

Key Points
Rapamycin significantly enhances lentiviral vector gene delivery to hematopoietic stem cells while preserving engraftment potential.
Rapamycin-mediated transduction enhancement is not accompanied by alterations in lentiviral integration profile.
Abstract
Transplantation of genetically modified hematopoietic stem cells (HSCs) is a promising therapeutic strategy for genetic diseases, HIV, and cancer. However, a barrier for clinical HSC gene therapy is the limited efficiency of gene delivery via lentiviral vectors (LVs) into HSCs. We show here that rapamycin, an allosteric inhibitor of the mammalian target of rapamycin complexes, facilitates highly efficient lentiviral transduction of mouse and human HSCs and dramatically enhances marking frequency in long-term engrafting cells in mice. Mechanistically, rapamycin enhanced postbinding endocytic events, leading to increased levels of LV cytoplasmic entry, reverse transcription, and genomic integration. Despite increasing LV copy number, rapamycin did not significantly alter LV integration site profile or chromosomal distribution in mouse HSCs. Rapamycin also enhanced in situ transduction of mouse HSCs via direct intraosseous infusion. Collectively, rapamycin strongly augments LV transduction of HSCs in vitro and in vivo and may prove useful for therapeutic gene delivery.
Introduction
Genetically modified hematopoietic stem cells (HSCs) offer an attractive therapeutic strategy for the treatment of inherited and acquired hematologic disorders such as HIV infection and cancer. Multipotent HSCs are capable of engrafting and self-renewing in the recipient host to provide life-long hematopoietic reconstitution.1,2 Genetically modified HSCs would ideally exhibit similar properties as normal HSCs, with the exogenously provided genetic program carried throughout the hematopoietic system. Human HSCs are commonly identified by the CD34+ antigen, which marks a heterogeneous cell population with both short- and long-term engrafting cells. Gene delivery to HSCs for genetic modification has revolved around the use of γ-retroviral vectors and self-inactivating HIV-based lentiviral vectors (LVs) pseudotyped with the vesicular stomatitis virus G glycoprotein (VSV-G). LVs are advantageous over γ-retroviral vectors for 2 reasons: they are able to integrate into the genome of noncycling CD34+ cells,3-5 and they lower oncogenic risk by reducing propensity for integration near transcriptional start sites and eliminating long terminal repeat–based gene expression.6-8
Despite recent advances, barriers for optimized HSC gene therapy still exist. Key to successful HSC gene therapy is the development of methods that allow highly efficient gene delivery under ex vivo conditions that do not significantly alter biological properties of HSCs. We and others have reported modest LV transduction efficiency of 15% to 25% in nonobese diabetic/severe combined immunodeficiency–repopulating HSCs derived from human umbilical cord blood in the absence of cytokine stimulation.9,10 Short-term culture of CD34+ cells with appropriate HSC-supportive cytokines approximately doubled transduction efficiency over that achieved under nonstimulating conditions.11,12 However, prolonged culture with the same cytokines resulted in a loss of human cell engraftment in nonobese diabetic/severe combined immunodeficiency mice without further enhancement in transduction, suggesting a trade-off between transduction efficiency and engraftment potential during ex vivo culture.12 Other methods aimed at enhancing LV transduction in HSCs, such as increasing the multiplicity of infection (MOI), using sequential transductions, or employing alternative envelope pseudotypes, have resulted in limited success in laboratory settings.11,13 Clinically, similarly modest patient marking efficacy has been reported from studies assessing LV-based gene-replacement strategies to treat β-thalassemia, leukodystrophy, and Wiskott-Aldrich syndrome, unless employing very high MOIs and extended ex vivo culture.14-18 These reports imply that intrinsic barriers exist in HSCs that limit LV transduction efficiency.
Early events in the HIV-1 replication cycle are not well defined and are targeted by multiple cell-intrinsic restriction pathways.19 Autophagy, a lysosomal degradative pathway that maintains cellular homeostasis and supports survival during periods of stress, is emerging as an antiviral defense mechanism. Autophagy intersects with HIV-1 replication in an intricate manner.20 In macrophages, basal autophagy is required for efficient HIV-1 infection, and early autophagosomes promote Gag processing.21,22 However, in line with its antiviral properties, mature autophagosomes degrade HIV-1 virions in macrophages, and HIV-1 downregulates productive autophagy in T cells, dendritic cells, and macrophages to facilitate infection.23,24 Despite mounting evidence for a role of autophagy in HIV-1 replication in mature hematopoietic cells, it has not been investigated in HSCs. Furthermore, existing evidence points to a role for autophagy in HIV-1 protein synthesis and assembly, but it is not known whether autophagy affects early stages of HIV-1 replication such as entry and uncoating. In this study, we investigated the effect of rapamycin, a canonical inducer of autophagy via inhibition of the mammalian target of rapamycin (mTOR) complexes, in LV entry and integration in mouse and human HSCs. We found that rapamycin-induced mTOR inhibition, but not autophagy, significantly enhanced HSC transduction while preserving engraftment potential. Our finding that small-molecule inhibitors of mTOR could modulate LV transduction efficiency in HSCs is potentially applicable in clinical settings.
Methods
Human HSC transduction and mouse transplantation
Human cord blood CD34+ cells, under approved institutional protocol and in accordance with the Declaration of Helsinki, were isolated using the EasySep Human Cord Blood CD34 Positive Selection Kit (STEMCELL Technologies, Vancouver, BC, Canada). The general transduction conditions are shown in Figure 1A. CD34+ cells were cultured in Iscove modified Dulbecco medium containing 20% BIT 9500 (STEMCELL Technologies) and either not stimulated or prestimulated for 24 hours with 50 ng/mL each of human thrombopoietin, human granulocyte colony-stimulating factor, and human interleukin-6, 100 ng/mL human Flt-3 ligand, and 150 ng/mL human stem cell factor (SCF; Peprotech, Rocky Hills, NJ). After 24 hours of prestimulation, cells were transduced at 1 × 105 cells/mL for 12 hours in the presence of 4 μg/mL polybrene. Rapamycin, or dimethylsulfoxide (DMSO) only as a vehicle control, was included in the transduction mixture where indicated and washed out after transduction. Cells were prestimulated unless otherwise stated. Following transduction, cells were maintained in growth phase for 10 to 14 days in vitro using Iscove modified Dulbecco medium containing 10% fetal bovine serum, 50 ng/mL of interleukin-3 and interleukin-6, and 100 ng/mL SCF. Enhanced green fluorescent protein (EGFP) expression was assessed by flow cytometry. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were maintained at The Scripps Research Institute under approved institutional protocol. For transplantation, 8- to 10-week-old NSG mice were irradiated at 230 cGy using a cesium source and injected retro-orbitally with freshly transduced CD34+ cells immediately after the 12-hour transduction period, within 24 hours of irradiation. Mice were bled retro-orbitally every 4 weeks and sacrificed 19 weeks posttransplantation to assess engraftment and EGFP marking.
Figure 1.

Rapamycin increases in vitro LV transduction efficiency in human and mouse hematopoietic progenitors. (A) Scheme of human CD34+ and mouse Lin− cell transduction and assessment. (B) Human cord blood CD34+ cells were transduced with CG-UbiC-EGFP LV in the presence of rapamycin or DMSO only as a diluent control. Black triangles, DMSO only; red circles, 10 μg/mL rapamycin. Cells were analyzed 10 to 14 days posttransduction. (C) Human cord blood CD34+ cells were transduced with CG-UbiC-EGFP, MOI = 25, in the presence of various concentrations of rapamycin or (D) Torin 1, an active site mTOR inhibitor. In most cases, data are pooled from 4 to 5 independent experiments, each with different cord blood donors and done in duplicate. At lower MOI or rapamycin concentrations, 2 different donors in duplicated experiments were assessed. (E) MFI of EGFP from a representative titration series in panel C. (F) Fold change over DMSO-treated controls in integrated LV copy number per cell, determined by qPCR, in CD34+ cells transduced in the presence of various concentrations of rapamycin and analyzed 10 to 14 days posttransduction. Data are pooled from 2 to 5 independent experiments, each with different cord blood donors. (G) EGFP cell marking and (H) fold change in integrated LV copy number in mouse bone marrow Lin− cells transduced with RRL-MND-GFP at various MOIs in the absence (DMSO only) or presence of 1 or 5 μg/mL of rapamycin and analyzed 10 to 12 days posttransduction. Data shown are derived from 4 independent experiments comprising 48 donor animals. LV copy numbers are shown as fold change over the average LV copy number of the DMSO-treated controls. For all panel, lines represent group mean and error bars represent standard deviation. *P < .05, **P < .01, ***P < .001, and ****P < .0001 from a parametric 2-tailed unpaired Student t test. BM, bone marrow; hu, human; mu, murine; Rapa, rapamycin.
Mouse HSC transduction and mouse transplantation
C57BL/6 mice were maintained at Seattle Children’s Research Institute under approved institutional protocol. Bone marrow was harvested from 6- to 8-week-old C57BL/6 congenically marked (CD45.1+ or CD45.2+) mice, and Lin− cells were isolated using EasySep Mouse Hematopoietic Progenitor Cell Enrichment kit (STEMCELL Technologies). Cells were transduced at 2 × 106 cells/mL in StemSpan SFEM (STEMCELL Technologies) containing 2% fetal calf serum, 50 ng/mL mouse SCF, and 20 ng/mL mouse thrombopoietin (PeproTech) for 12 to 16 hours in the presence of 4 μg/mL polybrene. Rapamycin, or DMSO only as a vehicle control, was included in the transduction mixture where indicated and washed out after transduction. Following transduction, mice were transplanted with 1 × 106 transduced cells injected retro-orbitally into C57BL/6 congenically disparate mice after 2 × 450 cGy total body irradiation. An aliquot of transduced cells were cultured in vitro for 10 to 12 days for analysis of input transduction levels. Mice were sacrificed at 11 to 16 weeks posttransplantation for analyses of engraftment and EGFP marking.
Additional methodology is detailed in the supplemental Methods (available at the Blood Web site).
Results
Rapamycin enhances in vitro transduction efficiency in mouse and human HSCs
To study the potential impact of autophagy on LV transduction of HSCs, we performed transduction in the presence of rapamycin, an inducer of autophagy through allosteric inhibition of mTOR. Human cord blood CD34+ cells, a population that contains HSCs and hematopoietic progenitors, were stimulated with HSC-supportive cytokines in serum-free medium and transduced with third-generation EGFP-expressing LVs (CG-UbiC-EGFP) in the presence of rapamycin (Figure 1A). Transduction efficiency was evaluated by flow cytometry after in vitro culture in rapamycin-free, serum-containing medium for 10 to 12 days. Rapamycin enhanced in vitro transduction efficiency by a factor of 2 or greater over a range of MOIs at concentrations above 5 μg/mL (Figure 1B-C). Torin 1, an active site mTOR inhibitor,25 also enhanced transduction at concentrations above 1.25 μM (Figure 1D). Mean fluorescence intensity (MFI) (Figure 1E) and fold-change in LV copy number per cell (Figure 1F) increased concomitantly with EGFP marking frequency. We observed minimal differences in the viability of CD34+ cells in the presence of 5 to 20 μg/mL of rapamycin (supplemental Figure 1A). Growth rate of rapamycin-treated cells was initially retarded in a dose-dependent manner, consistent with the previously reported antiproliferative effect of rapamycin (supplemental Figure 1B),26,27 but returned to levels of control cells within 12 days. Rapamycin-treated cells exhibited reduced forward scattering by flow cytometry, characteristic of smaller cell size (supplemental Figure 1C). Moreover, continued treatment with rapamycin has been shown to reduce expansion, but not myeloid differentiation of human CD34+ cells.28 Assessment of colony-forming activity of CD34+ cells after prestimulation and 12-hour rapamycin treatment indicated that 10 μg/mL of rapamycin slightly depressed progenitor plating efficiency, whereas 20 μg/mL had a more substantive effect, relative to controls (supplemental Figure 2A). Cell-type distribution among colonies, consistent with differentiation, was not statistically altered among rapamycin treatment and control groups (supplemental Figure 2B-C).
Next, we investigated whether rapamycin-mediated transduction enhancement was limited by HSC activation status, cell source, or LV entry receptor. We transduced CD34+ cells with or without 24 hours of cytokine prestimulation and observed largely diminished transduction enhancement in the absence of cytokines (supplemental Figure 3A). To determine if transduction enhancement is limited by the source of HSCs, we transduced human bone marrow CD34+ cells, as well as mouse bone marrow lineage negative (Lin−) cells containing HSCs and hematopoietic progenitors. Similar to observations in cord blood cells, rapamycin enhanced transduction of human bone marrow CD34+ cells, especially when cells were stimulated with cytokines (supplemental Figure 3B). Rapamycin (1-5 μg/mL) also enhanced transduction and LV copy number in purified mouse Lin− cells across a range of MOI (Figure 1G-H). To determine if transduction enhancement is restricted to VSV-G pseudotyped LVs that use LDL receptors for entry, we generated LVs pseudotyped with Lassa virus glycoprotein (LASV), which mediates α-dystroglycan–dependent entry.29,30 Transduction with LASV-LVs was enhanced by a factor of 21 in the presence of rapamycin (supplemental Figure 3C).
Rapamycin enhances transduction efficiency while preserving engraftment of mouse and human primitive HSCs
To determine if rapamycin enhanced LV transduction of HSCs capable of long-term engraftment, we transduced mouse Lin− donor bone marrow cells, in the presence or absence of 5 μg/mL of rapamycin and transplanted equal numbers of transduced cells into lethally irradiated, congenically marked recipients. We assessed engraftment, EGFP expression, and LV copy number at 11 to 16 weeks posttransplantation. Rapamycin did not adversely affect the engraftment ability of transduced Lin− HSCs; indeed, bone marrow and spleen donor cell levels were slightly increased in the rapamycin-treated group compared with controls (Figure 2A). Consistent with our in vitro observations, rapamycin significantly enhanced the frequency of EGFP-marked donor cells and LV copy number per cell for both Lin−Sca+c-Kit+ HSCs and lineage-committed bone marrow progenitor cells (Figure 2B-C). In individual differentiated hematopoietic subsets in the spleen, rapamycin significantly enhanced the frequency and MFI (not shown) of EGFP-marked donor B cells, T cells, monocytes, and neutrophils (Figure 2D). Importantly, EGFP-marked bone marrow cells from primary recipients of rapamycin-treated HSCs were able to engraft secondary recipients at equivalent levels as control cells (supplemental Figure 4A), and these recipient animals exhibited increased EGFP frequency and MFI (not shown) in Lin− and lineage-committed bone marrow cells and in all splenic hematopoietic subsets, with a trend for increased LV copy number in Lin− HSCs (supplemental Figure 4B-C).
Figure 2.

Increased EGFP marking in mouse hematopoietic lineages arising from rapamycin-treated and LV-transduced Lin− cells transplanted into mice. Congenically marked mouse bone marrow Lin− cells were transduced for 16 hours with RRL-MND-GFP LV, MOI = 2.5, in the presence of 5 μg/mL rapamycin or DMSO. Cells were washed, and 1 × 106 cells per mouse were injected retro-orbitally into lethally irradiated congenically disparate recipients. Mice were sacrificed 11 (gray triangles or open red circles) or 16 (black triangles or closed red circles) weeks posttransplant. (A) Percent donor cell engraftment in bone marrow and spleen. (B) EGFP marking, assessed by flow cytometry, in total donor bone marrow cells and bone marrow HSCs (Lin−Sca-1+c-Kit+). (C) LV copy number per cell in Lin− and Lin+ bone marrow cells as determined by qPCR (described in “Methods”). (D) EGFP marking in splenic subsets analyzed with the following markers: B220+ B cells, B220−CD4+ or CD8+ T cells, B220−CD4−CD8−CD11b+GR1lo/neg monocytes, and B220−CD4−CD8−CD11b+GR1hi neutrophils. Each point represents 1 mouse. Cells were pregated on donor-derived cells for all subset assessments. For all panels, lines represent group mean and error bars represent standard deviation. *P < .05, **P < .01, and ***P < .001 from a parametric 2-tailed unpaired Student t test.
Next, we asked whether LV transduction of CD34+ cells treated in the absence or presence of 10 or 20 μg/mL rapamycin enhanced EGFP marking of HSCs capable of long-term engraftment and development in sublethally irradiated NSG mice. Assessment of an aliquot of CD34+ cells used for transplanting NSG mice, either expanded in liquid culture or from in vitro colony formation assay, demonstrated increased frequencies of EGFP marking (supplemental Figure 5A-B). At 19 weeks post-CD34+ cell transplantation and engraftment in NSG mice, we observed no significant differences in human cell bone marrow engraftment levels between the 10 μg/mL rapamycin treatment and control groups, and modestly lowered engraftment in the 20 μg/mL rapamycin treatment group, possibly resulting from fewer CD34+ cells injected per mouse (Figure 3A). Both rapamycin treatment groups showed increased EGFP marking in CD45+ human cells in the bone marrow (Figure 3B) and spleen (supplemental Figure 4C), as well as increased MFI and bone marrow LV copy number (Figure 3B and supplemental Figure 5C). Hematopoietic lineage analyses showed increased EGFP marking frequency in bone marrow CD34+ HSCs, as well as myeloid, B cells, and T cells from the bone marrow, spleen, and thymus (Figure 3C and supplemental Figure 5D). EGFP-marked primary NSG bone marrow cells were capable of engrafting sublethally irradiated secondary recipients and maintaining EGFP expression (supplemental Figure 5E).
Figure 3.

Increased EGFP marking in human hematopoietic lineages arising from rapamycin-treated and LV-transduced CD34+ cells transplanted into NSG mice. Human cord blood CD34+ cells were prestimulated for 24 hours in HSC-supportive cytokines and then transduced for 12 hours with CG-UbiC-EGFP, MOI = 25, in the presence of DMSO, 10 μg/mL rapamycin, or 20 μg/mL rapamycin. NSG mice each received 3 × 106 CD34+ cells (DMSO control or 10 μg/mL rapamycin treatment) or 2.6 × 106 CD34+ cells (20 μg/mL rapamycin treatment). Mice were sacrificed 19 weeks posttransplant. (A) Total hCD45+ engraftment levels in the bone marrow. (B) EGFP expression and MFI were measured by flow cytometry in live hCD45+ from the BM (left). LV copy number per cell as determined by qPCR (described in “Methods”) (right). (C) EGFP expression in bone marrow and splenic subsets, analyzed with the following human markers: CD34+ for HSCs, CD33+ for myeloid cells, CD19+ for B cells, and CD3+ for T cells. Cells were pregated on hCD45 for subsequent lineage analyses. Each point represents 1 mouse. For all panels, lines represent group mean and error bars represent standard deviation. *P < .05, ***P < .001, and ****P < .0001 from a parametric 2-tailed unpaired Student t test.
Rapamycin-mediated transduction enhancement requires mTOR engagement
We investigated the role of rapamycin-mediated autophagy induction and mTOR inhibition in transduction. We hypothesized that autophagy induction alone without mTOR regulation may be sufficient to bring about transduction enhancement. However, when CD34+ cells were treated with a Tat-beclin 1 peptide recently shown to activate autophagy via an mTOR-independent mechanism,31 we observed a dose-dependent decline in transduction (Figure 4A). Furthermore, we found that Lin− bone marrow cells from mice heterozygous for beclin 1 which have impaired autophagy,32 showed the same degree of rapamycin-mediated transduction enhancement as wild-type cells, further supporting the lack of autophagy involvement in transduction (Figure 4B). We also did not detect colocalization of LV with LC3B+ vesicles by confocal microscopy, suggesting a lack of association between incoming LVs and LC3B+ autophagosomes (supplemental Figure 6). We next examined whether mTOR inhibition by rapamycin, upstream of autophagy induction, is necessary by using FK506 to disrupt rapamycin-mTOR interaction.33 FK506 diminished transduction enhancement induced by rapamycin (Figure 4C), but not by Torin 1 (Figure 4D), whose binding to mTOR is unaffected by FK506,25 indicating that mTOR inhibition is required.
Figure 4.

Rapamycin enhances LV transduction via an mTOR-dependent, nonautophagy mechanism. (A) Human cord blood CD34+ cells were transduced with CG-UbiC-EGFP, MOI = 25, in the absence or presence of the autophagy-stimulating peptide Tat-beclin 1. Black triangles, Tat-beclin 1 scrambled peptide control; blue circles, Tat-beclin 1. Data are pooled from 2 independent experiments, each with different cord blood donors and done in duplicate. (B) Bone marrow Lin− cells from wild-type or beclin 1+/− mice were transduced with RRL-MND-GFP, MOI = 0.5, in the presence of 5 μg/mL rapamycin or DMSO. EGFP marking was assessed by flow cytometry 10 days posttransduction. Data points represent cells from individual donor mice. Black triangles, wild-type cells; blue circles, beclin 1+/− cells. (C) Human cord blood CD34+ cells were transduced with CG-UbiC-EGFP, MOI = 25, in the presence of combinations of either rapamycin or (D) Torin 1 with FK506, an FKBP12-binding compound. Black triangles, DMSO only; green squares, 10 μg/mL FK506. Data are duplicate transductions from 2 representative experiments with different cord blood donors. For all panels, lines represent group mean and error bars represent standard deviation. ****P < .0001 from a parametric 2-tailed unpaired Student t test.
Rapamycin-mediated transduction enhancement is not due to modulation of LV capsid interacting factors, cellular p21 protein, or cell-cycle delay
We next determined whether rapamycin downregulates known LV restriction pathways in hematopoietic cells. The HIV-1 capsid is a major target for cellular factor recognition during viral entry and regulates postentry transduction efficiency through interactions with restriction factors such as TRIM5 or cofactors such as human cyclophilin A (CypA).34 The G89V capsid mutation abolishes interaction with CypA, reducing LV transduction efficiency and rendering LVs insensitive to the capsid-CypA–disrupting drug cyclosporin A.35,36 However, the G89V mutation did not abolish transduction enhancement by rapamycin (supplemental Figure 7A). Another set of capsid mutations that render LVs independent of downstream effects of CypA binding37 moderately enhanced transduction efficiency but did not preclude further enhancement by rapamycin (supplemental Figure 7B). Next, we examined whether rapamycin modulates the p21 protein, recently reported to restrict HIV-1 in HSCs.38 Flow cytometric analysis revealed no change in intracellular p21 protein levels following rapamycin treatment (supplemental Figure 7C).
Rapamycin is known to reduce cell-cycle progression and cell proliferation as well as promote HSC quiescence.39 To determine if the effect of rapamycin on transduction was distinct from that on cell cycle and growth of CD34+ cells, we first examined cell-cycle distribution of CD34+ cells following rapamycin treatment. We found a delayed progression through G1, characterized by reduced RNA and DNA levels within 6 and 12 hours of treatment, respectively (supplemental Figure 7D). However, a delayed cell cycle is unlikely to account for increased LV transduction, as pretreating and washing out rapamycin immediately prior to transduction initiation, a time frame during which the inhibitory effects on cell cycle are likely retained, did not increase transduction above control (supplemental Figure 7E).
Rapamycin enhances LV postbinding endocytic entry
To identify the cellular pathway involved in rapamycin-mediated transduction enhancement, we investigated the stage(s) at which LV transduction was affected. Rapamycin did not increase the amount of surface-bound LVs following a 2-hour incubation at 4°C as shown by anti-p24 enzyme-linked immunosorbent assay, even though it did allow increased transduction when cells were then washed and shifted to 37°C (Figure 5A). Cell-surface levels of LDL receptors and α-dystroglycan, the respective receptors for VSV-G and LASV, were unchanged after rapamycin treatment (Figure 5B). Rapamycin enhanced transduction by LVs prebound to the cell surface for 2 hours at 4°C (Figure 5C), but not by LVs that had entered cells for 2 hours at 37°C (Figure 5D).
Figure 5.

Rapamycin does not affect LV binding but enhances LV uptake into the cytoplasm. (A) Human cord blood CD34+ cells were treated with CG-UbiC-EGFP, MOI = 25, at 4°C (to prevent endocytosis and limit binding to the cell surface) for 2 hours, in the presence of 10 μg/mL rapamycin or DMSO. After transduction 60 000 to 80 000 cells were lysed and p24 protein content was determined to assess LV binding (left), and the remaining ∼10 000 cells were cultured at 37° for 10 days and evaluated for EGFP marking to determine transduction efficacy (right). Data are shown for 2 independent experiments with different cord blood donors, each done in triplicate. (B) Human cord blood CD34+ cells were prestimulated and treated with rapamycin or DMSO for 12 hours, and cell-surface LDL receptor (LDLR) and α-dystroglycan (α-DG) levels were determined by flow cytometry. (C) Human cord blood CD34+ cells were treated with DMSO (black triangles) or 10 μg/mL rapamycin (red circles) after allowing LVs to either prebind to the cell surface for 2 hours at 4°C or (D) preinternalize for 2 hours at 37°C followed by cleaving off external LVs with 0.05% trypsin (to remove bound LVs that have not internalized). Control represents same cells transduced under standard conditions outlined in Figure 1A. Data are duplicate or triplicate transductions from 1 experiment representative of 3 experiments with different cord blood donors. (E) Human cord blood CD34+ cells were transduced in the presence or absence of indicated concentrations of rapamycin, with CG-UbiC-mCherry, MO I = 25, carrying the BLAM-Vpr protein. After a 6-hour transduction, cells were loaded with the BLAM substrate CCF4-AM, and LV entry was quantified by flow cytometric detection of cells exhibiting cleaved CCF4. Data are pooled from 4 independent experiments, each with different cord blood donors. (F) Human cord blood CD34+ cells were transduced with CG-UbiC-EGFP, MOI = 25, and the following HIV-1 reverse-transcription products were quantified by qPCR: strong-stop DNA (Early RT), full-length DNA (Late RT), and 2-long terminal repeat circles. (G) Ratios between each pair of adjacent reverse-transcription products from panel F were calculated. Black, DMSO only; red, 10 μg/mL rapamycin. Data are pooled from 4 independent experiments, each with different cord blood donors. For all panels, lines represent group mean and error bars represent standard deviation. *P < .05, **P < .01, ***P < .001, and ****P < .0001 from a parametric 2-tailed unpaired Student t test (A,C,D-E) or paired Student t test (F-G). LTR, long terminal repeat.
These findings suggested that rapamycin enhanced postbinding events. Consistent with this idea, we observed increased cytoplasmic entry of LV cores following rapamycin treatment using a BLAM-Vpr fusion assay (Figure 5E).40 Moreover, the amounts of LV reverse-transcription products quantified by quantitative polymerase chain reaction (qPCR) were increased by a factor of 2 to 3, mirroring the magnitude of enhancement in in vitro transduction (Figure 5F). Notably, the efficiency of reverse transcription remained unchanged, indicated by similar ratios between adjacent pairs of reverse-transcription products in the absence or presence of rapamycin (Figure 5G). Also consistent with an effect of rapamycin early in LV processing, genomic integration was not required for transduction enhancement, as rapamycin increased transduction by integration-deficient LVs in both human and mouse HSCs (supplemental Figure 8). Finally, we verified that rapamycin treatment did not provide an alternative entry pathway for VSV-G pseudotyped LVs by blocking rapamycin-mediated transduction in human and mouse HSCs with bafilomycin A1, an inhibitor of pH-dependent endocytosis (supplemental Figure 9). Together, these findings show that rapamycin did not increase LV binding to cell-surface receptors but enhanced postbinding, pH-dependent endocytic entry mediated by VSV-G.
Rapamycin does not alter LV chromosomal integration profile
Given the increased LV copy number per cell, we asked whether rapamycin altered genomic accessibility for LV integration, because rapamycin is known to alter global cellular transcription.41-43 We performed high-throughput retrovirus integration site (RIS) analysis44 on Lin− bone marrow cells harvested from multiple mice derived from in vivo experiments. In agreement with the increase in LV copy number, we observed a significant increase in the total number of unique RISs in the rapamycin-treated group compared with control (Figure 6A). There were no differences in the chromosomal frequency of insertion between treatment groups, although insertions in chromosomes 7 and 13 exhibited a near-significant change (P values of .077 and .076, respectively) in the rapamycin-treated group (Figure 6B). Genome-wide display of RIS in these chromosomes (Figure 6C) suggested that this difference reflects fewer retrieved sites in control mice rather than an altered pattern of integration. Analysis of LV insertion site profiles proximal to mouse genes using QuickMap45 revealed similar patterns of insertion within genes and near gene transcription start elements between rapamycin-treated and control animals (supplemental Table 1). Using a semiquantitative capture frequency analysis to determine the relative contribution of each unique RIS-bearing clone to the Lin− cell pool in vivo, we observed dominant clonal contributions, defined as any unique RIS representing ≥20% of the detectable gene-modified cell pool, in both groups (supplemental Table 2). However, there was no difference in the relative frequency of clonal dominance that correlated with treatment group in this limited data set. A minimum of 35 additional clones were detected in all mice displaying clonal dominance, and none of the mice demonstrated evidence for myelodysplastic syndrome or leukemia.
Figure 6.

In vivo LV genomic integration profile in rapamycin-treated mouse bone marrow Lin− cells. (A) Total number of unique LV integration sites (RIS) identified in BM Lin− cells from primary transplant recipient mice described in Figure 2. *P < .05 from a parametric 2-tailed unpaired Student t test. (B) Percentage of the total number of RIS mapped to each chromosome. White, DMSO only; red, 5 μg/mL rapamycin. Asterisk (*) denotes borderline significant differences between the frequency of integration in chromosomes 7 and 13 (P = .077 and .076, respectively). (C) Genome distribution of identified integration sites. The mouse genome is represented by chromosomes 1 to 19, x and y clockwise from top. Inner circles represent all mice from a given treatment group: rapamycin-treated mice (first inner circle, warm colors) and control mice (second inner circle, cool colors). Each colored dot represents an individual integration event and is color-coded by the mouse in which it was identified within the specified treatment group. Integrations mapped to chromosomes 7 and 13 are magnified (center). For all panels, lines represent group mean and error bars represent standard deviation.
Rapamycin enhances in situ HSC transduction via IO infusion
The feasibility of in situ gene delivery by direct intraosseous (IO) infusion has recently been demonstrated in mice and could be adapted for human clinical trials.46-48 We hypothesized that localized delivery of rapamycin along with LVs via IO infusion may enhance in situ transduction. We performed mouse IO infusion of LVs in the presence or absence of 250 μg/kg codelivered rapamycin and examined EGFP expression 62 days after treatment. Rapamycin significantly increased the percentage of EGFP-expressing bone marrow Lin−Sca-1+c-Kit+ cells from 4% to 17%, whereas MFI and LV copy number per cell remained unchanged (Figure 7).
Figure 7.
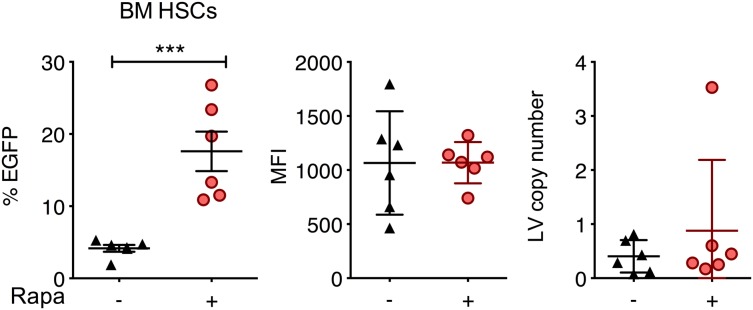
Rapamycin enhances in situ transduction of mouse bone marrow cells following IO infusion. C57BL/6 mice were treated with IO infusions of RRL-MND-GFP, 3.3 × 109 TU/mouse, in the absence or presence of 250 μg/kg rapamycin. Bone marrow cells were isolated 62 days after injection, and EGFP expression and MFI in bone marrow Lin−Sca-1+c-Kit+ cells were analyzed by flow cytometry. LV copy number per cell was determined by qPCR. Each point represents 1 mouse. Lines represent group mean, and error bars represent standard deviation. ***P < .001 from a parametric 2-tailed unpaired Student t test.
Discussion
Highly efficient gene delivery without compromising stem cell function is a holy grail in HSC gene therapy. Here, we show that a brief ex vivo rapamycin treatment efficiently enhances LV transduction of primitive human and mouse HSCs while preserving long-term and secondary engraftment. Transduction enhancement was reflected by concomitant increases in EGFP-marking frequency, MFI, and integrated LV copy number. Notably, when the same batch of transduced CD34+ cells was assayed in vitro and in vivo, we observed much higher transduction enhancement in vivo. This suggests that while rapamycin enhances LV transduction in both primitive and progenitor hematopoietic cells, the effect is much more pronounced in primitive HSCs and is dampened during differentiation. Furthermore, the cross-species observation of rapamycin-mediated transduction enhancement suggests the existence of an evolutionarily conserved pathway in HSCs in response to mTOR inhibition. Even though rapamycin modestly enhanced transduction efficiency in quiescent CD34+ cells, maximal enhancement was observed in the presence of HSC-supportive cytokines, suggesting involvement of additional cellular factors dependent on the activation state of HSCs.
Rapamycin-mediated transduction enhancement was dependent on mTOR inhibition and occurred via postbinding enhancement in LV endocytosis into the cytoplasm. Transduction enhancement occurred following treatment with either rapamycin or Torin 1, indicating that it can result from either direct or allosteric mTOR inhibition. Surprisingly, transduction enhancement appeared independent of rapamycin-induced autophagy. In fact, we observed little accumulation of cytoplasmic LC3B+ foci in CD34+ cells treated with 10 μg/mL rapamycin by confocal microscopy, suggesting that at these concentrations rapamycin may not be a potent autophagy inducer in HSCs. In contrast, treatment with Tat-beclin 1, which strongly promotes autophagy via an mTOR-independent mechanism,31 as was evidenced by increased autophagic vesicles in CD34+ cells (supplemental Figure 6), reduced LV transduction. Rather than enhancing autophagy, rapamycin functioned to enhance LV postbinding cytoplasmic entry events. Furthermore, we found that rapamycin did not affect LV cell-surface binding or efficiency of reverse transcription, but rather overcame the hurdle of delivering LV cores to the cytoplasm. This suggests that cytoplasmic entry may be a major restriction point in LV transduction of HSCs, analogous to observations using a cytoplasmic fusion-deficient HIV isolate, which can be rendered hyperinfectious by agents that disrupt the endocytic pathway to promote fusion.49 TORC1 has been shown to modulate endocytosis of plasma membrane proteins in yeast,50 but a role for mammalian mTOR complexes in endocytic regulation has yet to be established. It will be of interest to identify a physiological role(s) for this novel mTOR-regulated HSC endocytic program in future studies. Rapamycin-mediated transduction enhancement was observed for LVs pseudotyped with VSV-G or LASV, 2 envelope proteins that mediate clathrin-dependent, pH-dependent endocytosis. Further studies are needed to determine whether rapamycin enhances internalization via other entry pathways.
We investigated the role of other reported LV restriction pathways in HSCs and found that rapamycin-mediated transduction enhancement did not coincide with the action of capsid-binding factors or cellular p21 protein content. Mutating the LV capsid protein to abolish binding of human CypA or other factors had no effect on the magnitude of rapamycin-mediated transduction enhancement, and rapamycin did not alter p21 protein levels in CD34+ cells. This is in accordance with our finding that rapamycin enhances LV cytoplasmic entry, which is mechanistically upstream from the action of postentry restriction factors such as CypA, TRIM5, and p21.
Santoni de Sio et al reported that disruption of proteasome activity by MG132 enhanced LV transduction in CD34+ HSCs and other stem cells and that proteasome inhibition acted in conjunction with HSC-supportive cytokines for maximum relief of restriction, similar to our observations with rapamycin.13,51 The authors speculate that proteasome-mediated LV restriction targets postentry steps such as capsid uncoating and reverse transcription but did not directly examine the levels of cytoplasmic LV core or reverse-transcription products. Although little is known on the interplay between proteasome and mTOR in hematopoietic cells, it has been reported that mTOR activation requires proteasome function in muscle cells, and MG132 downregulates mTOR levels in tumor cells.52,53 Therefore, mechanistic overlap between mTOR and proteasome pathways in relieving LV restriction in HSCs is a possibility. On the other hand, Dueck and Guatelli54 reported maximum enhancement of HIV-1 infection using MG132 pretreatment rather than concurrent treatment, whereas we found that pretreatment with rapamycin abolished enhancement of LV transduction, suggesting different temporal actions of the 2 molecules. Further studies assessing the precise mechanisms whereby mTOR and proteasome inhibition relieve LV restriction are warranted.
Rapamycin reduces cell-cycle progression and cell proliferation in multiple cell types, which is the basis for its immunosuppressive and anticancer properties. Although rapamycin dampens cell-cycle progression of CD34+ cells, we observed rapid loss of transduction enhancement following rapamycin pretreatment and washout, suggesting that transduction enhancement likely occurs via a mechanism that quickly dissipates upon rapamycin withdrawal, distinct from cell-cycle delay. Nonetheless, cytokine stimulation was key for rapamycin enhancement, suggesting that additional cellular pathways independent of rapamycin activity were necessary. Apart from transduction enhancement, rapamycin may be beneficial for HSC gene therapy by promoting preservation of primitive HSCs, indicated by enhanced engraftment of CD34+ cells in NSG mice following ex vivo rapamycin treatment as previously reported.55 In this regard, we observed enhanced engraftment of rapamycin-treated mouse Lin− cells, but not human CD34+ cells, possibly due to the lower rapamycin concentration used to treat the former. Further adjustment of ex vivo transduction protocol may allow optimization of both transduction efficiency and engraftment ability in primitive HSCs.
Rapamycin is known to influence global transcriptional and translational profiles41-43 and could potentially impact LV integration site selection by altering chromosomal accessibility. Although rapamycin increased integrated LV copy number in Lin− mouse engrafting cells, we observed no significant alterations in chromosomal integration profile or LV insertional site usage. Consistent with the increase in total LV copy number, rapamycin-treated cells exhibited a broader range of integration sites and a trend toward decreased clonal dominance. Furthermore, none of the mice engrafted with rapamycin-treated cells displayed leukemia or oligoclonal reconstitution. However, analysis of larger numbers of recipient animals and integration sites will be required to verify these findings. The long-term safety of rapamycin-assisted gene delivery to human HSCs in clinical applications awaits detailed future examination.
Finally, rapamycin enhanced in situ HSC transduction via IO infusion, demonstrating its potential applicability to an alternative clinical transduction protocol that eliminates the need for ex vivo cell manipulation. We observed enhanced EGFP marking frequency, but not MFI or LV copy number, in in situ–transduced mouse bone marrow HSCs. This may be due to localized rapamycin concentration differing between the in situ and ex vivo settings or systemic effects of rapamycin. Thus, IO infusion may represent an attractive alternative to ex vivo transduction protocols by optimizing transduction frequency without increasing LV copy number per cell, further minimizing the risk of insertional mutagenesis. Further, our in situ findings suggest that fine-tuning rapamycin concentration in an ex vivo setting may allow enhanced transduction without concomitant increase in LV copy number per cell.
Taken together, our findings have major translational implications. Rapamycin treatment has the potential to significantly improve clinical efficacy of gene delivery to HSCs, possibly allowing the use of shorter ex vivo culture and/or fewer LV particles per cell to achieve clinically desirable LV copy numbers. In turn, this could be beneficial by reducing costs for clinical gene-delivery procedures. Our encouraging initial confirmation of the safety profile of rapamycin-assisted gene delivery, together with the practicality of rapamycin usage as a clinically approved drug, should open the door for future animal model and human studies.
Acknowledgments
The authors thank Olivia Garijo for excellent technical assistance in confocal microscopy, LV production, and hematopoietic cell culture; Nadia Ebrahim for excellent technical assistance in producing the CG-UbiC-mCherry plasmid; Dr. Nick Greco and the Cleveland Cord Blood Center, Cleveland, OH, for providing human cord blood; Dr. Lenny Shultz and Lisa Carney, The Jackson Laboratory, Bar Harbor, ME, for beclin 1+/− and control littermate mouse bone marrow; Drs. Skip Virgin and Tiffany Reese, Washington School of Medicine, St. Louis, MO, for Atg4B-null and control littermate mouse bone marrow; and Chris Tipper and Gabor Verse of Bluebird bio, INC, for suggestions and helpful advice.
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute, National Institute of General Medical Sciences, and National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers P50GM103368 (B.E.T.), R01HL116221 (B.E.T.), R01HL091219 (B.E.T.), R01AI084457 (D.J.R.), R01AI071163 (D.J.R.), PL1-HL092557 (D.J.R.), RL1-HL092553 (D.J.R.), R21-HL112148 (C.H.M.), HL098489 (H.-P.K.), HL084345 (H.-P.K.), and AI097100 (H.-P.K.), with additional funding from Canadian Institutes of Health Research doctoral research award 237503 (C.X.W.) and CHRP D12-SRI-355 (C.X.W.), core support from the Centers for AIDS Research (P30AI036214), and Seattle Children’s Center for Immunity and Immunotherapies. H.-P.K. also received support from the Markey Molecular Medicine Center at UW and he is the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research at Fred Hutchinson Cancer Research Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This is internal publication MEM #26030 from The Scripps Research Institute.
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.X.W. and B.E.T. conceptualized the study and designed the experiments; C.X.W. and B.E.T. performed or oversaw human HSC experiments; B.D.S., I.K., S.S., G.C., and D.J.R. performed or oversaw mouse HSC experiments; I.K. performed human and mouse HSC integrated LV copy-number assays; S.L. performed surface-bound LV p24 quantification; X.W. and C.H.M. performed or oversaw IO infusion experiments; J.A., A.A., and H.-P.K. performed or oversaw LV integration site analyses; and C.X.W., B.D.S., H.-P.K., C.H.M., D.J.R, and B.E.T. contributed to writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bruce E. Torbett, Department of Molecular and Experimental Medicine, The Scripps Research Institute, La Jolla, CA 92037; e-mail: betorbet@scripps.edu; and David J. Rawlings, Department of Pediatrics and Immunology, University of Washington School of Medicine, Seattle, WA 98101; e-mail: drawling@u.washington.edu.
References
- 1.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Maris M, Storb R. The transplantation of hematopoietic stem cells after non-myeloablative conditioning: a cellular therapeutic approach to hematologic and genetic diseases. Immunol Res. 2003;28(1):13–24. doi: 10.1385/IR:28:1:13. [DOI] [PubMed] [Google Scholar]
- 3.Uchida N, Sutton RE, Friera AM, et al. HIV, but not murine leukemia virus, vectors mediate high efficiency gene transfer into freshly isolated G0/G1 human hematopoietic stem cells. Proc Natl Acad Sci USA. 1998;95(20):11939–11944. doi: 10.1073/pnas.95.20.11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Case SS, Price MA, Jordan CT, et al. Stable transduction of quiescent CD34(+)CD38(-) human hematopoietic cells by HIV-1-based lentiviral vectors. Proc Natl Acad Sci USA. 1999;96(6):2988–2993. doi: 10.1073/pnas.96.6.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutton RE, Reitsma MJ, Uchida N, Brown PO. Transduction of human progenitor hematopoietic stem cells by human immunodeficiency virus type 1-based vectors is cell cycle dependent. J Virol. 1999;73(5):3649–3660. doi: 10.1128/jvi.73.5.3649-3660.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hematti P, Hong BK, Ferguson C, et al. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004;2(12):e423. doi: 10.1371/journal.pbio.0020423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewinski MK, Yamashita M, Emerman M, et al. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2006;2(6):e60. doi: 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montini E, Cesana D, Schmidt M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119(4):964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guenechea G, Gan OI, Inamitsu T, et al. Transduction of human CD34+ CD38- bone marrow and cord blood-derived SCID-repopulating cells with third-generation lentiviral vectors. Mol Ther. 2000;1(6):566–573. doi: 10.1006/mthe.2000.0077. [DOI] [PubMed] [Google Scholar]
- 10.Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science. 1999;283(5402):682–686. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- 11.Ailles L, Schmidt M, Santoni de Sio FR, et al. Molecular evidence of lentiviral vector-mediated gene transfer into human self-renewing, multi-potent, long-term NOD/SCID repopulating hematopoietic cells. Mol Ther. 2002;6(5):615–626. [PubMed] [Google Scholar]
- 12.Mazurier F, Gan OI, McKenzie JL, Doedens M, Dick JE. Lentivector-mediated clonal tracking reveals intrinsic heterogeneity in the human hematopoietic stem cell compartment and culture-induced stem cell impairment. Blood. 2004;103(2):545–552. doi: 10.1182/blood-2003-05-1558. [DOI] [PubMed] [Google Scholar]
- 13.Santoni de Sio FR, Cascio P, Zingale A, Gasparini M, Naldini L. Proteasome activity restricts lentiviral gene transfer into hematopoietic stem cells and is down-regulated by cytokines that enhance transduction. Blood. 2006;107(11):4257–4265. doi: 10.1182/blood-2005-10-4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 15.Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341(6148):1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiGiusto DL, Krishnan A, Li L, et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med. 2010;2(36):36ra43. doi: 10.1126/scitranslmed.3000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326(5954):818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 19.Blanco-Melo D, Venkatesh S, Bieniasz PD. Intrinsic cellular defenses against human immunodeficiency viruses. Immunity. 2012;37(3):399–411. doi: 10.1016/j.immuni.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5(6):527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell GR, Spector SA. Hormonally active vitamin D3 (1alpha,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J Biol Chem. 2011;286(21):18890–18902. doi: 10.1074/jbc.M110.206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kyei GB, Dinkins C, Davis AS, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186(2):255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanchet FP, Moris A, Nikolic DS, et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010;32(5):654–669. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borel S, Espert L, Biard-Piechaczyk M. Macroautophagy Regulation during HIV-1 Infection of CD4+ T Cells and Macrophages. Front Immunol. 2012;3:97. doi: 10.3389/fimmu.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thoreen CC, Kang SA, Chang JW, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mita MM, Mita A, Rowinsky EK. The molecular target of rapamycin (mTOR) as a therapeutic target against cancer. Cancer Biol Ther. 2003;2(4 Suppl 1):S169–S177. [PubMed] [Google Scholar]
- 27.Kay JE, Kromwel L, Doe SE, Denyer M. Inhibition of T and B lymphocyte proliferation by rapamycin. Immunology. 1991;72(4):544–549. [PMC free article] [PubMed] [Google Scholar]
- 28.Geest CR, Zwartkruis FJ, Vellenga E, Coffer PJ, Buitenhuis M. Mammalian target of rapamycin activity is required for expansion of CD34+ hematopoietic progenitor cells. Haematologica. 2009;94(7):901–910. doi: 10.3324/haematol.13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci USA. 2013;110(18):7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao W, Henry MD, Borrow P, et al. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science. 1998;282(5396):2079–2081. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- 31.Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wandless TJ, Michnick SW, Rosen MK, Karplus M, Schreiber SL. J Am Chem Soc. 1991;113(6):2339–2341. [Google Scholar]
- 34.Sokolskaja E, Berthoux L, Luban J. Cyclophilin A and TRIM5alpha independently regulate human immunodeficiency virus type 1 infectivity in human cells. J Virol. 2006;80(6):2855–2862. doi: 10.1128/JVI.80.6.2855-2862.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franke EK, Yuan HE, Luban J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 1994;372(6504):359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 36.Franke EK, Luban J. Inhibition of HIV-1 replication by cyclosporine A or related compounds correlates with the ability to disrupt the Gag-cyclophilin A interaction. Virology. 1996;222(1):279–282. doi: 10.1006/viro.1996.0421. [DOI] [PubMed] [Google Scholar]
- 37.Chatterji U, Bobardt MD, Stanfield R, et al. Naturally occurring capsid substitutions render HIV-1 cyclophilin A independent in human cells and TRIM-cyclophilin-resistant in Owl monkey cells. J Biol Chem. 2005;280(48):40293–40300. doi: 10.1074/jbc.M506314200. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Scadden DT, Crumpacker CS. Primitive hematopoietic cells resist HIV-1 infection via p21. J Clin Invest. 2007;117(2):473–481. doi: 10.1172/JCI28971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009;8(7):1003–1006. doi: 10.4161/cc.8.7.8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20(11):1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- 41.Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther. 2003;2(3):222–232. doi: 10.4161/cbt.2.3.360. [DOI] [PubMed] [Google Scholar]
- 42.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126(Pt 8):1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adair JE, Beard BC, Trobridge GD, et al. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci Transl Med. 2012;4(133):133ra57. doi: 10.1126/scitranslmed.3003425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Appelt JU, Giordano FA, Ecker M, et al. QuickMap: a public tool for large-scale gene therapy vector insertion site mapping and analysis. Gene Ther. 2009;16(7):885–893. doi: 10.1038/gt.2009.37. [DOI] [PubMed] [Google Scholar]
- 46.McCauslin CS, Wine J, Cheng L, et al. In vivo retroviral gene transfer by direct intrafemoral injection results in correction of the SCID phenotype in Jak3 knock-out animals. Blood. 2003;102(3):843–848. doi: 10.1182/blood-2002-12-3859. [DOI] [PubMed] [Google Scholar]
- 47.Worsham DN, Schuesler T, von Kalle C, Pan D. In vivo gene transfer into adult stem cells in unconditioned mice by in situ delivery of a lentiviral vector. Mol Ther. 2006;14(4):514–524. doi: 10.1016/j.ymthe.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Shin SC, Pan D, Rawlings DJ, Miao CH. Intraosseous delivery of lentiviral vectors expressing factor VIII under the control of the platelet-specific glycoprotein 1ba promoter leads to long-term correction of bleeding in hemophilia A mice. Mol Ther. 2013;21:S254. [Google Scholar]
- 49.Wei BL, Denton PW, O’Neill E, Luo T, Foster JL, Garcia JV. Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection. J Virol. 2005;79(9):5705–5712. doi: 10.1128/JVI.79.9.5705-5712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacGurn JA, Hsu PC, Smolka MB, Emr SD. TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell. 2011;147(5):1104–1117. doi: 10.1016/j.cell.2011.09.054. [DOI] [PubMed] [Google Scholar]
- 51.Santoni de Sio FR, Gritti A, Cascio P, et al. Lentiviral vector gene transfer is limited by the proteasome at postentry steps in various types of stem cells. Stem Cells. 2008;26(8):2142–2152. doi: 10.1634/stemcells.2007-0705. [DOI] [PubMed] [Google Scholar]
- 52.Ko JK, Choi CH, Kim YK, Kwon CH. The proteasome inhibitor MG-132 induces AIF nuclear translocation through down-regulation of ERK and Akt/mTOR pathway. Neurochem Res. 2011;36(5):722–731. doi: 10.1007/s11064-010-0387-9. [DOI] [PubMed] [Google Scholar]
- 53.Quy PN, Kuma A, Pierre P, Mizushima N. Proteasome-dependent activation of mammalian target of rapamycin complex 1 (mTORC1) is essential for autophagy suppression and muscle remodeling following denervation. J Biol Chem. 2013;288(2):1125–1134. doi: 10.1074/jbc.M112.399949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dueck M, Guatelli J. Evidence against a direct antiviral activity of the proteasome during the early steps of HIV-1 replication. Virology. 2007;361(1):1–8. doi: 10.1016/j.virol.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohrabaugh SL, Campbell TB, Hangoc G, Broxmeyer HE. Ex vivo rapamycin treatment of human cord blood CD34+ cells enhances their engraftment of NSG mice. Blood Cells Mol Dis. 2011;46(4):318–320. doi: 10.1016/j.bcmd.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]