyase product DNA in the absence of a reducing agent.
Figure 2. ALKBH1 covalently binds AP-containing DNA in the absence of NaBH4 reducing agent.

(A) Formation of an ALKBH1-DNA adduct. N-terminal His6-tagged ALKBH1 (2.5 μM), and EndoIII (10 U) were incubated with the radioactively labeled UDG-treated substrate oligo1+2 (1 μM) for 1 h at 37 °C in the absence and presence of the reducing agent NaBH4 (10 mM). Control lanes lack either lyase. The samples were denatured in a boiling water bath for 3 min prior to resolving by 12% SDS-PAGE and visualizing by autoradiography. The Schiff base intermediate formed with ALKBH1 and EndoIII was trapped with NaBH4 as expected, but ALKBH1 also formed an adduct with the DNA in the absence of the reducing agent. The positions of three protein molecular mass markers (in kDa) are indicated to the left of the gel. (B) AP lyase activity assays with and without proteinase K treatment. His6-tagged ALKBH1 (labeled A, 5 μM) and EndoIII (labeled E, 1 U/assay) were incubated with radioactively labeled UDG-treated oligo1+2 (1 μM) for 1 h at 37 °C. Half of the sample was subjected to proteinase K (prot K, 1 U) digestion at 65 °C for 30 min, whereas the other half was kept on ice. Control lanes did not contain either lyase. All samples were analyzed by denaturing 7 M urea PAGE and the 5′ radioactively labeled DNA species were detected by autoradiography. (C) Time course studies. ALKBH1 was incubated with UDG-treated oligo1+2 for the times indicated and examined directly by 12% SDS-PAGE to assess adduct formation.
ALKBH1 Covalently Binds the 5′ DNA Product in the Absence of Reducing Agent
To extend the above observations, the putative ALKBH1-DNA product adduct formed in the absence of reducing agent was further investigated. When ALKBH1 was incubated with radiolabeled AP-containing substrate and directly loaded onto a denaturing polyacrylamide gel, a signal was detected representing a large species that barely migrated into the matrix (Fig. 2B). In contrast, following digestion with proteinase K the samples released a DNA product with an apparent molecular mass slightly larger than that of EndoIII. This finding indicates that ALKBH1 forms a DNA-product adduct which is stable to the denaturing conditions of urea PAGE analysis and that the 5′-product can only be visualized by digesting the protein (Fig. 2B). The ALKBH1-DNA adduct was stable to boiling, β-mercaptoethanol, and SDS, as seen for a time-dependence series of samples analyzed by using SDS-PAGE (Fig. 2C). Taken together, these results provide additional evidence that a covalent bond is formed between ALKBH1 and the 5′-labeled DNA product.
To further characterize the ALKBH1-DNA adduct formed in the absence of a reducing agent, N-terminal His6-tagged ALKBH1 was incubated with different 32P-labeled AP-containing oligonucleotide substrates and the resulting species were examined. First, the electrophoretic behavior was compared for the adducts derived from single stranded oligonucleotides containing a single AP site located after 10 or 20 nucleotides. The resulting DNA-protein adducts were of different apparent sizes, with the species containing the larger 5′-product migrating more slowly (Fig. 3A, left panel). This confirms that ALKBH1 forms a bond to the 5′-product, i.e. to the fragment containing the α,β-unsaturated aldehyde. A second series of substrates consisted of double stranded DNA containing two abasic sites in close proximity on opposing strands and located at different distances from the radiolabeled end. ALKBH1 again formed protein-DNA adducts of different sizes with the different substrates, further confirming adduct formation with the 5′ product (Fig. 3A, right panel). In addition, the adducts derived from the double stranded substrates migrated at the same or slower rates compared to the adducts from single stranded substrates. These results are consistent with at least partial retention of double stranded DNA in the adducts, especially for the species containing the longer DNA fragment. To corroborate these findings, ALKBH1 was incubated with double stranded DNA containing one AP site on the labeled strand or on the opposite strand. Independent of the location of the AP site a signal was seen for the ALKBH1-DNA product adducts and these species were the same size, indicating that the DNA was double stranded in the bound adducts rather than being separated during SDS-PAGE (Fig. 3B). Each of these species migrated more slowly than the adduct formed using the DNA with two opposing AP sites close to the labeled position, thus demonstrating that ALBKH1 binds to the double stranded 5′-products following cleavage of the dual AP-containing DNA oligonucleotide.
Figure 3. Demonstration of an adduct between ALKBH1 and the 5′-DNA product.
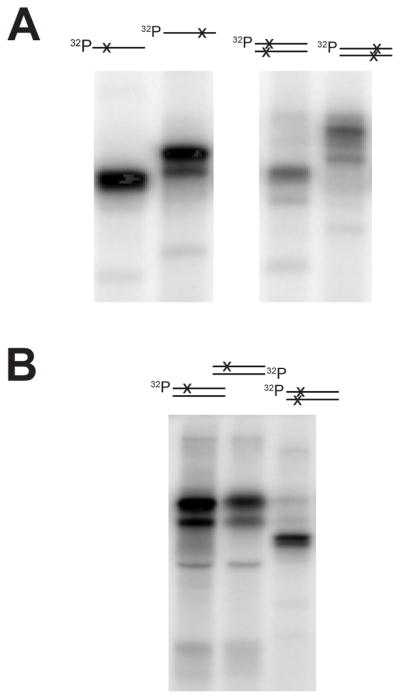
(A) Comparison of the DNA-ALKBH1 adducts with single and double stranded substrates. N-terminal His6-tagged ALKBH1 (5 μM) was incubated with DNA substrates (1 μM) for 1 h at 37 °C. UDG-treated oligo3 or oligo4 was used directly as single stranded species (left) or formed into double stranded oligo3+4 species, as schematically indicated above the autoradiographs (the AP sites are indicated by x and the location of the radiolabel is shown). After adduct formation, the DNA-protein adduct was analyzed by 12% SDS-PAGE and visualized by autoradiography. (B) Comparison of the DNA-ALKBH1 adducts with double stranded DNA containing one or two AP sites. His6-tagged ALKBH1 (5 μM) was incubated with 1 μM substrate containing one AP site, the UDG-treated oligo3+5 radioactively labeled on either oligo3 (lane 1) or oligo5 (lane 2), or substrate containing two AP sites in close proximity on opposing strands (UDG-treated oligo3+4, lane 3) for 1 h at 37 °C. The adducts were then analyzed as described in panel A.
To define the extent of reductant-independent ALKBH1-DNA adduct generation, several experiments were carried out using different DNA samples, different DNA and protein concentrations, and different times of incubation, with the electrophoretic analysis retaining the remaining substrate on the gels (data not shown). As a representative example, incubation of 1 μM 5′-labeled single stranded AP-containing DNA with 5 μM ALKBH1 for 60 min resulted in 40–45% of the label becoming bound to the protein. These experiments establish that a substantial proportion of the 5′-product of DNA forms an adduct with the enzyme. Indeed, we suggest the covalent linkage is essentially stoichiometric and attribute the incomplete binding to the slow rate of the reaction (Fig. 2C and see below) and to the presence of some inactive enzyme from aggregation, perhaps associated with its heterologous production.
The Amino-Terminal Region of ALKBH1 Attaches to the 5′ DNA Product
Having established that ALKBH1 forms a covalent bond to its AP lyase 5′ DNA product, we examined the protein region involved in this linkage by using different chemical and enzymatic cutters. First, large peptides were generated from the protein-DNA adduct by use of BNPS-skatole, a reagent which cleaves after tryptophan residues (located at residues 88, 112, 114, 144, 170, 179, and 327). As illustrated in the right two lanes of Figure 4A, N-terminal His6-tagged and untagged ALKBH1 free of DNA remained relatively intact after 72 h incubation in 70% acetic acid plus 0.1% phenol (i.e., the BNPS-skatole cleavage conditions) when examined by Tris-Tricine PAGE. When these proteins were incubated in the same conditions with the reagent (first and third lanes), the two forms of ALKBH1 generated similar peptide patterns except for two major bands labeled 1 and 2 (at about 12 and 15 kDa) in the tagged sample, or labeled 3 and 4 (at smaller apparent sizes) in the untagged sample. These results suggest that both of these bands include the amino terminal region, with and without the His6-tag, respectively. N-terminal sequencing of the lower band of the untagged protein yielded the sequence Gly-Ser-His-Met-Gly-Lys-Met-Ala, containing three linker residues and the authentic ALKBH1 amino terminus, thus confirming this interpretation. Significantly, each of these bands, but no others, shifted in position when the ALKBH1 samples were incubated with the AP-containing DNA substrate prior to BNPS-skatole treatment (Fig. 4A, bands labeled with the respective number and ′). This finding suggests the involvement of these peptides in adduct formation. The increased mobility of peptides derived from the protein-DNA adduct compared to the non-adduct peptides is presumed to be due to the extra negative charge of the bound DNA. Taken together, these results indicate that a residue near the N-terminus of ALKBH1 (i.e., within the first 88 residues) forms a covalent bond to the 5′-DNA product.
Figure 4. The amino-terminal region of ALKBH1 forms a covalent bond to the 5′-DNA product of AP-containing DNA.

(A) BNPS-skatole cleavage of ALKBH1. N-terminal His6-tagged and thrombin-treated (untagged) ALKBH1 samples (5 μM) were incubated in the absence of substrate or with AP oligo4 (7.5 μM) for 1 h at 37 °C. Samples to be cleaved with BNPS-skatole were subjected to the reagent (1 mM) in 70% acetic acid containing 0.1% phenol for 72 h, whereas the control proteins were incubated in the same conditions without the reagent. The samples were analyzed by using a 10–16% Tris-Tricine gel and visualized by Coomassie staining. The migration positions of molecular mass markers are shown to the left. The numbers next to the bands indicate peptides involved in protein-DNA adduct formation. (B) LysC digest of selected Lys variants of ALKBH1. Selected Lys variants of N-terminal His6-tagged ALKBH1 were incubated with 32P-labelled UDG-treated oligo4 for 1 h at 37 °C, denatured at 95 °C, and subjected to LysC digestion (0.2 U) overnight. The samples were analyzed by 15% SDS-PAGE and the DNA-peptide adducts were visualized by autoradiography. A band of interest (see text) is marked *. (C) The His-tag is not required for adduct formation. His6-tagged and untagged ALKBH1 samples were incubated with UDG-treated oligo1+2. Aliquots were removed at the indicated time intervals, frozen, and analyzed by 6% PAGE. (D) C-terminal His6-tagged ALKBH1 has AP-lyase activity and forms the enzyme-product adduct. N- and C-terminal His6-tagged ALKBH1 as well as N-terminal His6-tagged ALKBH1 treated with thrombin were incubated with AP-oligo1+2 for 1 h at 37 °C. One series of samples was treated with proteinase K (1 U) for 30 min at 65 °C, whereas the other half was kept on ice. All samples were analyzed by 18% native PAGE and the DNA visualized by ethidium bromide staining.
To better define the site of protein-DNA attachment, His6-tagged ALKBH1 and each of its 22 Lys-to-Ala variants were incubated with radioactively labeled AP-containing DNA substrate then digested with LysC, which cleaves C-terminal of lysine residues, and analyzed by SDS-PAGE. An autoradiograph of a subset of the samples (Fig. 4B) demonstrated that the K3A ALKBH1 variant lacked the most rapidly migrating band (marked *), consistent with the protein-DNA adduct being formed by a residue in the vicinity of Lys3. Lys3 itself, however, is unlikely to be involved in the linkage since the extent of adduct formation when using K3A ALKBH1 is similar to that of the wild-type (WT) and other variant enzymes. A more plausible explanation is that a residue from a peptide terminating at the Lys3-Met4 cleavage site is responsible for protein-DNA adduct formation. In other words, the cross-link requires either a residue in the amino terminus (including the His-tag) or an amino acid in a peptide starting at residue 4 and extending to possibly Lys55, Lys61 or Lys64 (LysC does not cleave at all Lys residues and these small differences could not be distinguished on the gel). A peptide ending at Lys25 can be excluded since the K25A variant shows the same signals as the WT protein (the K25A substitution would shift from LysC peptide 4–25 to peptide 4–55 or longer, leading to a change in band position, but this is not seen).
To gain further insight into the role of the amino terminus in protein-DNA adduct formation, N-terminal His6-tagged and untagged ALKBH1 samples were incubated with AP-containing DNA and examined over time by PAGE. As shown by the rate of loss of free substrate and the rate of increase of the protein-DNA adduct in Fig. 4C, the enzyme preparations behaved nearly identically (and were consistent with the time course of Fig. 2C) demonstrating that the His6-tag is not required for adduct formation. Extending analysis of N-terminal tagged protein, preliminary experiments were carried out with a distinct His6-tagged ALKBH1 that was produced in Sf9 insect cells [16]; that species also showed protein-DNA adduct formation without reductant (data not shown). Significantly, the His-tags and their linker sequences differ in the two systems, with a Gly-Ser-Pro-Gly-Leu-Asp sequence encoded by the baculovirus vector corresponding to Gly-Ser-His encoded in the bacterial construct. Because it is unlikely that nucleophilic residues are similarly placed in both fusion proteins, an internal residue of ALKBH1 (i.e., a residue in the peptide starting at Met4) expressed in the insect cell is probably responsible for linking to the DNA product. As final confirmation that the N-terminal His6-tag and its linker did not participate in covalent linkage to product DNA, a form of ALKBH1 with the tag shifted to the C-terminus was examined. The C-terminal His6-tagged product exhibited lyase activity and formed a covalent linkage to AP-containing DNA in the absence of reducing agents (Fig. 4D). In sum, the data are consistent with covalent attachment between the 5′ product of the substrate DNA and the amino terminal region of authentic ALKBH1; i.e., within a peptide starting at Met4 and extending to Lys55, Lys61 or Lys64.
Confirmation that ALKBH1 is an AP Lyase
The formation of a reductant-independent covalent adduct of ALKBH1 and its cleaved product DNA is not typical of other AP lyases. By contrast, protein-DNA adducts are known to occur in non-lyase proteins such as DNA recombinases, topoisomerases, and the meiotic protein Spo11 [25, 26]. The latter proteins catalyze phosphotransfer reactions in which a nucleophilic side chain attacks the phosphate backbone of the DNA substrate, forming a covalent bond to part of the DNA while releasing the remaining DNA with a free hydroxyl group. These observations led us to reevaluate the mechanism of DNA cleavage by ALKBH1. Previous investigations ruled out a hydrolase mechanism and favored a lyase activity [16] on the basis of three lines of evidence: (i) the inability of ALKBH1 to act on tetrahydrofuran-containing DNA, similar to EndoIII and distinct from ApeI, (ii) size analysis of the 5′ DNA product (released after proteinase K digestion) that was similar to the product from EndoIII and different from that of ApeI, and (iii) the ability to trap a protein-DNA adduct with reductant. These findings did not eliminate the possibility of a phosphotransfer reaction at the phosphorus positioned 3′ of the AP lesion, thus linking the protein to the 5′ product and releasing a 3′ product with a 5′ hydroxyl group.
To provide additional confirmation that ALKBH1 catalyzes an AP lyase reaction, the phosphorylation status of the newly created 5′ terminus of the 3′ product was assessed. A phosphate is found at this position when AP sites are cleaved by either hydrolases, like Ape1, or lyases, like EndoIII (Fig. 1). In contrast, a phosphotransfer reaction that links the protein to the 5′ product would release a 3′ product lacking phosphate at its 5′ terminus. To identify the 3′ product phosphorylation status, we examined the susceptibility of this product to phosphorylation (Fig. 5). N-terminal His6-tagged or untagged ALKBH1 as well as EndoIII and Ape1 were incubated with unlabeled single stranded oligonucleotides containing an AP site. One portion of each sample was directly incubated with PNK and [γ-32P]-ATP while a second aliquot of each sample was treated with Antarctic alkaline phosphatase, heated to inactivate the phosphatase, and then incubated with PNK and [γ-32P]-ATP. For each AP-DNA cleaving enzyme tested, the 3′ product was readily detected by phosphorimager analysis after treatment with both phosphatase and kinase, but not when treated with the kinase alone. As a useful internal control, the 5′ product was readily labeled without prior phosphatase treatment for EndoIII, Ape1, and ALKBH1. As previously found [16], the Ape1 product is smaller in size than the products of EndoIII or ALKBH1. Unlike the case when using the double stranded substrate in Figure 2B, much greater amounts of free 5′ product were detected for the ALKBH1 reaction with the single stranded substrate without proteinase K treatment; however, inclusion of proteinase K led to increased release of this product. Furthermore, proteinase K digestion was required to detect the 5′ product associated with the ALKBH1 protein-DNA adduct in the phosphatase-treated sample. These results provide clear evidence that ALKBH1 produces a 3′-product containing a phosphate on its 5′ end, thus verifying its classification as a lyase.
Figure 5. ALKBH1’s AP lyase activity yields a 5′-phosphorylated 3′ product.

N-terminal His6-tagged (ALKBH1His) or untagged ALKBH1 (5 μM), Ape1 (A, 10 U), and EndoIII (E, 10U) were incubated with unlabeled, single stranded AP-containing oligo4 (1 μM) for 1 h at 37 °C and either used directly or treated with proteinase K (2 U). One aliquot of each sample was incubated with Antarctic alkaline phosphatase (5 U) for 30 min at 37 °C followed by an inactivation step at 65 °C for 30 min, whereas a second aliquot was left on ice. All samples were incubated with 10 U of PNK plus [γ-32P] ATP and examined by denaturing 20% PAGE gel. The signals were detected by exposing the dried gel to a phosphorimager.
If ALKBH1 uses lyase chemistry to cleave at AP sites and subsequently forms a covalent attachment of the 5′ product α,β-unsaturated aldehyde to the enzyme, then a similar linkage may occur for protein incubated with the 5′ product that is separately produced. To test this possibility, AP containing oligonucleotide was incubated with EndoIII, resulting in complete conversion to the free products, prior to addition of ALKBH1; incubation of this sample led to the formation of some covalently linked product (Fig. 6). The low efficiency of this linkage may relate to the provision of free product rather than the enzyme attaching to the product as it is formed. Nevertheless, these studies provide further evidence that ALKBH1 exhibits lyase activity and, significantly, it forms a covalent adduct with the 5′ product containing the α,β-unsaturated aldehyde.
Figure 6. ALKBH1 forms a covalent bond with the α,β-unsaturated aldehyde.
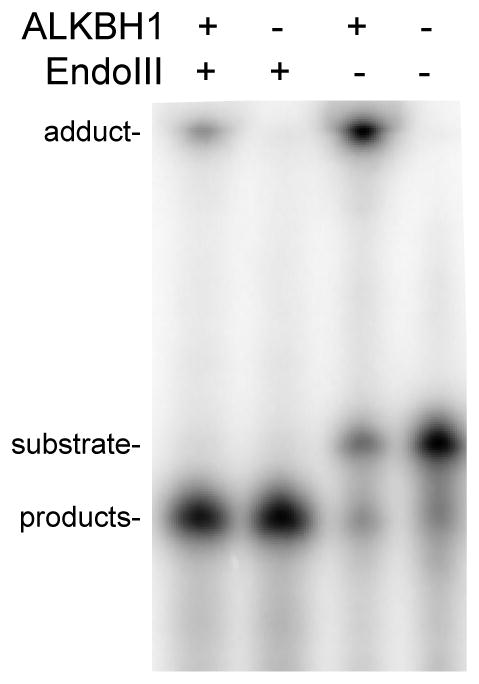
ALKBH1 forms an adduct with the product of the AP lyase activity of EndoIII. Radioactively labeled UDG-treated oligo1 (1 μM) was incubated with EndoIII (1 U) for 1 h at 37 °C. This sample was directly examined or His6-tagged ALKBH1 (5 μM) was added to the pre-cleaved products and allowed to form the adduct for 1 h at 37 °C. Adduct formation of ALKBH with radioactively labeled UDG-treated oligo1 and a lane lacking either enzyme are shown as additional controls. All protein-DNA adducts were analyzed by denaturing 7 M urea PAGE (20%).
Identification of the ALKBH1 AP Lyase Active Site Lysine
The stable protein-DNA substrate adduct formed between ALKBH1 and AP-containing DNA in the presence of a reducing agent [16] is consistent with the AP lyase active site Lys forming the typical Schiff base intermediate with the open-ring form of the abasic site. To identify the active site Lys in N-terminal His6-tagged ALKBH1, each of its 22 Lys residues was individually substituted by Ala using site-directed mutagenesis. When incubated with AP-containing DNA, all purified variants retained the ability to form a Schiff base that could be trapped with NaBH4 (data not shown). These results resemble those for selected other AP and dRP lyases where variants lacking the primary Lys nucleo-phile nevertheless could be reductively trapped as DNA-protein adducts; these earlier cases were due to imine formation using alternate side chains that substitute for the missing primary residue [27, 28].
In further studies to identify the most likely primary Lys of ALKBH1, activity assays were carried out with the isolated variant proteins. Compared to the N-terminal His6-tagged wild type (WT) enzyme, the K133A variant showed the greatest loss of activity, followed by the K25A protein; thus, Lys133 or Lys25 is most likely the primary residue involved in catalysis by the WT enzyme (Fig 7A). Given the precedence for opportunistic Lys residues being able to substitute for the primary nucleophiles in other AP lyases [27, 28], selected double variants of the K133A protein were created. Again, all samples tested retained the ability to trap a Schiff base (data not shown) and all variants were active (Fig. 7B). The K25A/K133A protein retained about 2/3 of the activity measured in the K133A variant; hence Lys25 did not appear to be a critical residue. Of greater interest is the K133A/K154A ALKBH1, which exhibited the lowest activity, namely 40% of the K133A single variant protein. These results provide evidence that Lys133 serves as the original nucleophile and Lys154 (possibly along with other residues) is capable of functionally substituting for Lys133.
Figure 7. Characterization of ALKBH1 Lys variants and elucidation of the AP lyase active site Lys residue.

(A) AP lyase activity of single Lys variants of ALKBH1. All 22 Lys residues in N-terminal His6-tagged ALKBH1 were individually substituted by Ala using site-directed mutagenesis and the purified proteins were analyzed for AP lyase activity (5 μM variant protein and 1 μM double stranded UDG-treated oligo1+2 for 1 h at 37 °C, proteinase K (0.2 U) digested for 30 min at 65 °C, and the products visualized by ethidium bromide treatment after 18% PAGE). The 100% activity of WT ALKBH1 corresponds to cleavage of 81% of the substrate. (B) AP lyase activity of ALKBH1 double variants. Selected variants of K133A ALKBH1 were constructed, purified, and analyzed as above. The activity of K133A ALKBH1 was set to 100%.
DISCUSSION
Our results demonstrate that ALKBH1 forms a covalent adduct with the 5′ DNA product of its AP lyase activity in the absence of the reducing agent NaBH4. The novelty of this reaction is clear when compared to other reported protein-DNA interactions:
Several AP/dRP lyases, such as poly(ADP-ribose) polymerase-1, histone H4, Fpg, and polymerase γ release their products slowly [13, 15, 29, 30] indicating a stabilized imine intermediate; however, their stabilities do not extend to denatured samples (e.g., boiling with β-mercaptoethanol and SDS) as found here. To be resistant to these denaturing conditions, a covalent bond is required. One report identified covalent adducts between OGG1 and AP sites in DNA in the absence of reductants [31], but the unidentified linkages were shown to be sensitive to heating in the presence of 0.1 M acetic acid. By contrast, ALKBH1 peptide-DNA adducts were stable for 72 h in 70% acetic acid as illustrated by the results in Figure 4A.
EndoIII, polymerase β, histone H4 and, to a lesser extent, other glycosylases form stable amide bonds with their cleaved 5′-DNA products when acting on substrates containing 2-deoxyribonolactone, the oxidized form of the abasic site [32, 33]. Significantly, the chemical reaction forming covalent linkages with this lactone does not apply to the AP lyase product and cannot account for the covalent binding to ALKBH1.
Nucleoside-diphosphate kinase NM23-H2 possesses lyase activity, forms a stable protein-DNA adduct that is heat-induced, and links via the lyase active site Lys to the 3′ product of DNA cleavage [34, 35]. In ALKBH1, the adduct forms with the 5′ DNA product and the linkage does not involve the lyase active site Lys.
Topoisomerases and recombinases use a nucleophile to attack the phosphodiester backbone; thus generating a phosphodiester linkage with the 5′-product and forming a 3′ product with a 5′ hydroxyl group before reversing the chemistry to rejoin the DNA fragments [25]. Contrary to this type of covalent linkage, ALKBH1 releases a 5′-phosphorylated 3′ product; thus arguing against a phosphotransferase mechanism for this enzyme.
The site of covalent linkage between ALKBH1 and its 5′ product has been localized to the amino terminal region of the protein (likely between residues 4 and 64) on the basis of chemical and enzymatic cleavage results. This attachment is independent of the His-tag since it occurs whether using protein with an N-terminal or C-terminal His6-tag, and when using non-tagged protein. Significantly, the site of linkage does not involve the lyase active site Lys residue, located at position 133. While the lyase chemistry is not abolished in any of the 22 variants, the K133A protein cleaves only 60% of the substrate compared to WT enzyme using the stated assay conditions indicating that Lys133 is most likely the primary imine-forming residue involved in catalysis. When this residue is mutated, however, other Lys residues are capable of fulfilling its role. While the rather modest loss of activity for K133A ALKBH1 is atypical compared to related situations, the occurrence of so-called opportunistic Lys residues has been reported for several examples in the literature [27, 28, 36, 37]. The K133A/K154A variant of ALKBH1 exhibits much lower activity than either of the single variants, consistent with Lys154 being positioned in such a manner so as to catalyze the reaction when Lys133 is mutated. Considering the Lys rich region in the middle of ALKBH1 (with Lys residues at positions 116, 120, 125, 133, 137, 148, 154, 158, and 167), we hypothesize that several Lys residues are able to form an imine with C1′ of the AP site and facilitate catalysis. Notably, this region of the protein is distinct from the region that covalently attaches to product DNA.
We propose two plausible hypotheses to explain the chemistry of covalent linkage in ALKBH1 that is independent of its demethylase or lyase activities (Fig. 8). In one case, an enzyme nucleophile is positioned near the lyase active site to immediately add to the β-elimination product, analogous to the Michael addition of proteins with α,β-unsaturated products of lipid peroxidation [38]. In other words, after ALKBH1 catalyzes the AP lyase reaction a nucleophile located near the enzyme amino terminus binds to the α,β-unsaturated aldehyde by attacking the C3′ atom of the deoxyribose ring. Such adducts are known to be stabilized by formation of a cyclic hemiacetal, with further stabilization provided by dehydration [38]. We postulate that similar stabilization steps may occur for ALKBH1. As an alternative possibility, a nucleophile in the amino terminal region of ALKBH1 may participate in a phosphotransfer reaction with the phosphate immediately preceding the α,β-unsaturated aldehyde, with release of the five-carbon unit. Further studies are required to resolve these testable hypotheses.
Figure 8. Hypothetical mechanisms for the covalent attachment of ALKBH1 to the AP DNA product.

(A) Overview of interaction between DNA and ALKBH1, with its separate Fe-containing demethylase and Lys-dependent AP lyase active sites. The line connecting protein and DNA indicates a covalent linkage between ALKBH1 and the 5′ DNA product. (B) One plausible mechanism for generating the covalent linkage uses a nucleophile to attacks the C3′ atom of the α,β-unsaturated aldehyde. The resulting adduct then forms a more stable cyclic hemiacetal which may be further stabilized by dehydration. (C) An alternative hypothesis has the nucleophile being involved in a phosphotransfer reaction with the phosphate preceding the α,β-unsaturated aldehyde with release of the five-carbon unit.
We conclude by speculating on the potential functional relevance, if any, of ALKBH1’s lyase activity and its ability to covalently attach to the 5′ product. It is perhaps not surprising that ALKBH1 possesses AP lyase chemistry in addition to its demethylase activity since several DNA repair and DNA binding enzymes also catalyze β-elimination reactions. The recent finding of AP/dRP lyase activity in Ku, for example, widens its role beyond recognizing DSB ends and recruiting end-processing proteins to removing AP sites at DNA ends in vivo [12]. Similarly, AP/dRP lyase activities extend the range of functions associated with the non-histone chromatin modifying enzyme HMGA2 [37] as well as histone H4 [15], whose Lys-rich tail is able to introduce DSB into DNA. Given these precedents, it is reasonable to postulate that the AP lyase activity of ALKBH1 may have an in vivo role. The covalent bond between ALKBH1 and its product explains why the enzyme catalyzes only a single turnover, but additional studies are needed to identify the specific residue(s) involved in attachment. The purpose of this activity is unclear, and it may represent the generation of a toxic product. Alternatively, one can speculate that this may be the first step in a “hand off” mechanism involving a subsequent enzyme that provides a self-defense mechanism in vivo in order to protect the genome from undesired DNA breaks as well as preventing unnecessary end-processing of the cleavage site. For instance, Spo11, involved in meiotic recombination, binds irreversibly to its 3′-product by forming a phosphodiester bond and is released only by endonuclease cleavage of a small oligonucleotide. On the basis of this behavior, Spo11 is suggested to catalyze a “suicide” reaction [26, 39]. Further studies will shed light on whether a similar situation exists with ALKBH1.
Acknowledgments
We thank Dr. Kathy Meek for advice and assistance with 32P experiments and Blair Murphy for help with protein purification.
FUNDING
This work was supported by the National Institutes of Health [R21AI79430 to T.A.M. and GM063584 to R.P.H.].
Abbreviations used
- ALKBH1
AlkB homolog 1
- AP
apyrimidinic/apurinic or abasic
- BER
base excision repair
- dRP
deoxyribophosphate
- DSB
double-strand break
- EndoIII
Escherichia coli endonuclease III
- Fpg
formamidopyrimidine DNA glycosylase
- PNK
T4 polynucleotide kinase
- UDG
Uracil DNA glycosylase
- WT
wild-type
Footnotes
AUTHOR CONTRIBUTION
Tina A. Müller designed and conducted the experiments, analysed the data, and co-wrote the paper. Megan M. Andrzejak assisted with the majority of the experiments. Robert P. Hausinger designed the study, assisted with data analysis, and co-wrote the paper.
References
- 1.Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 2.Lhomme J, Constant JF, Demeunynck M. Abasic DNA structure, reactivity, and recognition. Biopolymers. 1999;52:65–83. doi: 10.1002/1097-0282(1999)52:2<65::AID-BIP1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 4.Dianov GL, Sleeth KM, Dianova II, Allinson SL. Repair of abasic sites in DNA. Mutat Res. 2003;531:157–163. doi: 10.1016/j.mrfmmm.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476:73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 7.Piersen CE, McCullough AK, Lloyd RS. AP lyases and dRPases: commonality of mechanism. Mutat Res. 2000;459:43–53. doi: 10.1016/s0921-8777(99)00054-3. [DOI] [PubMed] [Google Scholar]
- 8.Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham RP, Ahern H, Xing D, Thayer MM, Tainer JA. Structure and function of Escherichia coli endonuclease III. Ann N Y Acad Sci. 1994;726:215–222. doi: 10.1111/j.1749-6632.1994.tb52818.x. [DOI] [PubMed] [Google Scholar]
- 10.Sidorkina OM, Laval J. Role of the N-terminal proline residue in the catalytic activities of the Escherichia coli Fpg protein. J Biol Chem. 2000;275:9924–9929. doi: 10.1074/jbc.275.14.9924. [DOI] [PubMed] [Google Scholar]
- 11.Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, Wallace SS. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci USA. 2010;107:4925–4930. doi: 10.1073/pnas.0908307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, Hasty P, Ramsden DA. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature. 2010;464:1214–1217. doi: 10.1038/nature08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khodyreva SN, Prasad R, Ilina ES, Sukhanova MV, Kutuzov MM, Liu Y, Hou EW, Wilson SH, Lavrik OI. Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1) Proc Natl Acad Sci USA. 2010;107:22090–22095. doi: 10.1073/pnas.1009182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hegde V, Wang M, Deutsch WA. Human ribosomal protein S3 interacts with DNA base excision repair proteins hAPE/Ref-1 and hOGG1. Biochemistry. 2004;43:14211–14217. doi: 10.1021/bi049234b. [DOI] [PubMed] [Google Scholar]
- 15.Sczepanski JT, Wong RS, McKnight JN, Bowman GD, Greenberg MM. Rapid DNA-protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc Natl Acad Sci USA. 2010;107:22475–22480. doi: 10.1073/pnas.1012860108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller TA, Meek K, Hausinger RP. Human AlkB homologue 1 (ABH1) exhibits DNA lyase activity at abasic sites. DNA Repair (Amst) 2010;9:58–65. doi: 10.1016/j.dnarep.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurowski MA, Bhagwat AS, Papaj G, Bujnicki JM. Phylogenomic identification of five new human homologs of the DNA repair enzyme AlkB. BMC Genomics. 2003;4:48. doi: 10.1186/1471-2164-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falnes PØ, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 19.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 20.Westbye MP, Feyzi E, Aas PA, Vagbo CB, Talstad VA, Kavli B, Hagen L, Sundheim O, Akbari M, Liabakk NB, Slupphaug G, Otterlei M, Krokan HE. Human AlkB homolog 1 is a mitochondrial protein that demethylates 3-methylcytosine in DNA and RNA. J Biol Chem. 2008;283:25046–25056. doi: 10.1074/jbc.M803776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ougland R, Lando D, Jonson I, Dahl JA, Moen MN, Nordstrand LM, Rognes T, Lee JT, Klungland A, Kouzarides T, Larsen E. ALKBH1 is a histone H2A dioxygenase involved in neural differentiation. Stem Cells. 2012 doi: 10.1002/stem.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korvald H, Falnes PO, Laerdahl JK, Bjoras M, Alseth I. The Schizosaccharomyces pombe AlkB homolog Abh1 exhibits AP lyase activity but no demethylase activity. DNA Repair (Amst) 2012;11:453–462. doi: 10.1016/j.dnarep.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci USA. 2002;99:16660–16665. doi: 10.1073/pnas.262589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schägger H. Tricine-SDS-PAGE. Nat Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 25.Grindley ND, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Annu Rev Biochem. 2006;75:567–605. doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- 26.Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Roy R. Mutation at active site lysine 212 to arginine uncouples the glycosylase activity from the lyase activity of human endonuclease III. Biochemistry. 2001;40:13617–13622. doi: 10.1021/bi011053b. [DOI] [PubMed] [Google Scholar]
- 28.Prasad R, Beard WA, Chyan JY, Maciejewski MW, Mullen GP, Wilson SH. Functional analysis of the amino-terminal 8-kDa domain of DNA polymerase β as revealed by site-directed mutagenesis. DNA binding and 5′-deoxyribose phosphate lyase activities. J Biol Chem. 1998;273:11121–11126. doi: 10.1074/jbc.273.18.11121. [DOI] [PubMed] [Google Scholar]
- 29.Kuznetsov NA, Zharkov DO, Koval VV, Buckle M, Fedorova OS. Reversible chemical step and rate-limiting enzyme regeneration in the reaction catalyzed by formamidopyrimidine-DNA glycosylase. Biochemistry. 2009;48:11335–11343. doi: 10.1021/bi901100b. [DOI] [PubMed] [Google Scholar]
- 30.Pinz KG, Bogenhagen DF. Characterization of a catalytically slow AP lyase activity in DNA polymerase γ and other family A DNA polymerases. J Biol Chem. 2000;275:12509–12514. doi: 10.1074/jbc.275.17.12509. [DOI] [PubMed] [Google Scholar]
- 31.Nazarkina ZK, Khodyreva SN, Marsin S, Lavrik OI, Radicella JP. XRCC1 interactions with base excision repair DNA intermediates. DNA Repair (Amst) 2007;6:254–264. doi: 10.1016/j.dnarep.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Kroeger KM, Hashimoto M, Kow YW, Greenberg MM. Cross-linking of 2-deoxyribonolactone and its β-elimination product by base excision repair enzymes. Biochemistry. 2003;42:2449–2455. doi: 10.1021/bi027168c. [DOI] [PubMed] [Google Scholar]
- 33.Zhou C, Greenberg MM. Histone-catalyzed cleavage of nucleosomal DNA containing 2-deoxyribonolactone. J Am Chem Soc. 2012;134:8090–8093. doi: 10.1021/ja302993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Postel EH. Cleavage of DNA by human NM23-H2/nucleoside diphosphate kinase involves formation of a covalent protein-DNA complex. J Biol Chem. 1999;274:22821–22829. doi: 10.1074/jbc.274.32.22821. [DOI] [PubMed] [Google Scholar]
- 35.Postel EH, Abramczyk BM, Levit MN, Kyin S. Catalysis of DNA cleavage and nucleoside triphosphate synthesis by NM23-H2/NDP kinase share an active site that implies a DNA repair function. Proc Natl Acad Sci USA. 2000;97:14194–14199. doi: 10.1073/pnas.97.26.14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prasad R, Beard WA, Strauss PR, Wilson SH. Human DNA polymerase β deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J Biol Chem. 1998;273:15263–15270. doi: 10.1074/jbc.273.24.15263. [DOI] [PubMed] [Google Scholar]
- 37.Summer H, Li O, Bao Q, Zhan L, Peter S, Sathiyanathan P, Henderson D, Klonisch T, Goodman SD, Droge P. HMGA2 exhibits dRP/AP site cleavage activity and protects cancer cells from DNA-damage-induced cytotoxicity during chemotherapy. Nucleic Acids Res. 2009;37:4371–4384. doi: 10.1093/nar/gkp375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug Metab Rev. 2006;38:651–675. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- 39.Neale MJ, Pan J, Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–1057. doi: 10.1038/nature03872. [DOI] [PMC free article] [PubMed] [Google Scholar]