

Abstract
Neuroinflammation, a pathological hallmark in Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS), is characterized by activated microglia and infiltrating T cells at sites of neuronal injury. In PD and ALS, neurons do not die alone; neuronal injury is non-cell-autonomous and depends on a well-orchestrated dialogue in which neuronally secreted misfolded proteins activate microglia and initiate a self-propagating cycle of neurotoxicity. Diverse populations and phenotypes of CD4+ T cells crosstalk with microglia, and depending on their activation status, may influence this dialogue and promote either neuroprotection or neurotoxicity. A greater understanding of the T cell population mediating these effects as well as the molecular signals involved should provide targets for neuroprotective immunomodulation to treat these devastating neurodegenerative diseases.
Introduction
The central nervous system (CNS) has traditionally been considered immunologically privileged, but over the years there has been a dramatic re-evaluation of this tenet. Current data suggest that the CNS is both immune competent and actively interacts with the peripheral immune system. Neuroinflammation is now recognized to be a prominent feature of many classic neurodegenerative disorders and can mediate either neuroprotection or neurotoxicity [1]. However, controversy still exists and this highlights the importance of a more thorough understanding of the toxic versus protective effects of the immune responses in the CNS.
Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS) are neurodegenerative diseases that present predominantly as sporadic, and to a lesser extent, inheritable disorders. The etiologies of the sporadic forms are unknown, but our understanding of disease pathogenesis has gained considerable momentum following the discovery of familial and toxin-induced forms of disease leading to the development of animal models. In ALS, the discovery of mutations in Cu2+/Zn2+ superoxide dismutase (mSOD1) as the most common cause of familial ALS (fALS) led to the development of mSOD1 transgenic animal models and an exponential increase in our understanding of disease pathogenesis [2,3]. In PD, the discovery that neurotoxins could produce the symptoms and signs of Parkinsonism [4] prompted the development of animal models which have elucidated the molecular pathways of dopaminergic (DA) cell injury. Information gleaned from both ALS and PD tissues and animal models have suggested that mitochondrial dysfunction, increased reactive oxygen species (ROS), misfolded and aggregated proteins, and dysfunction of the ubiquitin proteosome pathway may be key events provoking neurodegeneration [3,5]. Yet in both PD and ALS, a detailed molecular understanding of what causes neuronal death is lacking. Elegant experiments in the mSOD1 mouse suggest that neurons do not die alone, but rather the process is non-cell-autonomous [6] and depends on the active participation of non-neuronal cells such as microglia, astrocytes, and T cells. In PD and ALS, microglial activation and T cell infiltration are significant pathological hallmarks at sites of significant neuronal injury, yet such neuroinflammation has been considered the consequence and not the cause of neuronal injury. The present review challenges this dogma and suggests that both innate and adaptive immune systems may respond to, as well as contribute to, the pathology and tissue destruction.
Parkinson's disease
PD is a chronic CNS progressive disorder characterized by tremor, rigidity, and bradykinesia (slowness of movement), due to compromise of the nigrostriatal pathway as well as other DA and non-DA neurons [5]. The pathological hallmarks of PD are the loss of the nigrostriatal DA neurons, especially those projecting to the putamen, and the presence of Lewy bodies, intraneuronal proteinaceous cytoplasmic inclusions [7]. The degree of DA terminal loss in the striatum appears to be more pronounced than the loss of substantia nigra (SN) DA neurons, suggesting that DA terminals are the primary target, resulting in a “dying back” process [5]. Degeneration of the ventral midbrain DA neurons is accompanied by activation of microglia (the innate immune cells of the central nervous system) and infiltration of T lymphocytes (cells of the adaptive immune system).
Microglia
Microglia, the resident immunocompetent cells within the CNS, display functional plasticity during activation involving changes in cell number, morphology, surface receptor expression, and production of growth factors and cytokines [8]. The changes reflect altered activation states induced by signals arising from injured neurons and surrounding glia (Figure 1). As with macrophages, microglia may exhibit a classically activated M1 phenotype or an “alternatively activated” M2 phenotype [reviewed in 9,10] (Box 1). Classically activated M1 microglia secrete increased pro-inflammatory cytokines (TNF-α and IL-1β) and potent ROS [superoxide radicals (O2•−) and nitric oxide (NO)], and reduced neurotrophic factors. At least three different M2 phenotypes have been identified based upon their gene expression profile and cover a continuum of functional states [11]. The M2-“alternatively activated” (M2a) phenotype, induced by IL-4 or IL-13 with increased expression of arginase-1, FIZZ1, and Ym1, is distinguished from two others that include the M2b phenotype, induced by immune complexes and Toll-like receptor (TLR) agonists, and the M2-“deactivated” (M2c) phenotype that is induced by IL-10 or TGF-β with increased expression of CCR2 and scavenger receptors [12]. M2 microglia can enhance neurotrophic factor release, such as insulin like growth factor 1 (IGF-1), reduce pro-inflammatory cytokines, and assist in inflammation resolution.
Figure 1.
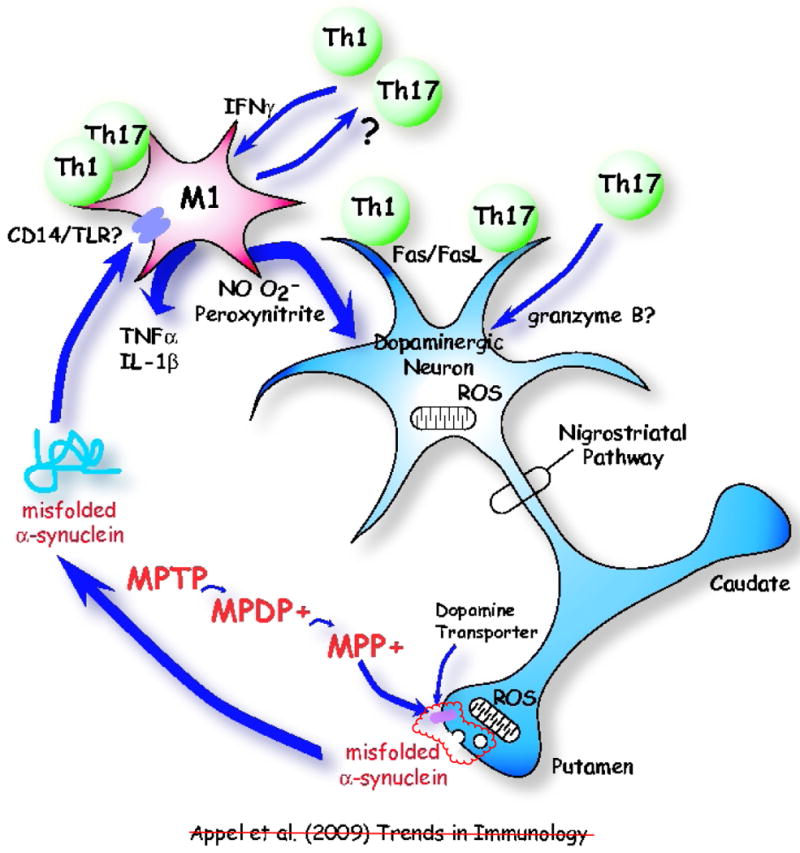
Communication between a neuron and the immune system in a chemically induced model of Parkinson's disease. The neurotoxin MPTP is first converted to MPDP+ and then to MPP+ by non-dopaminergic cells; e.g. glia or serotonergic neurons [5]. MPP+ is concentrated in dopaminergic cells via pre-synaptic dopamine transporters (DAT). In the presence of MPP+, reactive oxygen species (ROS) produced by mitochondria in substantia nigra (SN) dopaminergic neurons induce misfolded α-synuclein, which is subsequently released in vesicles at pre-synaptic terminals. The misfolded α-synuclein in turn activates microglia, possibly via a receptor, transforming microglia into a neurotoxic M1 microglial phenotype. Misfolded α-synuclein is released from presynaptic terminals in the striatum and activates microglia in proximity to dopaminergic cell somas in the substantia nigra. It is likely that α-synuclein is also released from dopaminergic cell bodies and activates local microglia, as well as being released from presynaptic dopaminergic terminals and activating adjacent microglia. Potential receptors for misfolded α-synuclein on microglia include CD14/Toll-like receptors (TLRs), and the scavenger receptors. The M1 microglia influence Th1 and perhaps Th17 T cells, which in turn maintain the M1 microglial phenotype via production of IFN-γ. The M1 microglia release ROS nitric oxide (NO) and superoxide radicals (O2•−) form the highly reactive and toxic peroxynitrite. These ROS compound the injury to dopaminergic neurons. M1 microglia also release pro-inflammatory cytokines, such as TNF-α and IL-1β and amplify the toxic inflammation. T cells can also directly injure dopaminergic neurons by signally through the Fas/FasL system.
Box 1. General concepts and properties of polarized macrophages and microglia.
Macrophage activation has been shown to be plastic, rapid, and fully reversible, suggesting that macrophage populations are dynamic and may first take part in inflammation and then participate in its resolution. The classically activated macrophage (M1) is characterized by the secretion of proinflammatory mediators and the release of reactive oxygen and nitrogen intermediates. M1 macrophages can release or express:
| Cytokines | Chemokines | Cell Surface Markers |
| IL-1 | CCL2 | MHC class II |
| IL-6 | CCL3 | CD80 |
| IL-12 | CCL5 | CD86 |
| IL-23 | CXCL8 | IL-1R |
| TNF-α | CXCL9 | CCR7 |
| CXCL10 | ||
| CXCL11 | ||
| CXCL16 |
Three different induction methods lead to distinct alternatively-activated M2 macrophage subsets, each with a typical cytokine/chemokine and cell surface marker profile, although recent data indicate that a certain degree of versatility exists between the subsets. M2 macrophages cover a continuum of functional states classified as M2a, induced by IL-4 or IL-13, M2b, induced by immune complexes and TLR agonists, and M2c, induced by IL-10 and glucocorticoids. M2a macrophages can release or express:
| Cytokines | Chemokines | Cell Surface Markers |
| IL-1ra | CCL17 | MHC class II |
| IL-10 | CCL18 | CXCR1 |
| IL-12 | CCL22 | CXCR2 |
| IL-23 | CCL24 | CD23 |
| CD163 |
M2b macrophages can release or express:
| Cytokines | Chemokines | Cell Surface Markers |
| IL-1 | CCL1 | MHC class II |
| IL-6 | CD80 | |
| IL-10 | CD86 | |
| TNF-α |
M2c macrophages can release or express:
| Cytokines | Chemokines | Cell Surface Markers |
| IL-1m | CCL16 | CCR2 |
| IL-10 | CCL18 | CD14 |
| TGF-β | CXCL13 | CD150 |
Cumulative data demonstrate that microglia (CNS-resident macrophages) also have distinct phenotypic states, and in line with other tissue macrophage populations, may exert either a toxic or protective effect on neurons depending on the physiologic conditions. Microglia colonize the CNS during early development and sample the extracellular space through continuous extension, retraction and remodeling of their cellular processes. In response to injury, microglia contribute to the neuroinflammatory response and undergo rapid morphological and functional activation which includes phagocytosis, antigen presentation, as well as the production and secretion of reactive oxygen species (ROS), cytokines and growth factors. Adapted from reference [94].
The CNS also has important neuroprotective mechanisms that can directly inhibit peripheral and central immune responses, but these are less well studied. One potential mechanism involves CD200, which is highly expressed on neurons and can downregulate immune activity through binding of the microglial receptor CD200R. CD200 and CD200R belong to the immunoglobulin superfamily. CD200 transmits an immunoregulatory signal through the CD200R to regulate microglial activation [13].
In PD, activated microglia are prominent, surrounding DA neurons and exhibiting a classically activated M1 phenotype [14,15]. Increased lipid peroxidation as well as carbonyl- and nitrotyrosine-modified proteins are also observed in CNS nigral tissue [16]. Activated microglia surrounding DA neurons are noted in post-mortem tissues of human subjects that had previously developed symptoms and signs of Parkinsonism following exposure to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [4] (also see Box 2 for further details). Because of the relationship between neuronal CD200 and microglial CD200R, it has been proposed that CD200-CD200R signaling is closely correlated to microglia activation in PD, and has the potential of modulating the pathogenesis and progression of PD [17].
Box 2. The Metabolism of MPTP.
In 1982, young intravenous drug users developed a rapidly progressive Parkinsonian syndrome due to the use of a street preparation of 1-methyl-4-phenyl-4-propionoxypiperidine (MPPP), an analog of the narcotic meperidine (Demerol). MPTP, a neurotoxic contaminant inadvertently produced during the illicit synthesis of MPPP, produces an irreversible and severe syndrome characterized by all of the features of PD, including tremor, rigidity, slowness of movement, postural instability, and freezing. The highly lipophilic MPTP crosses the blood-brain barrier within minutes where it is oxidized to 1-methyl-4-phenyl-2,3-dihydropyridinium (MPDP+) by monoamine oxidase B (MAO-B) in glia and serotonergic neurons, the only cells that produce this enzyme. MPDP+ is then converted to the active toxin MPP+, probably by spontaneous oxidation, and released by an unknown mechanism into the extracellular space. MPP+ is a high-affinity substrate for the dopamine transporter (DAT), as well as for noradrenaline and serotonin transporters, on neurons. Inside neurons, MPP+ can bind to the vesicular monoamine transporter-2 (VMAT2) which translocates MPP+ into synaptosomal vesicles, it can be concentrated within the mitochondria by a mechanism that relies on the mitochondrial transmembrane potential, or it can remain in cytosol to interact with cytosolic enzymes. Vesicular sequestration of MPP+ appears to protect cells from neurodegeneration by sequestering the toxin and preventing it from accessing mitochondria. However, once inside the mitochondria, MPP+ impairs oxidative phosphorylation by inhibiting complex I of the mitochondrial electron transport which leads to decreases in tissue ATP content. MPTP administration leads to the accumulation and nitration of α-synuclein in the cytosol of DA neurons. Adapted from reference [95].
The MPTP Mouse Model of PD
The discovery that MPTP induces Parkinsonian features in humans has lead to the development of MPTP-induced Parkinsonian animal models for defining the potential role of innate and adaptive immune systems in the pathophysiology of DA neuronal injury. In MPTP-injected mice, M1 microglia are associated topographically and temporally with DA neurodegenerative changes (Figure 1). These activated microglia release increased levels of NO and O2•− through the induction of NO synthase and NADPH-oxidase [18,19]. MPTP-treated mutant mice defective in NO synthase or NADPH-oxidase secrete less NO or O2•− and exhibit less neuronal loss with reduced levels of nitrotyrosine and carbonyl modified proteins than their MPTP treated wild-type (WT) littermates [18,19]. Such data suggest that in the MPTP model, activated microglia injure DA neurons possibly by peroxynitrite generated from O2•− and NO. Further support for this suggestion has come from in vitro studies which have demonstrated that activated microglia can directly induce DA neuron death [20].
Chemokines are chemotactic cytokines that play a key role in chemotaxis and activation of immune cells such as microglia. In the CNS, these chemokines are released by glial cells during an inflammatory response, and in addition to their chemotactic functions, they can play a role in neuromodulation and cell survival [21]. In the MPTP model of murine PD, monocyte chemoattractant protein-1 (CCL2) is upregulated in the striatum and the ventral midbrain. Astrocytes are the predominant source of CCL2 in the striatum and the SN, and DA neurons in the SN constitutively express CCL2 [22]. MPTP treatment results in decreased CCL2 expression in the surviving DA neurons. However, deletion of the genes encoding CCL2 and CCR2, its major receptor, did not affect MPTP-induced striatal dopamine depletion. These results suggest that recruitment of resident microglia or peripheral monocytes following MPTP treatment does not protect against striatal dopamine loss in this model of PD.
In the MPTP model, the initiation of neuronal injury has been attributed to MPP+ mediated blockade of mitochondrial complex I. In mouse striatal slices and isolated DA neurons, the blockade of complex I increases the production of ROS, and enhances oxidation and nitration of DA neuronal proteins [23-25]. A prominent neuronal protein undergoing oxidation is α-synuclein (α-syn). This protein is enriched in synaptic vesicles at pre-synaptic terminals of DA neurons, and accumulates as misfolded and aggregated protein in familial PD patients with mutations in the α-syn (SNCA) gene or when the WT α-syn protein is overexpressed [26,27]. These misfolded aggregates, including conformationally altered α-syn by dopamine treatment, are poorly degraded by chaperone-mediated autophagy, and block degradation of other substrates [28]. Inhibition of autophagy further enhances misfolding of α-syn and contributes to the selective degeneration of PD DA neurons.
A key question in the MPTP model is whether the mitochondrial dysfunction, increased ROS, and misfolded α-syn within DA neurons are sufficient to cause DA neuron death [29]. Recent evidence suggests that misfolded α-syn-induced cytotoxicity is non-cell-autonomous and requires the participation of non-neuronal cells such as microglia. Both normal monomeric and aggregated forms of α-syn are secreted from DA neurons and in turn, the aggregated α-syn activates microglia and upregulates NADPH oxidase and production of ROS [29-31]. Inhibitors of microglial production of NO and O2•− attenuate the in vitro DA neurotoxicity [32]. Expression of the human α-syn protein in transgenic mice resulted in accumulation of insoluble α-syn aggregates in nigral neurons and rendered DA neurons more vulnerable to lipopolysaccharide (LPS)-induced microglia-mediated neurotoxicity [32]. Thus, DA neurons exposed to MPP+ have increased accumulation and secretion of misfolded and aggregated α-syn thereby activating microglia to release free radicals and proinflammatory cytokines, and initiating a self-propagating cycle of DA neuronal injury. The increased release of misfolded and aggregated α-syn caused further oxidation and release of DA neuron α-syn, and further amplification of DA neurodegeneration.
Role of T cells in PD patients and animal models of PD
The potential role of T cells in PD pathogenesis has been less extensively investigated. T cells were originally documented to be present in the SN of PD patients [14]. Subsequent reports have confirmed the presence of CD4+ and CD8+ T cells in the SN of patients with PD [33]. In the murine MPTP model, T cells can enhance cytotoxicity since SCID mice, which lack T cells, are relatively resistant to MPTP-induced degeneration of SN neurons [34]. Similarly, MPTP-induced DA cell death is markedly attenuated in the absence of mature T cells in two other different immunodeficient mouse strains (Rag1-/- and Tcrb-/- mice) [33]. Attenuation of MPTP-induced DA cell death is noted in mice lacking CD4+ T cells as well as in Rag1-/- mice reconstituted with Fas ligand (FasL)-deficient splenocytes; i.e., splenocytes with impaired cell-mediated killing ability [33]. MPTP-induced DA cell death is not attenuated in mice lacking CD8+ T cells [33]. Furthermore, transfer of whole T cell populations from mice immunized with nitrated α-syn (i.e. misfolded) accelerates MPTP-induced DA cell loss [34]. Collectively, these data indicate that the deleterious activity of MPTP involves infiltrating CD4+ T cells with signals mediated through the Fas/FasL pathway.
At present, the specific CD4+ T cell subpopulation(s) (e.g. Th1, Th17) that modulates cytotoxicity in MPTP-treated mice is unknown. Whether microglia dictate the specific T cell phenotype or T cells dictate the specific microglial phenotype (i.e. M1 vs. M2) in MPTP mice is also unknown. Activated microglia and the cytokine milieu they generate may promote T cell differentiation into various functional lineages. M1 cells promote, whereas M2 cells decrease, proliferation and function of CD4+ Th1 cells [35]. Conversely, T cells dictate microglial anti- or pro-inflammatory phenotypes. CD4+ Th1 cells secrete IFN-γ, which in turn activates M1 microglia (or promotes their differentiation) and provokes loss of DA neurons [36]. Th17 cells may also contribute to neurotoxicity possibly through the secretion of IL-17 or the release of granzyme B, a cytolytic enzyme (see Figure 1) [37]. Activated T cells can also injure neurons directly through cell contact-dependent mechanisms involving FasL, LFA-1, and CD40 but not major histocompatibility class (MHC) class I; i.e. independent of peptide presentation by neurons [38].
T cells can also be neuroprotective as demonstrated in MPTP injected mice where the passive transfer of T cells obtained from mice immunized with the immunomodulatory drug glatiramer acetate (GA) is neuroprotective [39-41] (Figure 2). The injection of GA-derived T cells attenuates the MPTP-induced SN cell loss, possibly by increasing secretion of the anti-inflammatory cytokines IL-4 and IL-10, as well as TGF-β, and decreasing microglial release of proinflammatory factors. GA is classically used in the treatment of multiple sclerosis (MS) where it has been documented to induce Th2-polarized immune responses which may mediate neuroprotection through the release of IL-4, suppressing the production and release of free radicals from activated microglia [42,43]. An alternative neuroprotective effect of GA might be to modulate the differentiation and survival of Th17 cells. This may operate by altering STAT3 phosphorylation and subsequent expression the RORγ transcription factor necessary for the differentiation and survival of Th17 cells [44]. However, it has not been definitively established whether the neuroprotective functions of GA in the MPTP models derive from enhanced Th2 or CD4+CD25+ regulatory T (Treg) cell functions, or reduced Th17 or Th1 functions.
Figure 2.
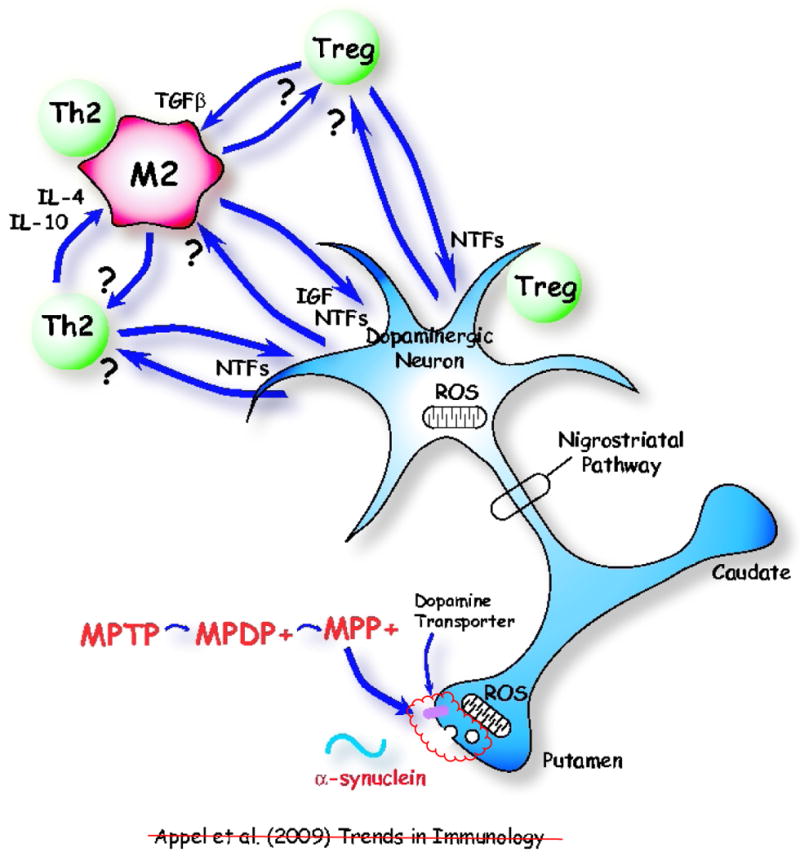
An immunologically protective model following adaptive transfer of T cells in MPTP-induced Parkinson's disease. Th2 and T regulatory (Treg) cells maintain a neuroprotective M2 microglial phenotype through the release of IL-4, IL-10 and TGF-β. Treg cells suppress microglial synthesis and release of reactive oxygen species (ROS) induced by misfolded α-synuclein. Th2 and Treg cells might also protect neurons directly by cell contact-dependent mechanisms or by releasing brain-derived neurotrophic factor (BDNF) and other neurotrophic factors (NTFs). M2 microglia release insulin-like growth factor-1 (IGF-1) and other NTFs, which protect dopaminergic neurons in the substantia nigra (SN). M2 microglia might also positively influence Th2 and Treg cells. Injured neurons may themselves induce M2 microglia and attract Th2 and Tregs cells via release of chemokines.
Although initial investigations attribute most GA activity to a preferential Th2 response, recent reports indicate that GA treatment can also exert immunomodulatory activity on monocytes/macrophages and dendritic cells [45]. In vitro, GA induces the transcription and production of secreted IL-1 receptor antagonist (sIL-1Ra), an inhibitor of the pro-inflammatory mediator IL-1β, in human monocytes and is considered an important regulator of inflammation and the overall immune response mediated by IL-1. Of relevance to chronic inflammation in PD, GA was shown to strongly diminish the expression of IL-1β and enhance the production of sIL-1Ra from monocytes in a model of T cell induced monocyte activation. These results suggest that a potential mechanism for GA neuroprotection may include enhancing the levels of circulating sIL-1Ra, thereby directly affecting monocytes and biasing the response towards a less inflammatory profile.
Neuroprotection may be mediated by Treg cells. In the PD model, increased protection against MPTP-induced DA cell death was reported following adoptive transfer of activated Treg cells whereas CD4+CD25- effector T (Teff) cells did not provide protection from MPTP-induced DA cell loss [46]. In vitro, Treg cells suppress microglial synthesis and release of ROS induced by misfolded α-syn, while Teff cells exacerbate microglial release of ROS [47]. The functional state of CD4+ T cells is highly dependent on their cytokine milieu, and it is not clear in MPTP-injected mice how long Treg cells remain functional in a milieu of M1 activated microglia secreting proinflammatory cytokines and ROS. Furthermore, it is uncertain in the MPTP model whether Treg cells mediate neuroprotection by suppressing cytotoxic T cells such as Th1 or Th17, by converting proinflammatory M1 microglial phenotypes into anti-inflammatory M2 microglia, or by direct neurotrophic factor support. Nevertheless, it is clear that in models of PD, both microglia and T cells are important participants in mediating cytotoxicity and neuroprotection. Therefore, a greater understanding of the initiating signals and the specific milieu dictating innate and adaptive immune responses might provide opportunities for novel therapeutic approaches in patients with PD.
Amyotrophic Lateral Sclerosis
ALS, commonly known as Lou Gehrig's disease, is an adult-onset, rapidly progressive disorder that selectively destroys upper and lower motoneurons, resulting in various degrees of weakness and atrophy of limb musculature, spasticity, dysarthria (slurred speech), and dysphagia (difficulty swallowing), leading to death typically within 4-6 years [3,48]. At present, therapy is mainly symptomatic, and fails to halt disease progression. Denervation at the neuromuscular junction is observed prior to motoneuron cell loss in the spinal cord, suggesting that motoneuron pathology begins at the distal axon and proceeds in a “dying back” process, similar to that observed in PD [5,49]. As in patients with PD, neuroinflammation is a prominent pathological feature of sporadic ALS (sALS), characterized by activated microglia and infiltrating T cells at sites of spinal cord motoneuron injury [50-52]. In addition, increased monocytes/macrophages, dendritic cells, and monocyte chemoattractive protein-1 (CCL2) are present in ALS spinal cord tissue [45]. Spinal cord tissue from ALS patients also has increased mRNA levels for specific dendritic cell markers which are associated with shortened lifespans after diagnosis, suggesting that dendritic cells may contribute to motoneuron injury [52]. The increased CCL2 is of considerable importance since it is known to enhance the trafficking of T cells into the CNS [53]. Depending on their phenotype and activation status, T cells may cross-talk with neurons and microglia, and either damage or protect neurons from stressful stimuli [54]. However, it is unclear whether microglia and/or T cells are neuroprotective, cytotoxic, or possibly both, in ALS. As in PD, animal models of ALS have provided clues to the possible role of the immune system in motoneuron injury and cell death (Figure 3).
Figure 3.
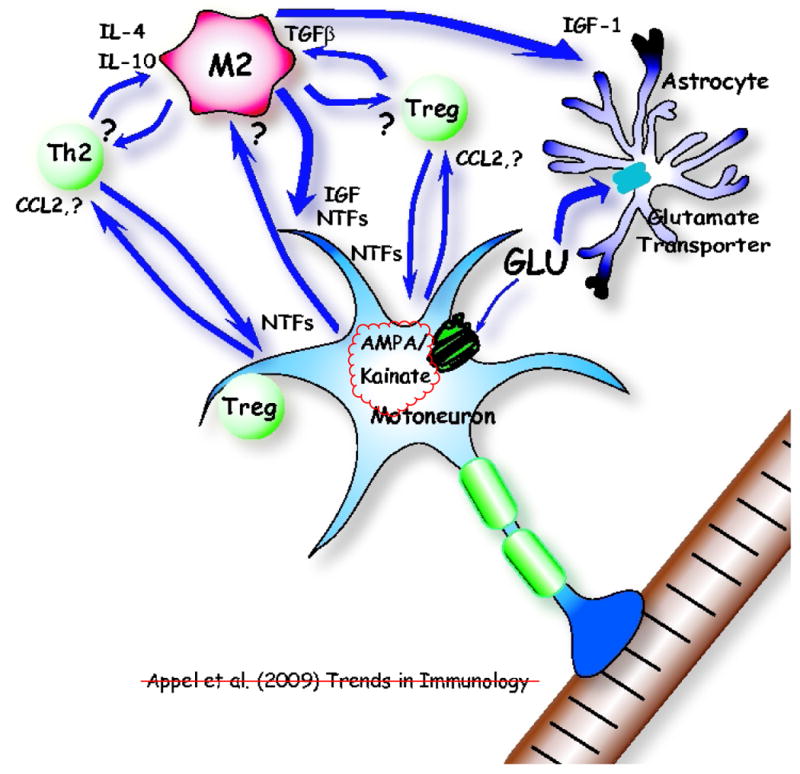
The innate and adaptive immune systems help maintain motoneuron homeostasis in an early phase of amyotrophic lateral sclerosis (ALS) as well as in the facial axotomy model. Initially, injured neurons attract and maintain neuroprotective M2 microglia by an unknown signal and attract Th2 and T regulatory (Treg) cells by chemokines, possibly CCL2. The first response to neuronal injury might be neuroprotective, and then with sustained neuronal stress, there is a transformation to a cytotoxic response; the M2 microglial phenotype may well be the CNS initial default state in both Parkinson's disease (PD) and ALS. M2 microglia secrete insulin-like growth factor 1 (IGF-1) and other neurotrophic factors (NTFs), which also enhance the neuroprotective properties of astrocytes; the astrocytic glutamate transporters are functional and actively remove glutamate (GLU). Th2 and Treg cells secrete NTFs, directly protecting motoneurons. IL-4, IL-10, and TGF-β secreted by Th2 and Treg cells help maintain M2 microglia, while M2 microglia also influence Th2 and Treg cells. Treg cells have been shown to directly differentiate macrophages toward the M2 state. M2 microglia may induce suppressive Treg cells (iTreg).
mSOD1 animal model of ALS
SOD1 is a ubiquitously expressed cytosolic enzyme that converts highly reactive and toxic O2•− molecules to H2O2; peroxidases then detoxify H2O2 to form water. SOD1 mutations cause fALS and transgenic animals overexpressing different SOD1 mutations (mSOD1) develop a chronic progressive motoneuron disease similar to human fALS [2,55-57] (Box 3). T cells, activated microglia, immunoglobulins, and dendritic cells are present in the spinal cords of mSOD1 mice [58-60]. Activated microglia are observed at an early age prior to signs of disease onset. Numerous studies of mSOD1 mice also suggest immune activation of microglia with increased iNOS expression, NOX2 expression, IL-10 expression, and increased IL-6, TNFα, and CCL2 expression [60-65]. Although it is not clear if these responses are protective or injurious, there is clear evidence of an early immune response in the transgenic mSOD1 animal models of fALS (Figure 4).
Box 3. A brief history of SOD1.
Superoxide dismutase 1 (SOD1) binds copper and zinc ions and is one of three isozymes responsible for removing free superoxide radicals (O2•−) in the body. The enzymatic action of SOD1, a 153-amino-acid ubiquitously expressed homodimer, is to convert O2•−, which is produced primarily by errors of oxidative phosphorylation in mitochondria, to water and hydrogen peroxide. Catalysis by SOD1 is mediated in two asymmetric steps by an essential copper atom, which is alternately reduced and oxidized by superoxide:
Cu1+-SOD1 may also use hydrogen peroxide (H2O2) as a substrate to produce the reactive hydroxyl radical:
In 1993, the discovery of missense mutations in the SOD1 gene in familial cases of ALS (fALS) directed research efforts toward identifying the mechanisms by which mutant SOD1 (mSOD1) confers selective motoneuron toxicity; currently, more than 130 different mutations distributed throughout SOD1 polypeptide have been linked to familial ALS. A distinct manganese superoxide dismutase (SOD2) is resident in mitochondria, yet only mutations in the soluble cytoplasmic SOD1 enzyme cause fALS. An initial hypothesis suggested that mSOD1 had reduced SOD1 enzymatic activity, promoting accumulation of toxic superoxide radicals. However, this hypothesis was not validated because several mSOD1 retain some, if not all, enzymatic activity. Furthermore, deletion of wild-type SOD1 does not cause an ALS phenotype. Thus, mSOD1 is now thought to confer a toxic “gain-of-function” independent of enzymatic activity, possibly due to the misfolding of the mSOD1 protein. This toxic “gain-of-function” underlies the many current hypotheses for the pathobiology of fALS, none of which is solely responsible for the pathogenesis of disease. What is clear is that mSOD1 does cause oxidative damage, accumulation of intracellular aggregates, endoplasmic reticulum stress, mitochondrial dysfunction, defects in axonal transport, growth factor deficiency, astroglial cell pathology, and glutamate excitotoxicity. To this extensive list, neuroinflammation, characterized by activated microglia and infiltrating T cells, should be added because it is clearly involved in the pathophysiology of this devastating neurodegenerative disease. Adapted from reference [96].
Figure 4.
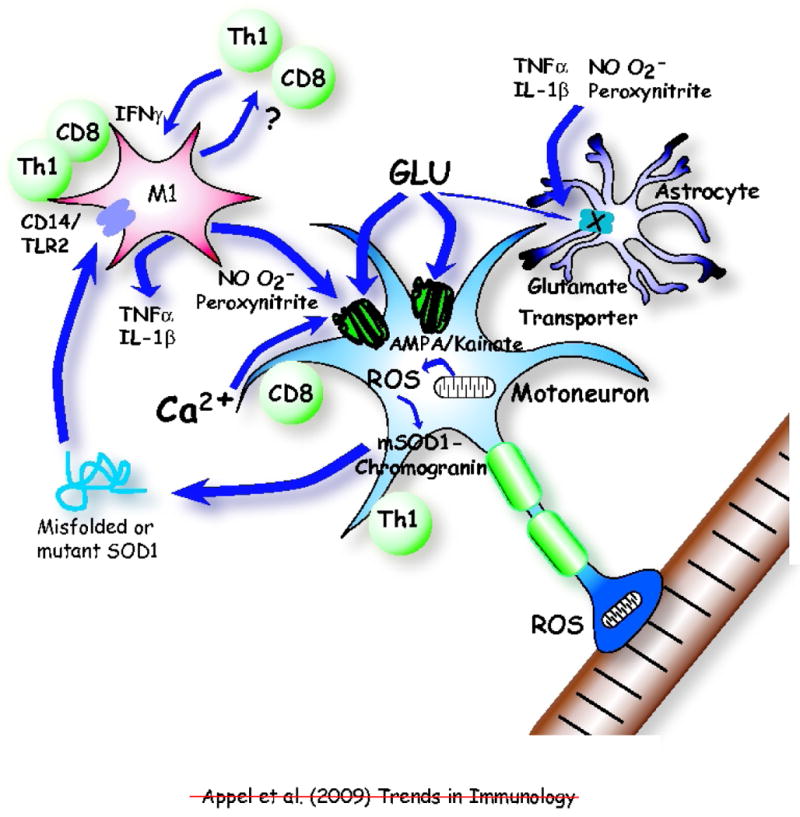
An immunological shift from protection to neurotoxicity accelerates disease in amyotrophic lateral sclerosis (ALS). Initial presentation of mutant superoxide dismutase (mSOD1) or oxidized and misfolded wild-type (WT) SOD1 to T cells might occur in the periphery. Later in disease, as the concentration of extracellular mutant or misfolded SOD1 accumulates, this now rogue protein activates the CD14/Toll-like receptors (TLRs) on microglia transforming them from a protective M2 phenotype to a neurotoxic M1 phenotype. Microglia may also re-present the rogue protein to T cells (Th1 and CD8). In vitro, the M1 microglia release the pro-inflammatory TNF-α and IL-1β cytokines and the reactive oxygen species (ROS) nitric oxide (NO) and superoxide (O2•−), which may form the highly reactive and toxic peroxynitrite. These ROS alter the motoneuron AMPA/kainate channel, resulting in increased glutamate (GLU) susceptibility and subsequent calcium (Ca2+) influx cytotoxicity. Extracellular glutamate concentrations may increase due to reduced numbers and abilities of the glutamate transporters expressed by astrocytes to remove glutamate resulting in further glutamate toxicity. The pro-inflammatory cytokines and ROS may also render the astroglial glutamate transporters ineffective further compounding the Ca2+-induced neurotoxicity. The M1 microglia also influence Th1 and CD8 T cells, which in turn maintain M1 microglial phenotype through the release of IFN-γ and may also exert direct toxic effects on motoneurons.
Role of microglia in mSOD1 models
Glutamate is the most abundant excitatory neurotransmitter in the CNS and AMPA/kainite receptors are the major glutamate receptors on motoneurons. Glutamate is normally non-toxic, but excessive levels of glutamate or modification of AMPA/kainite receptors result in increased influx of calcium and neuronal death. Activated microglia can induce motoneuron cell death in vitro by releasing free radicals and increasing the susceptibility of the motoneuron AMPA/kainate receptor to the toxic effects of glutamate [66]. Furthermore, microglia expressing mSOD1 are more readily activated and induce more motoneuron cell death than WT microglia [67]. To document that microglia contributed to motoneuron injury in vivo, we used PU.1-/- transgenic mice that are unable to develop myeloid and lymphoid cells. Following bone marrow transplantation (BMT), the entire CNS parenchyma is colonized with microglia of donor-derived origin [68]. In this model, mSOD1 microglia did not initiate disease when all other cells in the mouse, including motoneurons, did not express mSOD1. However, we have demonstrated that replacing mSOD1 microglia with WT microglia slowed motoneuron loss, prolonged disease duration (the time from disease onset until death), and extended survival of mSOD1 mice [68]. Reduced expression of mSOD1 in microglia or macrophages also slowed disease progression and prolonged survival [69]. These data suggest that WT microglia or microglia expressing less mSOD1 may exhibit a different phenotype and promote neuroprotection in ALS mice. Therefore, it is likely that microglia may have both neuroprotective and neurotoxic functions depending on the different states of microglial activation that are elicited by local factors and stimuli.
The fact that mSOD1 expression in both microglia and motoneurons enhances cytotoxicity raises the possibility that proteins or other signals released from motoneurons might activate microglia and contribute to neurotoxicity, just as misfolded and aggregated α-syn released from stressed DA neurons may activate microglia and initiate a self-propagating amplification of DA cell injury. In the ALS model, the signal could be mSOD1 itself, since mSOD1 protein bound to chromogranin (a secretory granule protein) is secreted from neural cells and triggers microgliosis and neuronal death in mixed spinal cord cultures [70]. Although mSOD1 protein does not directly injure motoneurons, it may activate microglia and trigger release of ROS and induce motoneuron injury and cell death through a CD14/TLR pathway [71]. Of possible relevance to sALS, WT SOD1 acquires binding and toxic properties of mSOD1 through oxidative damage, and may activate microglia and induce motoneuron death in spinal cord cultures [72]. Furthermore, an altered SOD1 species was detected within the spinal cords of sALS patients that must have originated from misfolded WT SOD1, possibly unifying a shared pathophysiologic pathway between sALS and fALS [73].
Role of T cells in the mSOD1 animal model
To determine whether T cells modulate the microglial-motoneuron dialogue in mSOD1 mice, we bred mSOD1 mice with mice lacking functional T cells or CD4+ T cells [60]. The totally unexpected result was that motoneuron disease accelerated, suggesting that CD4+ T cells are neuroprotective, and not cytotoxic as noted in the MPTP experiments. Furthermore, the early relatively slow phase of disease progression was eliminated in the absence of functional CD4+ T cells, accompanied by increased proinflammatory and cytotoxic factors, decreased anti-inflammatory and neurotrophic factors and decreased survival. BMT reconstituted mice with CD4+ T cells, restored the early slowly progressive phase of disease, prolonged survival, and suppressed cytotoxicity. The immunomodulatory effects of the CD4+ neuroprotective T cells appear to enhance an M2 activated microglial phenotype with increased anti-inflammatory factors and neurotrophic factors [74]. Transgenic mice with the same SOD1 mutation, but bred with different T cell deficient (Tcr3-/-) mice, confirmed that T cells play an endogenous neuroprotective role [75]. Reconstituting mSOD1 mice with ex vivo expanded Treg or Teff cells from WT donor mice delayed the loss of motor function and enhanced survival [76]. Our own preliminary data are in accord with these results and suggest that CD4+ T cells are neuroprotective in mSOD1xRAG2-/- mice during the stable phase and that CD4+ T cells obtained from mSOD1 mice are even more protective than CD4+ T cells from WT mice [74].
These studies suggest that Treg or Th2 cells, in conjunction with M2 microglia, contribute to neuroprotection in ALS models [74]. If Treg cells are responsible, it could be attributable to secretion of TGFβ and IL-10 or suppression of cytotoxic T cell function and down-regulation of proinflammatory cytokine production. Treg cells have been shown to directly differentiate macrophages toward the M2 state, upregulating the mannose and scavenger receptors and increasing phagocytic capacity [77]. Thymus-derived “natural Treg” (nTreg) cells can decrease M2 cell HLA-DR class II expression and reduce their capacity to produce proinflammatory mediators in response to LPS (a TLR4 ligand) [77]. M2 cells may induce Tregs cells (i.e. iTregs) with a strong suppressive function, and Treg cell function has been shown to be tightly controlled by the maturation state of dendritic cells [78]. Maintenance of M2 macrophage activation also requires CD4+ Th2 cells [79]. Furthermore, microglia that encounter anti-inflammatory Th2 cells, or IL-4, support neuronal survival via production of IGF-1 [43,80]. Other murine studies suggest that neurodegeneration is augmented by the presence of both Treg and Teff cells, but either alone is neuroprotective [81,82]. In the later rapidly progressing phases of disease, microglia have an M1 phenotype expressing increased proinflammatory cytokines and reactive oxygen generating enzymes [74]. At present, it is unclear whether at these later stages the neuroprotective CD4+ T cells (e.g. Treg or Th2) have been transformed into more cytotoxic CD4+ T cells (e.g. Th1 or Th17 cells); our preliminary evidence suggests this is not the case. Neurons themselves cannot be excluded as indirect mediators of disease, since they may have a crucial role in governing the T cell response and CNS inflammation [83,84].
Neuroprotective T cells in the facial axotomy model
Studies with facial nerve axotomy have provided insight into the neuroprotective effects of T cells. Following facial axotomy i.e. severing of facial axons, microglia surrounding the facial nerve cell body are activated and T cells are subsequently recruited to the motoneuron soma in the brain stem [85]. Specific evidence for T cells being neuroprotective are derived from studies in which immunodeficient mice lacking functional T- and B- cells, have significantly reduced facial motoneuron (FMN) survival [86]. Reconstitution of these mice with WT whole splenocytes restores FMN survival to WT levels. However, T cells have also been reported to have divergent effects in intact and immunodeficient mice [87]. Nevertheless, T-cells, possibly STAT-6 mediated CD4+ differentiated T-cells that secrete brain derived neurotrophic factor (BDNF) rescue FMN after axotomy [88-91]. CD4+ T cell neuroprotection depends on both resident microglia and bone marrow-derived antigen presenting cells [92]. The activation of microglia, causing them to function as antigen-presenting cells, and hence to express MHC class II proteins, is clearly correlated with a better outcome following CNS injury [93].
Although the neuroprotective signals released from motoneurons undergoing axotomy have not been clearly defined, the overall response is neuroprotective since the final consequence of axotomy in the adult rodent is the functional restoration of the facial motoneuron, its axon, and its synaptic connections. However, important physiologic information remains unknown, especially the first signals produced by injured motoneurons that initiate a neuroprotective response. Therefore, characterization of the cell-contact dependent and independent signaling involved in the T cell-microglia, neuron-microglia, and T cell-neuron dialogues are likely to be of significant therapeutic value [84].
Concluding Remarks
Neuroinflammation, characterized by activated microglia and infiltrating T cells, is a prominent pathological feature in the neurodegenerative diseases PD and ALS; the evidence presented in this review clearly suggests that the immune system is involved in the pathophysiology of these two devastating neurodegenerative diseases. Studies of experimental models suggest that activated microglia and CD4+ T cells contribute significantly to disease progression but probably not to disease initiation. Data from rodent axotomy models and early stages of disease in mSOD1 mice indicate that the first response to injury, provided by the surrounding predominantly M2 microglia and infiltrating CD4+ T cells, is neuroprotective. However, in murine models of PD and later stages of disease in mSOD1 mice, this neuroprotective response is either not initiated or does not persist and is transformed into a cytotoxic response, possibly due to continued neuronal stress and signaling. It is presently unknown why and how neuroprotection is transformed into cytotoxicity. In both the MPTP model of PD and the mSOD1 model of ALS, misfolded and aggregated proteins, either α-syn in PD models and mutant or oxidized SOD1 in ALS models, are secreted from neurons, promoting pro-inflammatory M1 microglia and cytotoxic T cells, and amplifying neuronal injury. In sporadic human PD and ALS, i.e., in the absence of known mutations, it is unknown what initiates disease onset, but a similar set of temporal and mechanistic events may transform neuroprotective microglia and T cells into cytotoxic cells thereby accelerating disease progression. Thus, the dialogues among microglia, T cells, and neurons suggest that the immune response is not merely a consequence of injury, but actively influences and significantly contributes to the balance between neuroprotection and neurotoxicity. What is clear is that neurodegeneration is non-cell-autonomous, with both the innate and adaptive immune systems contributing to neuronal viability and disease progression. A greater understanding of what dictates the presence of cytotoxic or neuroprotective immunomodulation and how to limit cytotoxicity and enhance neuroprotection would help identify appropriate targets for immune-based therapies in neurodegenerative diseases.
Acknowledgments
This study was supported by grants from the NIH (NS048950), the Muscular Dystrophy Association. We dedicate this manuscript to the late Dr. George Kozmetsky and to our courageous PD and ALS patients for their commitment to developing meaningful therapies for these devastating diseases. The authors declare they have no conflicting financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carson MJ, et al. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Boillée S, et al. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Langston JW, et al. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 5.Dauer W, Przedborski S. Parkinson's disease: Mechanisms and Models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 6.Clement AM, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 7.Spillantini MG, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 8.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 9.Geissmann F, et al. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol Cell Biol. 2008;86:398–408. doi: 10.1038/icb.2008.19. [DOI] [PubMed] [Google Scholar]
- 10.Martinez FO, et al. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 11.Benoit M, et al. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 12.Michelucci A, et al. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Koning N, et al. Distribution of the immune inhibitory molecules CD200 and CD200R in the normal central nervous system and multiple sclerosis lesions suggests neuron-glia and glia-glia interactions. J Neuropathol Exp Neurol. 2009;68:159–167. doi: 10.1097/NEN.0b013e3181964113. [DOI] [PubMed] [Google Scholar]
- 14.McGeer PL, et al. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. 1988;76:550–55. doi: 10.1007/BF00689592. [DOI] [PubMed] [Google Scholar]
- 15.Hunot S, et al. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hald A, Lotharius J. Oxidative stress and inflammation in Parkinson's disease: is there a causal link? Exp Neurol. 2005;193:279–290. doi: 10.1016/j.expneurol.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 17.Wang XJ, et al. CD200-CD200R regulation of microglia activation in the pathogenesis of Parkinson's disease. J Neuroimmune Pharmacol. 2007;2:259–264. doi: 10.1007/s11481-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 18.Liberatore GT, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 19.Wu DC, et al. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:145–150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le W, et al. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson's disease. J Neurosci. 2001;21:8447–8455. doi: 10.1523/JNEUROSCI.21-21-08447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ambrosini E, Aloisi F. Chemokines and glial cells: a complex network in the central nervous system. Neurochem Res. 2004;29:1017–1038. doi: 10.1023/b:nere.0000021246.96864.89. [DOI] [PubMed] [Google Scholar]
- 22.Kalkonde YV, et al. Chemokines in the MPTP model of Parkinson's disease: Absence of CCL2 and its receptor CCR2 does not protect against striatal neurodegeneration. Brain Res. 2007;1128:1–11. doi: 10.1016/j.brainres.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 23.Nicklas WJ, et al. MPTP, MPP+ and mitochondrial function. Life Sci. 1987;40:721–729. doi: 10.1016/0024-3205(87)90299-2. [DOI] [PubMed] [Google Scholar]
- 24.Lotharius J, et al. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci. 1999;19:1284–1293. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenamyre JT. Glutamatergic influences on the basal ganglia. Clin Neuropharmacol. 2001;24:65–70. doi: 10.1097/00002826-200103000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 27.Farrer M. Comparison of kindreds with parkinsonism and α-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Vicente M, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cookson MR. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HJ, et al. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 32.Gao HM, et al. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–7698. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brochard V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benner EJ, et al. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verreck FA, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco) bacteria. Proc Natl Acad Sci U S A. 2004;101:4560–4565. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mount MP. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J Neurosci. 2007;27:3328–3337. doi: 10.1523/JNEUROSCI.5321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kebir H, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giuliani F, et al. Vulnerability of human neurons to T cell-mediated cytotoxicity. J Immunol. 2003;171:368–379. doi: 10.4049/jimmunol.171.1.368. [DOI] [PubMed] [Google Scholar]
- 39.Kipnis JJ, et al. Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci U S A. 2002;99:15620–15625. doi: 10.1073/pnas.232565399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz M, Kipnis JJ. Protective autoimmunity and neuroprotection in inflammatory and noninflammatory neurodegenerative diseases. J Neurol Sci. 2005;233:163–166. doi: 10.1016/j.jns.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 41.Benner EJ, et al. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:9435–9440. doi: 10.1073/pnas.0400569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duda PW. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest. 2000;105:967–976. doi: 10.1172/JCI8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao W, et al. Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J Neurochem. 2006;99:1176–1187. doi: 10.1111/j.1471-4159.2006.04172.x. [DOI] [PubMed] [Google Scholar]
- 44.Chen C, et al. Regulatory properties of copolymer I in Th17 differentiation by altering STAT3 phosphorylation. J Immunol. 2009;183:246–253. doi: 10.4049/jimmunol.0900193. [DOI] [PubMed] [Google Scholar]
- 45.Burger D, et al. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1β in human monocytes and multiple sclerosis. Proc Natl Acad Sci U S A. 2009;106:4355–4359. doi: 10.1073/pnas.0812183106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reynolds AD, et al. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J Leukoc Biol. 2007;82:1083–1094. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- 47.Reynolds AD, et al. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J Immunol. 2009;182:4137–4149. doi: 10.4049/jimmunol.0803982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haverkamp LJ, et al. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain. 1995;118:707–719. doi: 10.1093/brain/118.3.707. [DOI] [PubMed] [Google Scholar]
- 49.Fischer LR, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Engelhardt JI, Appel SH. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol. 1990;47:1210–1216. doi: 10.1001/archneur.1990.00530110068019. [DOI] [PubMed] [Google Scholar]
- 51.Engelhardt JI, et al. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol. 1993;50:30–36. doi: 10.1001/archneur.1993.00540010026013. [DOI] [PubMed] [Google Scholar]
- 52.Henkel JS, et al. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- 53.Huang DR, et al. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kerschensteiner M, et al. Neuro-immune crosstalk in CNS diseases. Neuroscience. 2009;158:1122–1132. doi: 10.1016/j.neuroscience.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 55.Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 56.Wong PC, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 57.Howland DS, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alexianu ME, et al. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. doi: 10.1212/wnl.57.7.1282. [DOI] [PubMed] [Google Scholar]
- 59.Henkel JS, et al. The chemokine MCP-1 and the dendritic and myeloid cells it attracts are increased in the mSOD1 mouse model of ALS. Mol Cell Neurosci. 2006;31:427–437. doi: 10.1016/j.mcn.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 60.Beers DR, et al. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A. 2008;105:15558–15563. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall ED, et al. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 62.Almer G, et al. Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;72:2415–2425. doi: 10.1046/j.1471-4159.1999.0722415.x. [DOI] [PubMed] [Google Scholar]
- 63.Sasaki S, et al. iNOS and nitrotyrosine immunoreactivity in amyotrophic lateral sclerosis. Neurosci Lett. 2000;291:44–48. doi: 10.1016/s0304-3940(00)01370-7. [DOI] [PubMed] [Google Scholar]
- 64.Nguyen MD, et al. Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol. 2001;50:630–639. doi: 10.1002/ana.1256. [DOI] [PubMed] [Google Scholar]
- 65.Hensley K, et al. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J Neurochem. 2002;82:365–374. 1570. doi: 10.1046/j.1471-4159.2002.00968.x. Erratum in. [DOI] [PubMed] [Google Scholar]
- 66.Zhao W, et al. Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J Neuropathol Exp Neurol. 2004;63:964–977. doi: 10.1093/jnen/63.9.964. [DOI] [PubMed] [Google Scholar]
- 67.Xiao Q, et al. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J Neurochem. 2007;102:2008–2019. doi: 10.1111/j.1471-4159.2007.04677.x. [DOI] [PubMed] [Google Scholar]
- 68.Beers DR, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boillée S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 70.Urushitani M, et al. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–1018. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- 71.Zhao W, et al. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia. 2009 doi: 10.1002/glia.20919. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ezzi SA, et al. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem. 2007;102:170–178. doi: 10.1111/j.1471-4159.2007.04531.x. [DOI] [PubMed] [Google Scholar]
- 73.Gruzman A, et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104:12524–12529. doi: 10.1073/pnas.0705044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Henkel JS, et al. Alternative vs. classically activated microglial phenotypes throughout disease in the ALS mouse model. Soc for Neurosci Abstr 2009 [Google Scholar]
- 75.Chiu IM, et al. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A. 2008;105:17913–17918. doi: 10.1073/pnas.0804610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Banerjee R, et al. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS ONE. 2008;3:e2740. doi: 10.1371/journal.pone.0002740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tiemessen MM, et al. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Savage ND, et al. Human anti-inflammatory macrophages induce Foxp3+ GITR+ CD25+ regulatory T cells, which suppress via membrane-bound TGFbeta-1. J Immunol. 2008;181:2220–2226. doi: 10.4049/jimmunol.181.3.2220. [DOI] [PubMed] [Google Scholar]
- 79.Loke P, et al. Alternative activation is an innate response to injury that requires CD4+ T cells to be sustained during chronic infection. J Immunol. 2007;179:3926–3936. doi: 10.4049/jimmunol.179.6.3926. [DOI] [PubMed] [Google Scholar]
- 80.Schwartz M, et al. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29:68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 81.Avidan H, et al. Vaccination with autoantigen protects against aggregated beta-amyloid and glutamate toxicity by controlling microglia: effect of CD4+CD25+ T cells. Eur J Immunol. 2004;34:3434–3445. doi: 10.1002/eji.200424883. [DOI] [PubMed] [Google Scholar]
- 82.Kipnis J, Schwartz M. Controlled autoimmunity in CNS maintenance and repair: naturally occurring CD4+CD25+ regulatory T-Cells at the crossroads of health and disease. Neuromolecular Med. 2005;7:197–206. doi: 10.1385/NMM:7:3:197. [DOI] [PubMed] [Google Scholar]
- 83.Liu Y, et al. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518–525. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 84.Tian L, et al. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009;30:91–99. doi: 10.1016/j.it.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 85.Raivich G, et al. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Serpe CJ, et al. Exacerbation of facial motoneuron loss after facial nerve transaction in severe combined immunodeficient (scid) mice. J Neurosci. 1999;19(RC7):1–5. doi: 10.1523/JNEUROSCI.19-11-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ha GK, et al. Endogenous T lymphocytes and microglial reactivity in the axotomized facial motor nucleus of mice: effect of genetic background and the RAG2 gene. J Neuroimmunol. 2006;172:1–8. doi: 10.1016/j.jneuroim.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 88.Serpe CJ, et al. CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain Behav Immun. 2003;17:393–402. doi: 10.1016/s0889-1591(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 89.Serpe CJ, et al. Brain-derived neurotrophic factor supports facial motoneuron survival after facial nerve transaction in immunodeficient mice. Brain Behav Immun. 2005;19:173–180. doi: 10.1016/j.bbi.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 90.Cahn MY, et al. T cell-mediated facial motoneuron survival after injury: Distribution pattern of cell death and rescue throughout the extent of the facial motor nucleus. J Neuroimmunol. 2006;181:93–99. doi: 10.1016/j.jneuroim.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 91.Deboy CA, et al. Immune-mediated neuroprotection of axotomized mouse facial motoneurons is dependent on the IL-4/STAT6 signaling pathway in CD4(+) T cells. Exp Neurol. 2006;201:212–224. doi: 10.1016/j.expneurol.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 92.Byram SC, et al. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shaked I, et al. Early activation of microglia as antigen-presenting cells correlates with T cell-mediated protection and repair of the injured central nervous system. J Neuroimmunol. 2004;146:84–93. doi: 10.1016/j.jneuroim.2003.10.049. [DOI] [PubMed] [Google Scholar]
- 94.Benoit M, et al. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 95.Dauer W, Przedborski S. Parkinson's disease: Mechanisms and Models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 96.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]