

Abstract
Type-III interferons (IFN-λ, IFNL) are the most recently described family of IFNs. This family of innate cytokines are increasingly being ascribed pivotal roles in host–pathogen interactions. Herein, we will review the accumulating evidence detailing the immune biology of IFNL during viral infection, and the implications of this novel information on means to advance the development of therapies and vaccines against existing and emerging pathogens. IFNLs exert antiviral effects via induction of IFN-stimulated genes. Common single nucleotide polymorphisms (SNPs) in the IFNL3, IFNL4 and the IFNL receptor α-subunit genes have been strongly associated with IFN-α-based treatment of chronic hepatitis C virus infection. The clinical impact of these SNPs may be dependent on the status of viral infection (acute or chronic) and the potential to develop viral resistance. Another important function of IFNLs is macrophage and dendritic cell polarization, which prime helper T-cell activation and proliferation. It has been demonstrated that IFNL increase Th1- and reduce Th2-cytokines. Therefore, can such SNPs affect the IFNL signaling and thereby modulate the Th1/Th2 balance during infection? In turn, this may influence the subsequent priming of cytotoxic T cells versus antibody-secreting B cells, with implications for the breadth and durability of the host response.
Keywords: B cells, hepatitis C virus, immune response, innate immunity, interferon-lambda, receptor, respiratory viruses, T cells
GENERAL ASPECTS OF INTERFERONS (IFNS)
Since their inception, IFNs have displayed ever increasing diversity and demonstrated activity far more refined than the original observation of the ability to ‘interfere' with viral replication.1 Each IFN family member mediates important anti-viral activity via engagement with their respective IFN receptor. All IFNs share an α-helical bundle structure and broadly function in a similar manner however significant effector differences exist.2 IFNs are divided into three types: type-I mainly represented by IFN-α3,4,5 and -β,6,7 type-II by IFN-γ,8 and type III consulting the recently discovered IFN-lambda (IFNL) family.9,10,11 We propose that IFNL occupies a key position modulating the host response to pathogens. Comprehensive understanding of the immunological impact may become increasingly important to understand host-virus interactions of existing and emerging pathogens, with application to the development of novel therapies and vaccine strategies.
Type-I IFNs
The group of type-I IFNs includes 13 highly homologous IFN-α genes, as well as single genes encoding IFN-β, -ε, -κ and -ω. All type-I IFNs are encoded on chromosome 92 and almost all cell types can elicit a type-I IFN response. Binding of any of the type-I IFNs to a receptor subunit leads to the corecruitment of a second subunit and dimerization of the IFN-α/β receptor complex (IFNAR).12 The receptor subunits are associated with Janus-activated kinases (JAK1) and tyrosine kinase 2. Their interaction leads to the phosphorylation of signal transducer and activator of transcription (STAT)-1 and -2. In specific cell types, STAT-3, -4, -5 and -6 can also be phosphorylated.13,14,15 STAT-1/2 heterodimer interactions initiate the corecruitment of INF regulatory factor (IRF)-9 and consequent binding to the intra-nuclear IFN-stimulated response element. In contrast, a STAT-1 homodimer binds to the IFN-γ activated site enhancer elements in promoters of dozens of IFN-stimulated genes (ISG) and induces transcription.15,16,17
Type-II IFNs
IFN-γ is the sole representative of type-II IFNs. IFN-γ is not induced via direct sensing of virus fragments but rather through stimulation of epitope-specific T cells and natural killer cells. Similar to IFNAR, the receptor for IFN-γ (IFNGR) is a heterodimer of IFNGR1 and IFNGR2.18 The receptor subunits are associated with JAK1 and JAK2 tyrosine kinases. The interaction induces phosphorylation of STAT-1. IFNGRs are broadly expressed on many cells and induce anti-viral ISGs.18,19
Type-III IFNs
The IFNL family is the most recently discovered group of IFNs, comprising four homologous members: IFNL1 (interleukin (IL)-29), IFNL2 (IL-28A), IFNL3 (IL-28B)9,10 and the very recently described IFNL4.20,21 All are encoded on chromosome 19 (19q13.13 region). Formally, IFNLs belong to the IL-10 family of cytokines containing IL-10, IL-19, IL-20, IL-22, IL-24 and IL-26. The IFNL coding region has five exons and four introns. Their positions in relation to the open reading frames are conserved for all IFNL and IL-10-related cytokines.22,23,24
The high degree of amino-acid sequence similarities within IFNL1–3 suggests a common ancestor gene.21 IFNL2 and 3 share 96% sequence similarity, whereas IFNL1 is less similar and also differs in terms of the pattern of disulfide bridges.25 IFNL4 arises as a consequence of a frameshift mutation generating a new gene not normally expressed, and demonstrates only a 40.8% similarity to IFNL3. Figure 1A shows a sequence alignment of all IFNLs. Figure 1B highlights the differences in side chains between IFNL1–3 in three-dimensional structures. Despite almost identical sequences, IFNL3 and IFNL2,9,26,27 have markedly different anti-viral activities. IFNL3 demonstrates the highest anti-viral activity as measured by HepG2 challenge with encephalomyocarditis virus. IFNL3 had a 16-fold lower half-maximal effective concentration (EC50) compared to IFNL2 and two-fold lower EC50 value relative to IFNL1.28 Additionally ISG induction (Mx1 and IRF9) by IFNL3 was significantly higher compared to IFNL1 and IFNL2.29
Figure 1.
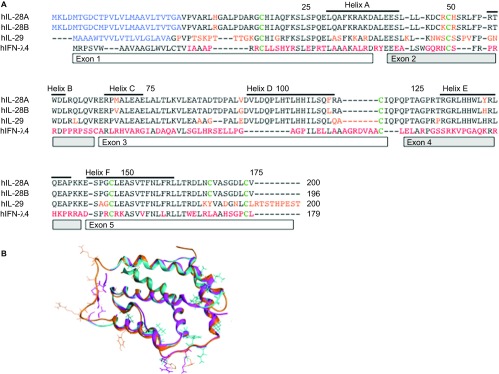
(A) Sequence alignment of IFNLs. An amino-acid sequence alignment of human IFNL1–4 is shown. Red letters indicate significant differences. Green letters indicate common cysteines. Helices and exons are indicated with boxes. (B) Sequence alignment overlay for different IFNLs. Amino acids of significance are highlighted.
EXPRESSION OF IFNLS
Almost any cell type is able to express IFNL1–3 mRNA in response to diverse viral infections such as Sindbis virus, dengue virus, vesicular stomatitis virus, encephalomyocarditis virus,9,10 respiratory syncytial virus,30,31 influenza virus, Sendai virus32,33and hepatitis C virus (HCV).34,35,36 High levels of IFNLs, but not IFN-α, were observed during viral infection of lung and liver tissues,36,37,38,39,40,41and IFNLs seem to be the major IFNs induced in airway epithelial cells during infection with respiratory viruses.42,43,44 The most potent producers of IFNLs however, seem to be myeloid and plasmacytoid dendritic cells (DCs).27,45,46,47,48
The expression kinetics of IFNL1–3 may be cell type dependent and have not been fully elucidated. However, our data suggests a clear difference in the expression kinetics between different IFNL family members during stimulation of fibroblasts and human peripheral blood mononuclear cells (PBMCs). IFNL3 showed a peak at 24-h after stimulation with cytomegalovirus (CMV), whereas IFNL1 peaked already at 6 h when measuring mRNA levels. Stimulation with poly(I:C) showed a significantly earlier peak of IFNL expression compared to infection with CMV.49
IFNL4 mRNA could be detected in primary human hepatocytes (with the required single nucleotide polymorphism (SNP)) 2–4 h after stimulation with poly(I:C) and after in vitro infection with HCV. However, a rapid downregulation of IFNL4 mRNA expression at 8 h was observed. In contrast, IFNL3 mRNA was still expressed 24 h after stimulation (Figure 2).21 The rapid downregulation after stimulation may suggest either a lack of an amplifying positive feedback loop, or rapid induction of a specific negative feedback mechanism. Interestingly, in liver biopsies from non-HCV-infected patients, a baseline mRNA expression of IFNL4 could be observed.21 Although this may suggest that IFNL4 is expressed in conditions unrelated to HCV, activation of this signaling pathway may happen through a wide spectrum of stimuli. DCs have not yet been specifically explored as a source of IFNL4.
Figure 2.

Expression dynamics of IFNs and ISGs during viral infection or inflammatory stimulation. Red line represents IFNL3, blue line IFN-α and black line IFNL4 expression dynamics. The lower panel illustrates the amount of ISG expression.
Purported explanations for the variance in expression kinetics and subsequent ISG expression (see below) may include: transcription factor differences in specific cell types; the presence of SNPs regulating IFNL expression (cell types have rarely been genotyped to determine the background of the original cell); regulatory feedback loops; receptor expression, and receptor binding affinity. In addition, various viruses may have evolved specific defence mechanisms based on the cytokine and IFN microenvironment which ultimately determines the viral tropism.50 The IFN-α signaling cascade is known to be modulated at numerous points51 and it is also very likely that co-evolutionary competition between IFNLs and different viruses has occurred. Of note, the replication of emerging respiratory viruses such as influenza H7N9 or Middle East respiratory syndrome coronavirus may be substantially controlled by IFNLs. Many ‘new' viruses have shown an ability to interfere with the IFN signaling pathways or involved antiviral ISGs.51,52,53,54 Multiple factors likely act to influence IFNL-associated immune responses and subsequently impact outcomes of infectious diseases.
Transcription factors
Both, type-I and -III IFNs are directly induced through sensing of viral infection via pattern recognition receptors such as Toll-like receptors (TLRs), retinoic acid-inducible gene 1 or melanoma differentiation-associated protein 5.17,51 For example: CpG-DNA (TLR-9 agonist) induces the expression of IFN-α, -β and IFNL1-3 in plasmacytoid DCs.27,33,46,47,55 Lipopolysaccharide (TLR-4 agonist) and poly(I:C) (TLR-3 agonist) induce IFN-β and IFNL1–3, but not IFN-α in myeloid DCs.33 Poly(I:C) can also induce IFNL4 expression in the appropriate genetic background.
The co-expression of type-I and -III IFNs is explained by common transcription factors and binding sites in the promoter regions. Specifically, binding sites for activator protein 1, IRF3, IRF7 and NF-κB were predicted in the promoter region of all IFNL genes.32,33,56,57,58,59,60,61,62 Further, the promoter regions of IFNL1-3 share very similar or the same transcriptional regulatory elements. Recently, another important transcription factor of IFNLs, Med23, was discovered.63 Med23 significantly upregulated the expression of IFNL mRNA and protein level by directly interacting with the transcription factor IRF7. The synergistic effect of Med23 and IRF7 on IFNL induction suggests that this may be a particularly important transcription factor. Transcription factor binding sites for IFNL4 have not yet been characterized in detail.
Single nucleotide polymorphisms
A series of SNPs near the IFNL3 and IFNL4 genes have been discovered and importantly are noted to be in high linkage disequilibrium.26,64 Although the SNPs are widely distributed and some are located considerable distances from coding sequences, they potentially impact the binding of transcription factors and methylation sites,65,66,67,68 thereby influencing the promoter output. The rs4803217 SNP in the 3′ untranslated region of the IFNL3 gene has been associated with mRNA stability.69,70 Some of these SNPs are possibly just in linkage with other functional SNPs (e.g., rs12979860 with ss469415590). Thus, deciphering the relative clinical importance of an individual SNP is rendered very difficult. Table 1 summarizes the locations and impact of published SNPs involving the IFNL signaling cascade.
Table 1. Selection of important SNPs in the IFNL signaling cascade.
| Gene | SNP | Nucleotides | Localization | Impact on expression (minor allele) | Impact on virus replication (minor allele) | Impact on adaptive immune functions (minor allele) | Reference |
|---|---|---|---|---|---|---|---|
| IFNL4a (IFNL3) | rs12979860 | Major: C/C Minor: C/T, T/T | — | Reducing IFNL3 | HCV: reduced IFN-α treatment response, lower rates of spontaneous clearance | 49, 71–75 | |
| Acute CMV: reduced replication | |||||||
| Noneb | rs8099917 | Major: T/T Minor: T/G, G/G | — | Reducing IFNL3 | HCV: reduced IFN-α treatment response, lower rates of spontaneous clearance | Acute CMV: less priming of CMV-specific T cells | 71–74 |
| Acute CMV: reduced replication | |||||||
| IFNL4a | rs10853727 | Major: A/A | ? | - | Higher post-vaccine measles titers | 76 | |
| (IFNL3) | Minor: A/G, G/G | ||||||
| IFNL4 | ss469415590 | Major: T/T | CDR | Frameshift, causing IFNL4 expression | HCV: reduced IFN-α treatment response, lower rates of spontaneous clearance | ? | 20, 21 |
| (rs368234815) | Minor: T/ΔG, ΔG/ΔG | ||||||
| IFNLR1 | rs10903035 | Major: A/A Minor: G/A, G/G | 3′ UTR | ? | HCV: reduced IFN-α treatment response, lower rates of spontaneous clearance | ? | 77 |
Originally some SNPs were assigned to or close to IFNL3; however, with the discovery of IFNL4,21 there are various changes. We show both locations, although the rs129 79860 and the rs10853727 have their ‘true' location in IFNL4.
Originally the SNP rs8099917 has been assigned to IFNL3; however, its true location is between IFNL4 and IFNL4P1.
?, not known.
The impact of the SNPs on IFNL expression is debated. Most studies have concluded that the SNPs rs12979860 and rs8099917 are associated with reduced IFNL3 expression during chronic HCV infection in liver biopsies,71,72,73,74 serum,75 and PBMCs.49 In addition, in CMV-infected fibroblasts and stimulated PBMCs, SNPs in rs12979860 were associated with lower IFNL3 gene expression.49 To the best of our knowledge, no study has shown a significant increase in IFNL3 expression due to a SNP. There is a high likelihood that SNPs may also affect the function of other genes in the locus. These variants have been associated with alteration in serum IFNL1 and IFNL2 levels and with HCV infection outcomes, both spontaneous resolution and IFN treatment responses.34,74,76
The impact of the ss469415590 SNP on the expression of IFNL4 is well defined. In the context of a delta-G polymorphism, the frameshift mutation generates a ‘new' gene.21 Based on the genotype distributions of the strongly linked rs12979860 SNP, we could assume that almost 40% of Caucasians express IFNL4.
IFN-lambda receptor expression
The IL-28R consists of two subunits. IFNL first binds to the IL-28RA (α-subunit; encoded on chromosome 1), followed by the corecruitment of the IL-10RB (β-subunit; encoded on chromosome 21).25,77,78,79,80 In a similar fashion to the IFNAR receptor–ligand interaction, the dimerization of the receptor leads to the activation of JAK1 and tyrosine kinase 2 and phosphorylation of STAT-1 and -2. STAT1, STAT2 and IRF9 combine to form the IFN-stimulated gene factor-3 transcription factor complex, which induces the expression of multiple ISGs.
IL-10RB shows a broad expression pattern,81 whereas in contrast, the IL-28RA subunit is more restricted. A high expression of IL-28RA mRNA could be found in lung, intestine, and liver tissues with epithelial cells considered to be the main responders to IFNL exposure. A few other immune cells express IL-28RA especially at the mRNA level (e.g., B cells, macrophages and plasmacytoid DCs), but conflicting protein expression data are reported in the literature. Expression in brain tissue was low.9,10,45,82,83,84,85 IL-28RA also exists as a membrane bound form and a secreted form lacking exon VI, representing different splice variants.10,82,86 The secreted receptor may bind and inhibit IFNLs.
Regulation of receptor expression
Important transcription factor binding sites have been identified within the promoter region of the IL-28RA gene (e.g., STAT1, AP-2 and p53).87 This points to an upregulation of IL-28RA gene expression during stimulation with various IFNs. During maturation of monocytes to macrophages a significant upregulation of IL-28RA surface expression could be observed.88 The expression of IL-28RA on hepatocytes seems to be different between cell lines such as Huh7 or HepG2 (responsive to IFNL) and primary hepatocytes.89,90,91,92 Primary hepatocytes show an initial low responsiveness to IFNL due to a low baseline mRNA expression of IL-28RA, however during treatment with IFN-α a marked upregulation of IL-28RA mRNA was observed.88,89 Interestingly this upregulation was significantly enhanced in patients with a minor allele SNP genotype (rs12979860 and rs80999917) in IFNL3. This suggests the existence of a potent regulatory feedback loop between IFNL and IL-28RA.89 Although somewhat speculative at the current point, this increase in IL-28RA mRNA may be mediated by IFNL4 expression in the appropriate genetic background. During CMV infection of fibroblasts, the mRNA of IL-28RA increased by about two-fold, but protein expression levels (western blots) remained stable.49 Though requiring further definitive elucidation, these findings support considerable interplay in initial IFN mediated anti-viral responses, the ultimate outcomes of which are heavily influenced by the genetic background of the host.
Like IFNAR, IL-28RA could be modulated during infection by viral proteins or host and viral microRNAs.93 Interestingly, multiple SNPs in IL-28RA have been described (rs10903032, rs10903034, rs10903035, rs11249002 and rs11249006). The rs10903035 SNP (GA or GG) is within the 3′ untranslated region of the IL-28RA mRNA sequence and was an independent risk factor for IFN-α treatment failure against HCV (odds ratio: 2.0; 95% confidence interval: 1.19–3.36).46,94 This SNP has a relatively high minor allele frequency of 41.1% (ref SNP NCBI database). According to the 1000genome tool of the NCBI, the risk allele ranges from 25% (Iberian population in Spain) to 55% (Southern Han Chinese). The mechanism of how the SNPs in the IL-28RA gene affect IFN-α treatment outcomes has not been studied.
Interestingly, the rs10903035 IL-28RA SNP has been associated with insulin resistance in HIV/HCV co-infected patients.95 Another, rs4649203 has been linked to a risk of psoriasis in four independent populations,96 and to a risk to develop systemic lupus erythematosus.97 Although the IFNL3 SNPs have not been associated with treatment response of IFN-β in multiple sclerosis,98 the IL-28RA SNP itself was associated with an increased risk for multiple sclerosis.99,100 These observations suggest important influences on adaptive immune cells, which are tightly connected to autoimmune disease.
The ligand–receptor complex
The ligand interface includes helix A, loop AB and helix F (Figure 1). The IL-28RA interface arises from the N-terminal domain and interdomain hinge region. The extracellular region of IL-28RA has two β-sandwich domains D1 and D2. The interaction region consists largely of van der Waals and hydrophobicity.25 The most important amino acids on the ligand for interaction with IL28RA are located in the AB loop: Lys49 and Arg51 in IFNL3 and Arg49 and His51 in IFNL2, respectively.11 Binding to IL-10RB is important via the helix D amino acids: Gly95 in IFNL3 and Val95 in IFNL2. Of note IFNL4 differs in the area corresponding to the D helix of IFNL3.21 Various mutations in the receptor binding sites of IFNL3 and IFNL1 have been explored in terms of functionality 11,25,77.
The observed disparities in inducible responses to IFNs may arise due to relative differences in IFNL subtype expression, different ligand binding affinities and competition among IFNs for the same receptor. Rather than the affinity to the individual subunits, the stability of the ternary interferon receptor complex might be central to explaining the differences between the IFNL cytokines, akin to that observed with IFN-α.101 To date, no complete model for the ligand–IL-28RA–IL-10RB complex exists.
MAJOR FUNCTION OF IFNLS: DIRECT ANTI-VIRAL CONTROL
Not surprisingly, IFNLs share many biological activities with IFN-α, in particular direct anti-viral effects.15,102,103 Differences with reference to IFN-γ may be explained by the expression dynamics of IFN-γ, which is only secreted by a subset of immune cells such as natural killer cells and pathogen-specific T cells. In addition, the IFNGR expression differs and therefore, these effects are not comparable to IFN-α and -λ.104,105,106
IFN-α-treated Huh7 cells demonstrate a short induction of STAT1 phosphorylation (30 min–4 h), followed by a sharp reduction in ISG expression, temporally associated with the induction of the anti-inflammatory ISG USP18 (ubiquitin carboxy-terminal hydrolase 18). IFNL on the other hand induces both a later and more sustained phosphorylation of STAT-1 over 24 h29,107 (more details discussed below). The antiviral activity of IFNLs has been demonstrated in cell cultures infected with murine CMV,83 HIV,40 influenza virus,108,109 hepatitis B virus91 and HCV.84,91,110 In addition, their role in direct antiviral effects in vivo has also been demonstrated in IL-28RA and STAT1 knockout animals, where a significant increase in viral replication was observed.108,109,111
As mentioned previously, binding of IFN type-I and -III to their respective receptors induce a variety of ISGs.15 Anti-viral ISGs interfere with viral replication at different stages.15,17 Important examples are ISG15, MX1, OAS1-3, and PKR (proteins of 15 kDa; the dynamin protein family of Mx GTP-ases; 2′5′-oligoadenylate synthetase; and protein kinase R, respectively). In contrast, the antiviral signaling is controlled by anti-inflammatory ISGs including USP18 and SOCS1-3 (suppressor of cytokine signaling), which interfere with the STAT signaling cascade. They function as part of a negative feedback loop to limit the extent and duration of the IFN response.112,113 This feedback mechanism seems to be inherently different for IFN-α and IFNLs (see details below).
Limited biological activity attributable to IFNL4 is only now emerging following its original description. IFNL4 can induce ISG expression in HepG2 cells and anti-viral effects are also described against HCV infected Huh7-lunet cells, as well as for coronaviruses in human airway epithelial cell culture.20 Blockade of IL-10RB with siRNA and blocking antibodies significantly reduced the activity of IFNL4. Compared to IFNL3, however, IFNL4 demonstrated an almost 10-fold lower EC50 in a human embryonic kidney (HEK293) cell luciferase reporter system.20 The efficacy of his-tagged IFNL4 secretion seems to be significantly lower compared to IFNL3, which may account for the lower EC50.20,21
Differences in ISG expression between IFN types
A high overlap of gene induction between type-I and -III IFNs exists. However, in hepatocytes treated with IFN-α and IFNL3, differential effects on ISG expression levels and temporal kinetics have been described.114,115,116 IFN-α stimulates a rapid peaked induction of ISGs, whereas IFNL3 induces a slower, but more sustained increase in gene expression (Figure 2). IFN-α induced genes are generally associated with leukocyte chemotaxis, IFN signaling and lymphocyte proliferation, whereas IFNL3-induced genes are associated with antigen presentation and IFN signaling.114 More specifically, 6 h after stimulation with IFN-α, 134 genes were upregulated, with only 13 genes after 24 h. In contrast, 6 h after stimulation with IFNL3, 40 genes were upregulated, with 580 genes upregulated after 24 h. Of particular note is the differential expression profile of certain immunomodulatory genes. USP18 and SOCS1 were downregulated over 24 h in IFN-α-treated hepatocytes (USP18: 16.5- to 4.2-fold; SOCS1: 2.3- to 0.5-fold). IFNL3 on the other hand upregulated USP18 and SOCS1 over 24 h (USP 18: 13.6- to 19.6-fold; SOCS1: 3.3- to 6.8-fold).114 USP18 was shown to be necessary and sufficient to induce differential desensitization by impairing JAK1 at the IFNAR.117,118,119 Although these expression profiles may be cell type-specific, the potent and sustained effects of USP18 upregulation in the context of a chronic infection may significantly affect IFN-α induced signaling.
RNA sequencing of HepG2 cells transfected with IFNL4 and treated with IFN-α and IFNL3 suggests that some genes known as markers of HCV-induced liver damage, such as rantes and fos, were specifically induced by IFNL4 but not by other IFNs. This suggests specific functional roles for IFNL4, distinct from IFN-α and IFNL3.21 During infection with HCV, the expression pattern of many ISGs significantly change, most likely due to immunomodulatory effects of HCV proteins and complex inhibitory effects of both IFN signaling pathways.115
IMPACT OF IFNL SNPS ON INNATE ANTI-VIRAL EFFECTS OF IFNL3 AND IFNL4
The role of IFNLs in the host response to virus has been most well studied in HCV infection. HCV infection has been classically treated with (pegylated) IFN-α-based therapy to stimulate a robust antiviral immune response. Baseline ISG expression is considered an important marker of successful IFN-α treatment of HCV.72,120 Interestingly, patients with chronic HCV infection and non-responsiveness to IFN-α treatment show a higher baseline hepatocyte ISG expression.121,122,123,124,125,126 Elevated basal ISG expression and treatment failure have both been linked to SNPs in the IFNL3/4 and IL-28RA genes. The clinical importance of these SNPs is best explored in HCV infection and discussed in detail below. Importantly, however, the role of SNPs in the IFNL signaling may also be highly important for existing ‘old' and ‘emerging' pathogens. Though beyond the scope of this review, it is also conceivable that such polymorphisms shaped the tropism of viruses and influenced viral evolution and will continue to do so.50,127
IFN-feedback mechanism
Insight into the higher baseline ISG expression was provided through genome wide association studies of patients with chronic HCV infection. SNPs in the IFNL3 gene were associated with a risk of progression to chronic infection and significantly lower rates of HCV clearance in response to IFN-α-based treatment.26,74,76,128 Many SNPs (in particular rs12979860 and rs8099917) in the IFNL3 gene region were associated with lower IFNL3 expression levels and higher baseline ISG expression,71,110 suggesting the existence of a regulatory feedback mechanism. IFNL treatment and expression in hepatocytes is associated with potent induction of ‘immunoregulatory' ISGs, such as USP18 and SOCS1, whose duration of expression is comparatively longer compared to IFN-α.29,107,114 These ISGs act in an inhibitory capacity to dampen pro-inflammatory cascades. Likewise, in liver biopsies from HCV-infected patients with a major allele genotype, A20 and ZC3H12A were upregulated.123 A20 is a key inhibitor of NFκB signaling. ZC3H12A destabilizes the mRNA of IL-6 and IL-12p40—both potent inducers of an inflammatory response.129 The enhanced expression of these immunoregulatory ISGs may lead to a cumulative reduction of anti-viral ISG expression, thereby modulating the innate immune response to viruses.121,122,123,124,130,131
The paradoxical situation of higher ISG expression in IFNL3 SNPs despite lower IFNL3 gene expression could be explained by (i) a disruption in the temporal regulatory aspects of type-I and -III IFN-signaling cascades (at the level of either ligand or receptor); or (ii) the actual expression of IFNL4 in the context of reduced IFNL3 by virtue of high IFNL SNP linkage. Based on the current knowledge and our work, we favor the former, as IFNL4 secretion seems to be low and the biological impact in comparison to other IFNL proteins would seem less potent. We postulate, that IFN-α induces a more potent but temporally limited induction of ISGs due to a negative feedback loop (via SOCS1 and USP18, see above). In contrast, IFNLs, and in particular IFNL3 induce a somewhat ‘weaker' but more sustained ISG expression comprising both anti-viral and immunoregulatory profiles (Figure 2).
Figure 3 illustrates a proposed hypothesis regarding the feedback mechanism between IFN-α and IFNL3/4 according to specific genotypic backgrounds and in response to differing infection scenarios; acute and chronic viral infection (Figure 3). During acute viral infection, the expression of both IFN-α and IFNLs is triggered. In the context of a major allele IFNL3 genotype, IFNL3 is highly expressed and may better inhibit the initial replication of a virus due to a longer lasting anti-viral effect of induced ISGs. This may culminate in virological control and ultimately affect spontaneous clearance of virus. Clearly such outcomes will be reliant on efficient virus-specific T-cell priming, where a key role for IFNL is also proposed.
Figure 3.
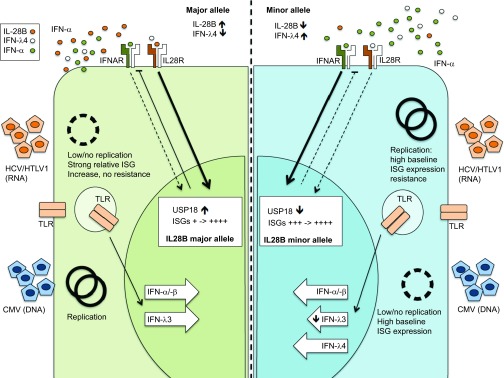
Hypothesis of IFNL3 single nucleotide polymorphism and impact on virus replication. The left panel shows a major allele genotype with a presumed fully functional IFNL3 expression during virus replication. The right shows a minor allele genotype with reduced IFNL3 expression, but increased IFNL4 expression. The genotype results in a significantly different ISG background expression which may be an advantage or disadvantage for different viruses.
In contrast, in the setting of a minor allele IFNL3 SNP, the amount of IFNL3, and subsequent anti-viral and modulatory ISG induction, is significantly lower. Thus, a sharp but limited IFN-α response predominates with high, short-lived ISG expression. This can foster breakthrough replication of viruses and subsequent selection of quasi-species resistant to ISGs (especially relevant to RNA viruses lacking polymerase proof reading capacity such as HCV). The constant viral replication in conjunction with impaired IFNL3 mediated regulatory activity amplifies IFN-α-mediated ISG expression further. In principle, the anti-viral properties of ISGs are similar to those of antiviral therapy where the threshold of replication is determined by a minimal inhibitory concentration.132,133,134,135 If the exposure of the virus towards ISGs is on occasion, as a consequence ‘subtherapeutic' mutation can occur with selection of quasi-species.
Aside from this proposed regulatory interplay between IFNs, the actual expression of a new player, IFNL4, could itself provide explanation for the elevated baseline ISG expression. It was recently shown that the expression of IFNL4 directly correlated with IFI44, IFIT1 and MX1 expression in hepatocytes.136 It remains to be seen if indeed the activity of this ‘new' gene product can directly account for the differing phenotypes observed.
At present, we would propose that the higher baseline expression of ISGs in patients with a minor allele genotype arises due to a combination of persistent activation of the IFN-α signaling cascade and a general reduction of inhibitory signals such as USP18 due to relative reductions in IFNL3 expression. In addition, it is likely that the newly discovered SNP in IL-28RA stands to affect all IFNL signaling, further disrupting the IFN-α and IFNL cross-talk. The clinical implications of this model are discussed in the next chapter.
Clinical impact of SNPs
The impact of SNPs in IFNL3 has clearly been defined for HCV infection.26,74,76,128 Minor allele carriage has been associated with high ISG background, and selection of quasi-species of ISG resistant HCV genomes131 (see above suggested model). Furthermore in this context exogenous IFN-α is incapable of effecting sufficient ISG induction from the relatively high baseline level that exists. Using the model of liver transplantation in HCV illustrates some of these aspects. Recently, it was shown that liver transplant recipients demonstrate an initial lower hepatitis C viral load if the donor liver graft has a minor allele genotype, and the recipient has a major allele genotype.137,138 In this specific constellation, a relatively ISG-susceptible virus (from the major allele recipient) has to replicate in a high ISG expression background (minor allele donor graft), which may result in improved initial control of replication. With iatrogenic immunosuppression, the adaptive immune response is impaired meaning viral replication control is achieved preferentially by the innate immune response. The reduced expression of IFNL3 (minor allele donor graft) allows a more potent IFN-α response with earlier control of the virus; however, with chronicity selection for resistant species ultimately occurs.
The role of various SNPs in the IFNL signaling cascade for other viruses is not as well established.139 In particular for the human T-lymphotropic virus 1, the impact of the minor allele genotype for IFNL3 (rs12979860) is unresolved following conflicting reports in terms of disease progression.140,141
Considering the above, it could be proposed that DNA viruses with accurate proof reading such as cytomegalovirus (during acute virus infection) would be at a disadvantage in a high ISG environment, as the development of escape mutants may take too long. Indeed, solid organ transplant recipients at high risk for primary (acute) CMV infection (donor sero-positive, recipient sero-negative) with rs8099917 minor allele genotype were in fact at decreased risk for CMV viremia in the first year post-transplant.49 A similar observation was made in hematopoietic stem cell transplant recipients where a trend for reduced risk of CMV replication and lower CMV viral loads was observed in a minor allele background.142 One potential explanation for this includes the combination of a higher anti-viral ISG background in the context of lower immunoregulatory activity. Without a doubt adaptive T-cell responses also make a telling contribution, which we will address in the following paragraphs.
EXPANDING ROLE FOR IFNL: FROM INNATE TO T- AND B-CELL MODULATION
Macrophages
Macrophages show marked phenotypic heterogeneity.143 Functional diversity results from a differentiation programme that is subject to environmental imprinting. Macrophages can regulate three different homeostatic activities—host defence, wound healing and immune regulation. Although a range of macrophages have been described, classically two types of macrophages can be differentiated: M1 and M2. The M1 phenotype is differentiated from the M0 phenotype in response to IFN-γ.144 M1 cells produce high amounts of IL-12 and IL-23, but low amounts of IL-10. In addition, effector molecules such as reactive oxygen species and inflammatory cytokines such as IL-1, tumor-necrosis factor-α and IL-6 are produced. A key feature is the induction of Th1 cells. M2 cells in contrast, are induced by IL-4 or IL-13. M2 cells do not present antigen to T cells and produce minimal amounts of pro-inflammatory cytokines. A key feature is the induction of Th2 cytokines and subsequent stimulation of T regulatory cells.145,146 Macrophages thus are positioned at the intersecting innate and adaptive immune responses continuously modulating the Th1 and Th2 cytokine balance during infection.
Interestingly, the differentiation cascade of monocytes to macrophages is influenced by IFNL.88 IFNL1 increases TLR-induced IL-12p40 production in human monocyte-derived macrophages. Furthermore, IFNL1-treated macrophages were more responsive to IFN-γ, because IFNL1 enhanced IFN-γ-induced IL-12p40 and tumor necrosis factor production by macrophages following R848 stimulation. In addition, IFNL1 upregulated, whereas IFN-α downregulated, the surface expression of the IFN-γ receptor α-subunit on macrophages, which affected the responsiveness of TLR-challenged macrophages to IFN-γ.88 This data highlights the diverse effects of IFN-α and IFNL1 in modulating macrophage populations. The influence of IFNL3/4 SNPs and IL-28RA genotypic background on macrophage polarization and function remains to be studied, but affecting macrophages in general will have a significant impact on host responses to infection (Figure 4).
Figure 4.
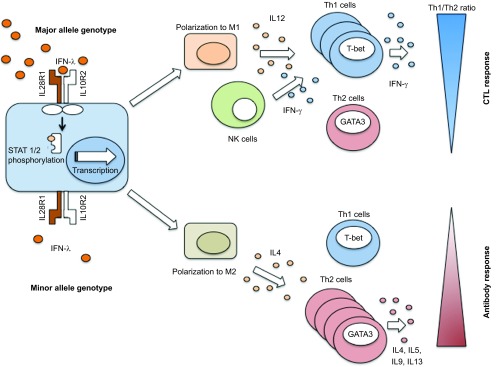
Impact of IFNL on Th1/Th2 cytokine profiling. The IFNL3 major allele genotype shows a predominant polarization to a macrophage M1 phenotype, whereas the minor allele genotype may show a relative increase of M2 phenotype. These macrophage phenotypes are associated with the downstream regulation and polarization of Th1 and Th2 cells. NK cells, natural killer cells.
Dendritic cells
Similar to macrophages, DCs function at the interface between innate and adaptive immune responses. In mice, CD8α+ DC promote T-cell skewing towards a Th1 lineage through the secretion of IL-12, while the CD8α− DC subsets were linked to Th2 differentiation.147 Human BDCA-3+ DC share several relevant characteristics with CD8α+ DC. The expression of IL-28RA on human DCs has been mostly seen on plasmacytoid DCs,46 while conflicting reports exist for expression of IL28RA on human monocyte derived DCs, possibly due to whether one looks at protein or mRNA.88,148 To the best of our knowledge, however, no functional differences have been studied in the context of differing IFNL genetic backgrounds.
Th1/Th2 balance
Virus-specific Th1 and cytotoxic T cells are crucial to control viral replication, whereas B cells receive important growth signals from Th2 cytokines.
Antigen-presenting cells such as DCs and macrophages play a decisive role in the differentiation of T-cell subsets into a Th1 or Th2 phenotype. IL-12 and IFN-γ are the critical cytokines initiating the downstream signaling cascade for the differentiation of Th1 cells.149 IL-12 is secreted in large amounts by antigen-presenting cells, in particular DCs and macrophages, following their activation through pathogen detection via pattern recognition receptors.150,151 IL-12, in turn, induces natural killer cells to produce IFN-γ. The major transcription factor to induce Th1 cells is T-bet.
IL-4 and IL-2 are critical for Th2 differentiation. The major transcription factor involved in Th2 lineage differentiation includes the IL-4-induced STAT6, which upregulates the expression of the master regulator GATA3.152,153,154,155 The expression of T-bet and GATA3 have opposing effects.156
Treatment of PBMCs with IFNLs have been shown to increase Th1 and suppress Th2 cytokine producing T cells.157,158,159,160,161 The IL-28R surface expression by immune cell fractions remains somewhat controversial. Though some authors have demonstrated mRNA expression of IL-28RA in T cells, this does not necessarily correlate with functional surface expression. In our hands, T cells do not express a surface IL-28RA protein (unpublished data). However, others have reported that purified naive and memory human CD4 T cells express IL-28RA and that these cells were responsive to IFNL1. The expression of Th2 cytokines (IL-4 and IL-13) was suppressed in CD4 T cells by IFNL1. Further GATA3 expression was reduced by IFNL1 treatment,157 and super-antigen stimulated cells pre-treated with IFNL1 showed significantly lower levels of IL-13. Strikingly, the IL-13 secretion was 100-fold more sensitive to IFNL1 treatment compared to IFN-γ. Interestingly, naive T cells exposed to allogeneic myeloid DC that had been matured in the presence of IFNL1, demonstrated a similar reduction in IL-13 production, IFN-γ secretion being unaffected.158 Taken together, this suggests that myeloid DC function may be significantly altered by IFNLs and have important downstream effects on the Th1/Th2 balance.
Likewise, IFNL3 treatment of lung DCs in a mouse model of asthma significantly increased Th1 cytokine and reduced Th2 cytokine expression.159 This activity may be of particular importance for immune-mediated lung diseases such as asthma. Indeed, in children with asthma, it was observed that those patients with a minor allele genotype had lower IFNL3 levels in BAL samples, but also significantly higher eosinophil granulocytes in sputa samples with higher Th2 cytokines.162,163,164,165,166
Keeping to the respiratory microenvironment blockade of the IL-28R markedly reduced the RSV-mediated suppression of CD4 T-cell proliferation, suggesting a specific role for IFNL signaling.167 We too observed a similar phenomenon with naïve CMV-specific T cells from sero-negative healthy blood donors demonstrating a reduced proliferation capacity when pre-treated with IFNL3 and stimulated with CMV lysate.49
The modulation of IFNL expression by SNPs in the IFNL3/4 gene region and IL28RA signaling cascade may potently affect Th1 and Th2 cytokine production and memory formation during viral infections. This remains to be fully explored but clearly represents a modulation of a critical component of the host response to pathogens.
B-cell activation
Th2 cytokines are known to promote optimal B-cell activation. Therefore, modulation of Th2 cytokines by IFNLs can profoundly affect B-cell functions (proliferation and antibody production). One study has examined the effects of IFNL1 and IFNL2 on the activation of B cells by anti-CD40 with/without cytokines. Treatment with IFNL1 or IFNL2 was able to modestly downregulate only IgG4 production in response to both anti-CD40 and IL-4.168
We would postulate that modulation of IFNL by virtue of a skew away from Th1 to Th2 could result in a propensity towards increased B-cell activation states in response to stimulation. Indeed, in a large cohort of healthy children vaccinated against measles, children with a minor allele genotype (rs10853727) had significantly higher post-vaccine antibody titers (median 807 mIU/mL versus 1727 mIU/mL).169 This actually suggests that the SNP potentially affecting IFNL4 expression may have a significant impact on antibody production in general; against pathogens, in response to vaccination, or in autoimmune diseases. This important association has to be explored in future studies.
SUMMARY
Infectious diseases are a perpetual threat with novel emerging entities a continuous challenge. Despite enormous advances in the understanding of the immunological processes involved in responses to pathogens, viral infections remain a theme of considerable unmet need. The discovery of IFNLs, and even more the determination of common SNPs, which affect their expression and biological function, has opened a new chapter and illustrates the intricate immunomodulatory activity, which critically determines infection outcomes. IFNL has direct antiviral functions, common to other IFNs; however, complex regulatory mechanisms exist which have implications on viral infection. Intriguingly, IFNL demonstrates activity at the interface of innate and adaptive immunity. Dynamic immune responses differ according to host IFNL3 genotypes with varied consequences for distinct viruses both those in existence and those now emerging. New data continue to emerge placing IFNL at the core of host responses effectively coordinating the fine-tuned activation of innate and adaptive immune responses. The long-term effects on the Th1/Th2 balance might have implications for the priming of T- and B-cell memory responses, but also for vaccine responses and autoimmune disease prevalence. Manipulation of IFNL signaling provides a means to specifically tailor and individualise therapeutic manipulations in favor of the host perhaps even taking into account the nature of the infecting pathogen. Future work will conclusively determine the role of IFNL SNPs in diverse viral infections outside of HCV. This signaling cascade appears optimally positioned to orchestrate or be manipulated to effect robust and durable responses to an ever-increasing array and complexity of pathogens.
Acknowledgments
We wish to thank Dr Rakesh Bhat (University of Alberta, Canada) for providing graphs of the ligand receptor interaction model. Adrian Egli's research was supported by the ‘Nachwuchsförderung' University of Basel, Switzerland (2013). Deanna M Santer's research was supported by Canadian Institutes of Health Research and Alberta Innovates Health Solutions postdoctoral fellowships. D Lorne Tyrrell's research was supported by the Li Ka Shing Institute of Virology and the Canadian Institutes of Health Research (MOP 126142).
References
- Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–267. [PubMed] [Google Scholar]
- Kotenko SV, Langer JA. Full house: 12 receptors for 27 cytokines. Int Immunopharmacol. 2004;4:593–608. doi: 10.1016/j.intimp.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Allen G, Fantes KH. A family of structural genes for human lymphoblastoid (leukocyte-type) interferon. Nature. 1980;287:408–411. doi: 10.1038/287408a0. [DOI] [PubMed] [Google Scholar]
- Seto MH, Harkins RN, Adler M, Whitlow M, Church WB, Croze E. Homology model of human interferon-alpha 8 and its receptor complex. Protein Sci. 1995;4:655–670. doi: 10.1002/pro.5560040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli M, Nagata S, Weissmann C. At least three human type alpha interferons: structure of alpha 2. Science. 1980;209:1343–1347. doi: 10.1126/science.6158094. [DOI] [PubMed] [Google Scholar]
- Karpusas M, Nolte M, Benton CB, Meier W, Lipscomb WN, Goelz S. The crystal structure of human interferon beta at 2.2-A resolution. Proc Natl Acad Sci USA. 1997;94:11813–11818. doi: 10.1073/pnas.94.22.11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senda T, Shimazu T, Matsuda S, et al. Three-dimensional crystal structure of recombinant murine interferon-beta. EMBO J. 1992;11:3193–3201. doi: 10.1002/j.1460-2075.1992.tb05396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ealick SE, Cook WJ, Vijay-Kumar S, et al. Three-dimensional structure of recombinant human interferon-gamma. Science. 1991;252:698–702. doi: 10.1126/science.1902591. [DOI] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Sheppard P, Kindsvogel W, Xu W, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- Gad HH, Hamming OJ, Hartmann R. The structure of human interferon lambda and what it has taught us. J Interferon Cytokine Res. 2010;30:565–571. doi: 10.1089/jir.2010.0062. [DOI] [PubMed] [Google Scholar]
- Kumaran J, Wei L, Kotra LP, Fish EN. A structural basis for interferon-alpha–receptor interactions. FASEB J. 2007;21:3288–3296. doi: 10.1096/fj.07-8585com. [DOI] [PubMed] [Google Scholar]
- Fasler-Kan E, Pansky A, Wiederkehr M, Battegay M, Heim MH. Interferon-alpha activates signal transducers and activators of transcription 5 and 6 in Daudi cells. Eur J Biochem. 1998;254:514–519. doi: 10.1046/j.1432-1327.1998.2540514.x. [DOI] [PubMed] [Google Scholar]
- Matikainen S, Sareneva T, Ronni T, Lehtonen A, Koskinen PJ, Julkunen I. Interferon-alpha activates multiple STAT proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and pim-1 genes in human T cells. Blood. 1999;93:1980–1991. [PubMed] [Google Scholar]
- Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaronson DS, Horvath CM. A road map for those who don't know JAK–STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Blouin CM, Lamaze C. Interferon gamma receptor: the beginning of the journey. Front Immunol. 2013;4:267. doi: 10.3389/fimmu.2013.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamming OJ, Terczynska-Dyla E, Vieyres G, et al. Interferon lambda 4 signals via the IFNlambda receptor to regulate antiviral activity against HCV and coronaviruses. EMBO J. 2013;32:3055–3065. doi: 10.1038/emboj.2013.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko SV. The family of IL-10-related cytokines and their receptors: related, but to what extent. Cytokine Growth Factor Rev. 2002;13:223–240. doi: 10.1016/s1359-6101(02)00012-6. [DOI] [PubMed] [Google Scholar]
- Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res. 2010;30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox BA, Sheppard PO, O'Hara PJ. The role of genomic data in the discovery, annotation and evolutionary interpretation of the interferon-lambda family. PLoS ONE. 2009;4:e4933. doi: 10.1371/journal.pone.0004933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miknis ZJ, Magracheva E, Li W, Zdanov A, Kotenko SV, Wlodawer A. Crystal structure of human interferon-lambda1 in complex with its high-affinity receptor interferon-lambdaR1. J Mol Biol. 2010;404:650–664. doi: 10.1016/j.jmb.2010.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Kutalik Z, Descombes P, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–1345. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- Hillyer P, Mane VP, Schramm LM, et al. Expression profiles of human interferon-alpha and interferon-lambda subtypes are ligand- and cell-dependent. Immunol Cell Biol. 2012;90:774–783. doi: 10.1038/icb.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellgren C, Gad HH, Hamming OJ, Melchjorsen J, Hartmann R. Human interferon-lambda3 is a potent member of the type III interferon family. Genes Immun. 2009;10:125–131. doi: 10.1038/gene.2008.87. [DOI] [PubMed] [Google Scholar]
- Bolen CR, Ding S, Robek MD, Kleinstein SH. Dynamic expression profiling of Type I and Type III Interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology. 2014;59:1262–1272. doi: 10.1002/hep.26657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann KM, Tran KC, Chi B, Rabin RL, Collins PL. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected] J Virol. 2004;78:4363–4369. doi: 10.1128/JVI.78.8.4363-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis I, Ye F, McNally B, Willette M, Flano E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol. 2013;87:3261–3270. doi: 10.1128/JVI.01956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR.Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo J Virol2006; 804501–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccia EM, Severa M, Giacomini E, et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. 2004;34:796–805. doi: 10.1002/eji.200324610. [DOI] [PubMed] [Google Scholar]
- Langhans B, Kupfer B, Braunschweiger I, et al. Interferon-lambda serum levels in hepatitis C. J Hepatol. 2011;54:859–865. doi: 10.1016/j.jhep.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Diegelmann J, Beigel F, Zitzmann K, et al. Comparative analysis of the lambda-interferons IL-28A and IL-29 regarding their transcriptome and their antiviral properties against hepatitis C virus. PLoS ONE. 2010;5:e15200. doi: 10.1371/journal.pone.0015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihm S, Frese M, Meier V, et al. Interferon type I gene expression in chronic hepatitis C. Lab Invest. 2004;84:1148–1159. doi: 10.1038/labinvest.3700135. [DOI] [PubMed] [Google Scholar]
- Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltz M, Klingstrom J. Alpha/beta interferon (IFN-alpha/beta)-independent induction of IFN-lambda1 (interleukin-29) in response to Hantaan virus infection. J Virology. 2010;84:9140–9148. doi: 10.1128/JVI.00717-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N, Iversen MB, Bartholdy C, et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol. 2008;180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- Hou W, Wang X, Ye L, et al. Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J Virol. 2009;83:3834–3842. doi: 10.1128/JVI.01773-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra PK, Bao L, Song K, et al. HCV infection selectively impairs type I but not type III IFN signaling. Am J Pathol. 2014;184:214–229. doi: 10.1016/j.ajpath.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T, Morrison TE, Funkhouser W, et al. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 2008;4:e1000240. doi: 10.1371/journal.ppat.1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitov MR, Laza-Stanca V, Edwards MR, et al. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy. 2009;64:375–386. doi: 10.1111/j.1398-9995.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- Jewell NA, Cline T, Mertz SE, et al. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol. 2010;84:11515–11522. doi: 10.1128/JVI.01703-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk K, Witte K, Witte E, et al. Maturing dendritic cells are an important source of IL-29 and IL-20 that may cooperatively increase the innate immunity of keratinocytes. J Leukoc Biol. 2008;83:1181–1193. doi: 10.1189/jlb.0807525. [DOI] [PubMed] [Google Scholar]
- Yin Z, Dai J, Deng J, et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol. 2012;189:2735–2745. doi: 10.4049/jimmunol.1102038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach H, Bathke B, Gilles S, et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med. 2010;207:2703–2717. doi: 10.1084/jem.20092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megjugorac NJ, Gallagher GE, Gallagher G. IL-4 enhances IFN-lambda1 (IL-29) production by plasmacytoid DCs via monocyte secretion of IL-1Ra. Blood. 2010;115:4185–4190. doi: 10.1182/blood-2009-09-246157. [DOI] [PubMed] [Google Scholar]
- Egli A, Levin A, Santer DM, et al. Immunomodulatory function of interleukin-28B during primary infection with Cytomegalovirus J Infect Dise-pub ahead of print 8 April 2014. 10.1093/infdis/jiu144 [DOI] [PubMed]
- McFadden G, Mohamed MR, Rahman MM, Bartee E. Cytokine determinants of viral tropism. Nat Rev Immunol. 2009;9:645–655. doi: 10.1038/nri2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze MG, He Y, Gale M., Jr Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- Dankar SK, Miranda E, Forbes NE, et al. Influenza A/Hong Kong/156/1997(H5N1) virus NS1 gene mutations F103L and M106I both increase IFN antagonism, virulence and cytoplasmic localization but differ in binding to RIG-I and CPSF30. Virol J. 2013;10:243. doi: 10.1186/1743-422X-10-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumpey TM, Szretter KJ, van Hoeven N, et al. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J Virol. 2007;81:10818–10821. doi: 10.1128/JVI.01116-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhang L, Geng H, et al. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell. 2013;4:951–961. doi: 10.1007/s13238-013-3096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol. 2004;173:1535–1548. doi: 10.4049/jimmunol.173.3.1535. [DOI] [PubMed] [Google Scholar]
- Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Molecular Cell. 1998;1:507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- Onoguchi K, Yoneyama M, Takemura A, et al. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282:7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- Osterlund PI, Pietila TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179:3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- Thomson SJ, Goh FG, Banks H, et al. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc Natl Acad Sci USA. 2009;106:11564–11569. doi: 10.1073/pnas.0904477106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin P, Vaccaro A, Civas A. The role of differential expression of human interferon—a genes in antiviral immunity. Cytokine Growth Factor Rev. 2009;20:283–295. doi: 10.1016/j.cytogfr.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Lee HC, Narayanan S, Park SJ, Seong SY, Hahn YS. Transcriptional regulation of IFN-lambda genes in hepatitis C virus-infected hepatocytes via IRF-3.IRF-7.NF-kappaB complex. J Biol Chem. 2014;289:5310–5319. doi: 10.1074/jbc.M113.536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths SJ, Koegl M, Boutell C, et al. A systematic analysis of host factors reveals a Med23-interferon-lambda regulatory axis against herpes simplex virus type 1 replication. PLoS Pathog. 2013;9:e1003514. doi: 10.1371/journal.ppat.1003514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggal P, Thio CL, Wojcik GL, et al. Genome-wide association study of spontaneous resolution of hepatitis C virus infection: data from multiple cohorts. Ann Intern Med. 2013;158:235–245. doi: 10.7326/0003-4819-158-4-201302190-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibert S, Roger T, Calandra T, et al. IL28B expression depends on a novel TT/-G polymorphism which improves HCV clearance prediction. J Exp Med. 2013;210:1109–1116. doi: 10.1084/jem.20130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaswamy S, Chatterjee S, Boopathi R, Mukherjee S, Bhattacharjee S, Kundu TK. A single nucleotide polymorphism associated with hepatitis C virus infections located in the distal region of the IL28B promoter influences NF-kappaB-mediated gene transcription. PLoS ONE. 2013;8:e75495. doi: 10.1371/journal.pone.0075495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J, Bohm S, Scholz M, et al. Combined effects of different interleukin-28B gene variants on the outcome of dual combination therapy in chronic hepatitis C virus type 1 infection. Hepatology. 2012;55:1700–1710. doi: 10.1002/hep.25582. [DOI] [PubMed] [Google Scholar]
- Smith KR, Suppiah V, O'Connor K, et al. Identification of improved IL28B SNPs and haplotypes for prediction of drug response in treatment of hepatitis C using massively parallel sequencing in a cross-sectional European cohort. Genome Med. 2011;3:57. doi: 10.1186/gm273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castellarnau M, Aparicio E, Parera M, et al. Deciphering the interleukin 28B variants that better predict response to pegylated interferon-alpha and ribavirin therapy in HCV/HIV-1 coinfected patients. PLoS ONE. 2012;7:e31016. doi: 10.1371/journal.pone.0031016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland AP, Horner SM, Jarret A, et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol. 2014;15:72–79. doi: 10.1038/ni.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Shirasaki T, Shimakami T, et al. Hepatic interferon-stimulated genes are differentially regulated in the liver of chronic hepatitis C patients with different interleukin 28B genotypes. Hepatology. 2014;59:828–838. doi: 10.1002/hep.26788. [DOI] [PubMed] [Google Scholar]
- Dill MT, Duong FH, Vogt JE, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140:1021–1031. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- Rallon NI, Soriano V, Naggie S, et al. Impact of IL28B gene polymorphisms on interferon-lambda3 plasma levels during pegylated interferon-alpha/ribavirin therapy for chronic hepatitis C in patients coinfected with HIV. J Antimicrob Chemother. 2012;67:1246–1249. doi: 10.1093/jac/dkr598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- Shi X, Pan Y, Wang M, et al. IL28B genetic variation is associated with spontaneous clearance of hepatitis C virus, treatment response, serum IL-28B levels in Chinese population. PLoS ONE. 2012;7:e37054. doi: 10.1371/journal.pone.0037054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haralambieva IH, Ovsyannikova IG, Kennedy RB, et al. Associations between single nucleotide polymorphisms and haplotypes in cytokine and cytokine receptor genes and immunity to measles vaccination. Vaccine. 2011;29:7883–7895. doi: 10.1016/j.vaccine.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Sousa MA, Berenguer J, Fernandez-Rodriguez A, et al. IL28RA polymorphism (rs10903035) is associated with insulin resistance in HIV/HCV-coinfected patients. J Viral Hepat. 2014;21:189–197. doi: 10.1111/jvh.12130. [DOI] [PubMed] [Google Scholar]
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, Hartmann R. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem. 2009;284:20869–20875. doi: 10.1074/jbc.M109.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboul J, Gardiner K, Monneron D, Uze G, Lutfalla G. Comparative genomic analysis of the interferon/interleukin-10 receptor gene cluster. Genome Res. 1999;9:242–250. [PMC free article] [PubMed] [Google Scholar]
- Josephson K, Logsdon NJ, Walter MR. Crystal structure of the IL-10/IL-10R1 complex reveals a shared receptor binding site. Immunity. 2001;15:35–46. doi: 10.1016/s1074-7613(01)00169-8. [DOI] [PubMed] [Google Scholar]
- Magracheva E, Pletnev S, Kotenko S, Li W, Wlodawer A, Zdanov A. Purification, crystallization and preliminary crystallographic studies of the complex of interferon-lambda1 with its receptor. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:61–63. doi: 10.1107/S1744309109048817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SI, Jones BC, Logsdon NJ, et al. Structure and mechanism of receptor sharing by the IL-10R2 common chain. Structure. 2010;18:638–648. doi: 10.1016/j.str.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte K, Gruetz G, Volk HD, et al. Despite IFN-lambda receptor expression, blood immune cells, but not keratinocytes or melanocytes, have an impaired response to type III interferons: implications for therapeutic applications of these cytokines. Genes Immun. 2009;10:702–714. doi: 10.1038/gene.2009.72. [DOI] [PubMed] [Google Scholar]
- Brand S, Beigel F, Olszak T, et al. IL-28A and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am J Physiol Gastrointest Liver Physiol. 2005;289:G960–G968. doi: 10.1152/ajpgi.00126.2005. [DOI] [PubMed] [Google Scholar]
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- Maher SG, Sheikh F, Scarzello AJ, et al. IFNalpha and IFNlambda differ in their antiproliferative effects and duration of JAK/STAT signaling activity. Cancer Biol Ther. 2008;7:1109–1115. doi: 10.4161/cbt.7.7.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumoutier L, Lejeune D, Hor S, Fickenscher H, Renauld JC. Cloning of a new type II cytokine receptor activating signal transducer and activator of transcription (STAT)1, STAT2 and STAT3. Biochem J. 2003;370:391–396. doi: 10.1042/BJ20021935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Luo Y, Wei J, He S. Integrative genomic analyses on IL28RA, the common receptor of interferon-lambda1, -lambda2 and -lambda3. Int J Mol Med. 2010;25:807–812. [PubMed] [Google Scholar]
- Liu BS, Janssen HL, Boonstra A. IL-29 and IFNalpha differ in their ability to modulate IL-12 production by TLR-activated human macrophages and exhibit differential regulation of the IFNgamma receptor expression. Blood. 2011;117:2385–2395. doi: 10.1182/blood-2010-07-298976. [DOI] [PubMed] [Google Scholar]
- Duong FH, Trincucci G, Boldanova T, et al. IFN-lambda receptor 1 expression is induced in chronic hepatitis C and correlates with the IFN-lambda3 genotype and with nonresponsiveness to IFN-alpha therapies. J Exp Med. 2014;211:857–868. doi: 10.1084/jem.20131557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli A, Zhang X, Yang J, et al. Distinct and overlapping genomic profiles and antiviral effects of interferon-lambda and -alpha on HCV-infected and noninfected hepatoma cells. J Viral Hepat. 2012;19:843–853. doi: 10.1111/j.1365-2893.2012.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits hepatitis B and C virus replication. J Virol. 2005;79:3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li J, Wang X, et al. Hepatitis C virus impairs TLR3 signaling and inhibits IFN-lambda 1 expression in human hepatoma cell line. Innate Immun. 2014;20:3–11. doi: 10.1177/1753425913478991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedger LM. microRNA control of interferons and interferon induced anti-viral activity. Mol Immunol. 2013;56:781–793. doi: 10.1016/j.molimm.2013.07.009. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sousa MA, Berenguer J, Rallon N, et al. IL28RA polymorphism is associated with early hepatitis C virus (HCV) treatment failure in human immunodeficiency virus-/HCV-coinfected patients. J Viral Hepat. 2013;20:358–366. doi: 10.1111/jvh.12041. [DOI] [PubMed] [Google Scholar]
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2, Strange A, Capon F et al A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cheng H, Zuo XB, et al. Association analyses identifying two common susceptibility loci shared by psoriasis and systemic lupus erythematosus in the Chinese Han population. J Med Genet. 2013;50:812–818. doi: 10.1136/jmedgenet-2013-101787. [DOI] [PubMed] [Google Scholar]
- Malhotra S, Morcillo-Suarez C, Brassat D, et al. IL28B polymorphisms are not associated with the response to interferon-beta in multiple sclerosis. J Neuroimmunol. 2011;239:101–104. doi: 10.1016/j.jneuroim.2011.08.004. [DOI] [PubMed] [Google Scholar]
- Lopez de Lapuente A, Alloza I, Goertsches R, et al. Analysis of the IL28RA locus as genetic risk factor for multiple sclerosis. J Neuroimmunol. 2012;245:98–101. doi: 10.1016/j.jneuroim.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Vandenbroeck K, Alvarez J, Swaminathan B, et al. A cytokine gene screen uncovers SOCS1 as genetic risk factor for multiple sclerosis. Genes Immun. 2012;13:21–28. doi: 10.1038/gene.2011.44. [DOI] [PubMed] [Google Scholar]
- Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G. The stability of the ternary interferon–receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J Biol Chem. 2008;283:32925–32936. doi: 10.1074/jbc.M806019200. [DOI] [PubMed] [Google Scholar]
- Kelly C, Klenerman P, Barnes E. Interferon lambdas: the next cytokine storm. Gut. 2011;60:1284–1293. doi: 10.1136/gut.2010.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumoutier L, Tounsi A, Michiels T, Sommereyns C, Kotenko SV, Renauld JC. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J Biol Chem. 2004;279:32269–32274. doi: 10.1074/jbc.M404789200. [DOI] [PubMed] [Google Scholar]
- Steed A, Buch T, Waisman A, Virgin HW., 4th Gamma interferon blocks gammaherpesvirus reactivation from latency in a cell type-specific manner. J Virol. 2007;81:6134–6140. doi: 10.1128/JVI.00108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNgamma signaling—does it mean JAK–STAT. Cytokine Growth Factor Rev. 2008;19:383–394. doi: 10.1016/j.cytogfr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Kearney S, Delgado C, Lenz LL. Differential effects of type I and II interferons on myeloid cells and resistance to intracellular bacterial infections. Immunol Res. 2013;55:187–200. doi: 10.1007/s12026-012-8362-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakhova OA, Kim KI, Luo JK, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordstein M, Kochs G, Dumoutier L, et al. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 2008;4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordstein M, Neugebauer E, Ditt V, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol. 2010;84:5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- Mahlakoiv T, Ritz D, Mordstein M, et al. Combined action of type I and type III interferon restricts initial replication of SARS-Coronavirus in the lung but fails to inhibit systemic virus spread. J Gen Virol. 2012;93:2601–2605. doi: 10.1099/vir.0.046284-0. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- Thomas E, Gonzalez VD, Li Q, et al. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology. 2012;142:978–988. doi: 10.1053/j.gastro.2011.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilg N, Lin W, Hong J, et al. Kinetic differences in the induction of interferon stimulated genes by interferon-alpha and IL28B are altered by infection with hepatitis C virus. Hepatology. 2014;59:1250–1261. doi: 10.1002/hep.26653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Birerdinc A, Estep M, Stepanova M, Afendy A, Baranova A. The impact of IL28B genotype on the gene expression profile of patients with chronic hepatitis C treated with pegylated interferon alpha and ribavirin. J Transl Med. 2012;10:25. doi: 10.1186/1479-5876-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M, Wang X, Yan M, et al. Alpha interferon induces long-lasting refractoriness of JAK–STAT signaling in the mouse liver through induction of USP18/UBP43. Mol Cell Biol. 2009;29:4841–4851. doi: 10.1128/MCB.00224-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS ONE. 2011;6:e22200. doi: 10.1371/journal.pone.0022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill MT, Makowska Z, Duong FH, et al. Interferon-gamma-stimulated genes, but not USP18, are expressed in livers of patients with acute hepatitis C. Gastroenterology. 2012;143:777–786. doi: 10.1053/j.gastro.2012.05.044. [DOI] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M, Oakeley EJ, Duong FH, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci USA. 2008;105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M, Krol J, Markiewicz I, Heim MH, Filipowicz W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat Med. 2009;15:31–33. doi: 10.1038/nm.1902. [DOI] [PubMed] [Google Scholar]
- Honda M, Sakai A, Yamashita T, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139:499–509. doi: 10.1053/j.gastro.2010.04.049. [DOI] [PubMed] [Google Scholar]
- Abe H, Hayes CN, Ochi H, et al. Inverse association of IL28B genotype and liver mRNA expression of genes promoting or suppressing antiviral state. J Med Virol. 2011;83:1597–1607. doi: 10.1002/jmv.22158. [DOI] [PubMed] [Google Scholar]
- Chen L, Borozan I, Feld J, et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128:1437–1444. doi: 10.1053/j.gastro.2005.01.059. [DOI] [PubMed] [Google Scholar]
- Feld JJ, Nanda S, Huang Y, et al. Hepatic gene expression during treatment with peginterferon and ribavirin: identifying molecular pathways for treatment response. Hepatology. 2007;46:1548–1563. doi: 10.1002/hep.21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau DT, Negash A, Chen J, et al. Innate immune tolerance and the role of kupffer cells in differential responses to interferon therapy among patients with HCV genotype 1 infection. Gastroenterology. 2013;144:402–413. doi: 10.1053/j.gastro.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manry J, Laval G, Patin E, et al. Evolutionary genetic dissection of human interferons. J Exp Med. 2011;208:2747–2759. doi: 10.1084/jem.20111680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- Uehata T, Akira S. mRNA degradation by the endoribonuclease Regnase-1/ZC3H12a/MCPIP-1. Biochim Biophys Acta. 2013;1829:708–713. doi: 10.1016/j.bbagrm.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Urban TJ, Thompson AJ, Bradrick SS, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52:1888–1896. doi: 10.1002/hep.23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa S, Sakamoto M, Miura M, et al. Comprehensive analysis for viral elements and interleukin-28B polymorphisms in response to peginterferon plus ribavirin therapy in hepatitis C virus 1B infection. Hepatology. 2012;56:1611–1621. doi: 10.1002/hep.25826. [DOI] [PubMed] [Google Scholar]
- Gullberg E, Cao S, Berg OG, et al. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011;7:e1002158. doi: 10.1371/journal.ppat.1002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima C, Polyak SJ, Ray R, et al. Hepatitis C virus-specific immune responses and quasi-species variability at baseline are associated with nonresponse to antiviral therapy during advanced hepatitis C. J Infect Dis. 2006;193:931–940. doi: 10.1086/500952. [DOI] [PubMed] [Google Scholar]
- Sarrazin C, Mihm U, Herrmann E, et al. Clinical significance of in vitro replication-enhancing mutations of the hepatitis C virus (HCV) replicon in patients with chronic HCV infection. J Infect Dis. 2005;192:1710–1719. doi: 10.1086/497142. [DOI] [PubMed] [Google Scholar]
- Rousseau CM, Daniels MG, Carlson JM, et al. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype c proteome: immune escape and viral load. J Virol. 2008;82:6434–6446. doi: 10.1128/JVI.02455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentenaar RJ, Gamadia LE, van DerHoek N, et al. Development of virus-specific CD4+ T cells during primary cytomegalovirus infection. J Clin Invest. 2000;105:541–548. doi: 10.1172/JCI8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte-Rojo A, Veldt BJ, Goldstein DD, et al. The course of posttransplant hepatitis c infection: comparative impact of donor and recipient source of the favorable IL28B genotype and other variables. Transplantation. 2012;94:197–203. doi: 10.1097/TP.0b013e3182547551. [DOI] [PubMed] [Google Scholar]
- Cisneros E, Banos I, Citores M, et al. Increased risk of severe hepatitis C virus recurrence after liver transplantation in patients with a T allele of IL28B rs12979860. Transplantation. 2012;94:275–280. doi: 10.1097/TP.0b013e31825668f6. [DOI] [PubMed] [Google Scholar]
- Russell CD, Griffiths SJ, Haas J. Interferon lambda genetic polymorphisms and viral infection: the tip of the iceberg. DNA Cell Biol. 2014;33:60–63. doi: 10.1089/dna.2013.2261. [DOI] [PubMed] [Google Scholar]
- Trevino A, Lopez M, Vispo E, et al. Development of tropical spastic paraparesis in human T-lymphotropic virus type 1 carriers is influenced by interleukin 28B gene polymorphisms. Clin Infect Dis. 2012;55:e1–e4. doi: 10.1093/cid/cis343. [DOI] [PubMed] [Google Scholar]
- Kamihira S, Usui T, Ichikawa T, et al. Paradoxical expression of IL-28B mRNA in peripheral blood in human T-cell leukemia virus type-1 mono-infection and co-infection with hepatitis C virus. Virol J. 2012;9:40. doi: 10.1186/1743-422X-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo D, Solano C, Gimenez E, et al. Effect of the IL28B Rs12979860 C/T polymorphism on the incidence and features of active cytomegalovirus infection in allogeneic stem cell transplant patients. J Med Virol. 2013;86:838–844. doi: 10.1002/jmv.23865. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthner R, Anders HJ. Interferon-regulatory factors determine macrophage phenotype polarization. Mediators Inflamm. 2013;2013:731023. doi: 10.1155/2013/731023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. 2013;93:875–881. doi: 10.1189/jlb.1012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–343. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohu K, Ohmori H, Wong WF, et al. The Runx3 transcription factor augments Th1 and down-modulates Th2 phenotypes by interacting with and attenuating GATA3. J Immunol. 2009;183:7817–7824. doi: 10.4049/jimmunol.0802527. [DOI] [PubMed] [Google Scholar]
- Mennechet FJ, Uze G. Interferon-lambda-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood. 2006;107:4417–4423. doi: 10.1182/blood-2005-10-4129. [DOI] [PubMed] [Google Scholar]
- Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 2000;14:1693–1711. [PubMed] [Google Scholar]
- Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- Zhu J, Guo L, Watson CJ, Hu-Li J, Paul WE. Stat6 is necessary and sufficient for IL-4's role in Th2 differentiation and cell expansion. J Immunol. 2001;166:7276–7281. doi: 10.4049/jimmunol.166.12.7276. [DOI] [PubMed] [Google Scholar]
- Zhu J, Min B, Hu-Li J, et al. Conditional deletion of Gata3 shows its essential function in TH1–TH2 responses. Nat Immunol. 2004;5:1157–1165. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- Lohoff M, Mak TW. Roles of interferon-regulatory factors in T-helper-cell differentiation. Nat Rev Immunol. 2005;5:125–135. doi: 10.1038/nri1552. [DOI] [PubMed] [Google Scholar]
- Dai J, Megjugorac NJ, Gallagher GE, Yu RY, Gallagher G. IFN-lambda1 (IL-29) inhibits GATA3 expression and suppresses Th2 responses in human naive and memory T cells. Blood. 2009;113:5829–5838. doi: 10.1182/blood-2008-09-179507. [DOI] [PubMed] [Google Scholar]
- Jordan WJ, Eskdale J, Srinivas S, et al. Human interferon lambda-1 (IFN-lambda1/IL-29) modulates the Th1/Th2 response. Genes Immun. 2007;8:254–261. doi: 10.1038/sj.gene.6364382. [DOI] [PubMed] [Google Scholar]
- Koltsida O, Hausding M, Stavropoulos A, et al. IL-28A (IFN-lambda2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol Med. 2011;3:348–361. doi: 10.1002/emmm.201100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Dai J, Eskdale J, Gallagher GE, Megjugorac NJ, Gallagher G. Interferon-lambda1 (interleukin-29) preferentially down-regulates interleukin-13 over other T helper type 2 cytokine responses in vitro. Immunology. 2008;125:492–502. doi: 10.1111/j.1365-2567.2008.02862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher G, Megjugorac NJ, Yu RY, et al. The lambda interferons: guardians of the immune-epithelial interface and the T-helper 2 response. J Interferon Cytokine Res. 2010;30:603–615. doi: 10.1089/jir.2010.0081. [DOI] [PubMed] [Google Scholar]
- Bullens DM, Decraene A, Dilissen E, et al. Type III IFN-lambda mRNA expression in sputum of adult and school-aged asthmatics. Clin Exp Allergy. 2008;38:1459–1467. doi: 10.1111/j.1365-2222.2008.03045.x. [DOI] [PubMed] [Google Scholar]
- Edwards MR, Johnston SL. Deficient interferon in virus-induced asthma exacerbations. Clin Exp Allergy. 2008;38:1416–1418. doi: 10.1111/j.1365-2222.2008.03064.x. [DOI] [PubMed] [Google Scholar]
- Baraldo S, Contoli M, Bazzan E, et al. Deficient antiviral immune responses in childhood: distinct roles of atopy and asthma. J Allergy Clin Immunol. 2012;130:1307–1314. doi: 10.1016/j.jaci.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Contoli M, Message SD, Laza-Stanca V, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- Russell CD, Griffiths SJ, Haas J. Interferon lambda genetic polymorphisms and viral infection: the tip of the iceberg. DNA Cell Biol. 2014;33:60–63. doi: 10.1089/dna.2013.2261. [DOI] [PubMed] [Google Scholar]
- Chi B, Dickensheets HL, Spann KM, et al. Alpha and lambda interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J Virol. 2006;80:5032–5040. doi: 10.1128/JVI.80.10.5032-5040.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummelshoj L, Ryder LP, Poulsen LK. The role of the interleukin-10 subfamily members in immunoglobulin production by human B cells. Scand J Immunol. 2006;64:40–47. doi: 10.1111/j.1365-3083.2006.01773.x. [DOI] [PubMed] [Google Scholar]