

Abstract
It is currently unclear if Merlin/NF2 suppresses tumorigenesis by activating upstream components of the Hippo pathway at the plasma membrane or by inhibiting the E3 ubiquitin ligase CRL4DCAF1 in the nucleus. We found that de-repressed CRL4DCAF1 promotes YAP and TEAD-dependent transcription by ubiquitylating and thereby inhibiting Lats1 and 2 in the nucleus. Genetic epistasis experiments and analysis of tumor-derived missense mutations indicate that this signaling connection sustains the oncogenicity of Merlin-deficient tumor cells. Analysis of clinical samples confirms that this pathway operates in NF2 mutant tumors. We conclude that de-repressed CRL4DCAF1 promotes activation of YAP by inhibiting Lats1 and 2 in the nucleus.
Introduction
The tumor suppressor Merlin/NF2 was identified in 1993 in patients affected by Neurofibromatosis type 2 (NF2) and was later found to be mutated in sporadic meningiomas, ependymomas, and schwannomas, a large fraction of malignant pleural mesotheliomas, and a small percentage of other tumor types. Merlin is a multifunctional protein, which shuttles between the cell cortex and the nucleus in a manner reminiscent of the cell adhesion and signaling component β-catenin. However, it remains unclear if Merlin suppresses tumorigenesis by activating anti-mitogenic signals at the cell cortex or in the nucleus and by what molecular mechanism (Li et al., 2012; and references therein).
Studies in drosophila suggest that Merlin combines with Expanded and Kibra at the cell cortex to activate the Hippo tumor suppressor pathway (Halder and Johnson, 2011). This pathway consists of a kinase cascade comprising the kinase Hippo (MST1/2 in mammals), the adaptor Salvador (Sav1), and the kinase Warts (Lats1/2). The final element, Warts/Lats, phosphorylates and inactivates the transcriptional co-activator Yorkie/YAP, suppressing Scalloped/TEAD-dependent transcription of genes involved in cell survival and proliferation (Hariharan and Bilder, 2006; Harvey and Tapon, 2007; Pan, 2010; Zhao et al., 2010a). In drosophila, the Hippo pathway restrains cell proliferation and promotes apoptosis to limit organ size and to suppress the development of tumorous overgrowths (Hariharan and Bilder, 2006; Harvey and Tapon, 2007; Pan, 2010; Zhao et al., 2010a). Genetic studies in mouse models and genomic analyses of human tumors indicate that Lats1/2 can function as a tumor suppressor and YAP as an oncogene, providing evidence that the function of the downstream segment of the Hippo pathway is evolutionarily conserved (Overholtzer et al., 2006; St John et al., 1999; Zender et al., 2006). Since mammalian Merlin can inhibit YAP and TEAD-dependent transcription and deletion of Yap suppresses liver overgrowth and tumorigenesis in mice carrying a conditional ablation of Nf2, it has been proposed that Merlin suppresses tumorigenesis by activating the Hippo pathway (Zhang et al., 2010; Zhao et al., 2007). However, since YAP is necessary for the expansion of bipotential liver progenitors during development, this result does not necessary imply that YAP signaling drives hepatocellular carcinoma development. In fact, analysis of TCGA and Cosmic datasets indicates that no Hippo pathway component is altered at a significant frequency in human liver cancer (Cerami et al., 2012). In addition, the upstream regulators of the Hippo pathway, and the way they are interconnected, have diverged after the separation of arthropods and chordates (Bossuyt et al., 2014), suggesting that Merlin inhibits Hippo signaling through divergent mechanisms in drosophila and mammals.
We have found that the de-phosphorylated, active conformer of mammalian Merlin suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4DCAF1 in the nucleus (Li et al., 2010). Intriguingly, CRL4DCAF1 controls an oncogenic program of gene expression that includes TEAD target genes, suggesting that Merlin controls Hippo signaling by inhibiting CRL4DCAF1 (Li et al., 2010). Here, we provide evidence that de-repressed CRL4DCAF1 targets Lats1 and 2 for ubiquitylation and inhibition in the nucleus and thus activates YAP-driven transcription and oncogenesis.
RESULTS
De-regulated CRL4DCAF1 induces activation of YAP
To examine if CRL4DCAF1 inactivates the Hippo signaling pathway, we examined NF2 mutant mesothelioma and Schwannoma cells. Meso-33 mesothelioma cells undergo a complete proliferation arrest in response to re-expression of Merlin (Li et al., 2010) and do not possess genetic abnormalities at the BAP1 locus, which is frequently mutated in malignant mesotheliomas devoid of NF2 mutations (Bott et al., 2011). As anticipated, re-expression of Merlin induced robust phosphorylation of YAP at its major negative regulatory site, Ser 127, which is phosphorylated by Lats 1 and 2 in these cells (Figure 1A, lanes 1 and 2). Notably, expression of DCAF1 reduced phosphorylation of YAP at the same site, suggesting that CRL4DCAF1 can promote YAP signaling (Figure S1A).
Figure 1.

CRL4DCAF1 controls YAP activation and TEAD-dependent transcription in Merlin-deficient cells. (A) Schematics depict wild type (WT) and Merlin-resistant (1417X) DCAF1 (top). Meso-33 cells were transfected with HA-Merlin alone or in combination with HA-DCAF1 WT or 1417X and subjected to immunoblotting. (B) Meso-33 cells were transfected with the indicated si-RNAs and examined by immunoblotting. (C) Nf2 mutant FC-1801 cells were transduced with the indicated sh-RNAs and subjected to immunofluorescent staining with anti-YAP/TAZ. Nuclei were stained with DAPI. Scale bar, 10μm. (D) FC-1801 cells were transduced with either a control sh-RNA, a sh-RNA targeting DCAF1, Merlin, or dominant negative TEAD2 followed by a TEAD reporter, and subjected to luciferase assay. Error bar, +/−SEM. (E) and (G) Meso-33 cells were transfected with the indicated si-RNAs and immunoblotted as indicated. (F) and (H) Meso-33 cells were transfected with the indicated si-RNAs and then with empty vector or Merlin and immunoblotted as indicated. See also Figure S1.
We have previously shown that wild type DCAF1 partially reverses the inhibition of proliferation induced by Merlin, whereas a mutant lacking the C-terminal Merlin-binding segment (DCAF1 1417X) completely reverses this process (Li et al., 2010). Intriguingly, whereas wild type DCAF1 alleviated the ability of Merlin to induce phosphorylation of YAP, DCAF1 1417X suppressed it almost completely, suggesting that Merlin inactivates YAP by inhibiting CRL4DCAF1 (Figure 1A). In agreement with this hypothesis, depletion of DCAF1 induced phosphorylation of YAP and suppressed expression of the YAP target genes CTGF (Figures 1B and S1B), BIRC5 and CYR61 (Figure S1B). Moreover, it induced translocation of YAP/TAZ from the nucleus (Figures S1C and S1D) and suppressed transcription from a TEAD-dependent reporter (Figure S1E). These results indicate that CRL4DCAF1 is necessary for activation of Yap in NF2 mutant mesothelioma cells.
To extend these findings, we examined FC-1801 mouse schwannoma cells, which were derived from Nf2Flox/Flox mice (Lallemand et al., 2009). In response to re-expression of Merlin, these cells undergo proliferation arrest and are no longer tumorigenic (Li et al., 2010). Expression of Merlin induced phosphorylation of YAP also in these cells (Figure S1F). Strikingly, whereas YAP/TAZ accumulated almost exclusively in the nuclei of these cells even if they were confluent, silencing of DCAF1 provoked almost complete YAP/TAZ extrusion to the cytoplasm in most cells (Figure 1C; data not shown). Moreover, silencing of DCAF1 inhibited transcription from a TEAD-dependent reporter, although not as completely as overexpression of Merlin or dominant negative TEAD (Figure 1D). Finally, in agreement with prior DNA microarray analyses (Li et al., 2010), overexpression of DCAF1 increased, and silencing of DCAF1 decreased, the expression of YAP target genes in FC1801 cells (Figure S1G). Thus, de-repression of CRL4DCAF1 activates YAP and induces TEAD-dependent transcription in NF2 mutant tumor cells.
CRL4DCAF1 activates YAP without inhibiting MST or Sav1
Unexpectedly, neither expression of Merlin nor silencing of DCAF1 increased phosphorylation of the activation loop of MST1 or MST2 (Figures S1H and S1I). In addition, neither of these manipulations promoted phosphorylation of Lats1 at the MST1/2 phosphorylation site Thr 1079. Rather, these manipulations decreased this phosphorylation (Figures S1H and S1I), presumably by activating the negative feedback loops that restrain flux through the Hippo pathway (Genevet et al., 2010; Hamaratoglu et al., 2006; Xiao et al., 2011). These results suggest that the deregulation of YAP induced by loss of Merlin and activation of CRL4DCAF1 is not due to decreased activation of Lats1 by MST1/2.
To corroborate this hypothesis, we examined if simultaneous depletion of MST1 and MST2 decreased phosphorylation of YAP in Merlin re-expressing or DCAF1-silenced Meso-33 cells. Notably, phosphorylation of YAP proceeded unabated upon both manipulations (Figures 1E and 1F). Moreover, this process was not affected by inactivation of the mammalian ortholog of Salvador, Sav1, an essential component of the core Hippo cassette (Lee et al., 2008; Tapon et al., 2002) (Figures 1G and 1H). Additional experiments revealed that silencing of Sav1 enhances TEAD-dependent transcription in Meso-33 cells, suggesting that the canonical Hippo pathway operates in these cells. However, expression of Merlin counteracted this process to a similar extent in control and Sav1-silenced cells (Figure S1J). As anticipated, silencing of DCAF1 and re-expression of Merlin failed to induce phosphorylation of YAP at Ser 127 in cells depleted of Lats1 and 2 (Figure S1K and S1L). These findings indicate that the loss of Merlin and ensuing de-repression of CRL4DCAF1 activate YAP independently of decreased flux through the core Hippo kinase cassette.
Lats1 and Lats2 are potential substrates of CRL4DCAF1
Motivated by the above findings, we examined the hypothesis that CRL4DCAF1 inhibits Lats1 and 2 by promoting their ubiquitylation. Flag-HA-DCAF1 associates efficiently with endogenous Lats1 and 2 but not with MST1 or 2 in HEK 293T cells (Figures 2A, 2B and S2A). Mutagenesis indicated that the C-terminal segment of DCAF1 interacts directly with the kinase domain of Lats1 (Figure S2B and S2C). Since we could not produce recombinant Lats2 in bacteria, we were unable to verify that the C-terminal segment of DCAF1 also interacts directly with the kinase domain of Lats2. However, the homology between the kinase domains of Lats1 and 2 suggests that this is the case. Since the C-terminal fragment of DCAF1 contains the WD40 domain, which is involved in substrate recruitment (Angers et al., 2006; Jin et al., 2006), we hypothesized that CRL4DCAF1 recruits Lats1 and 2 in order to direct their ubiquitylation.
Figure 2.

DCAF1 interacts with Lats1/2, which are ubiquitylated in vivo. (A) and (B) 293T cells transfected with empty vector or Flag-HA (FH)-tagged DCAF1 were immunoprecipitated with anti-Flag and immunoblotted as indicated. (C) and (D) Nickel precipitates and total lysates from 293T cells expressing HA-Lats1 or HA-Lats2 alone or in combination with His-Ubiquitin were immunoblotted with anti-HA. Asterisk points to a band that may correspond to Lats1/2 non-specifically bound to nickel beads. (E) Summary of ubiquitylation sites identified on Lats1/2 by mass spectrometry. See also Figure S2 and Table S1.
To examine if Lats1 and 2 are ubiquitylated in vivo, 293T cells were transfected with His-ubiquitin and HA-tagged Lats1 or Lats2, treated with the proteasome inhibitor MG132, lysed in a guanidinium chloride-containing buffer, and subjected to pull down with Nickel beads. Anti-HA immunoblotting revealed that Lats1 is poly-ubiquitylated under these conditions (Figure 2C). Lower amounts of ubiquitylated Lats1 were recovered in the absence of MG132 (Li and Giancotti, unpublished results), in consonance with the hypothesis that poly-ubiquitylation targets Lats1 for degradation through the proteasome. In contrast, Lats2 appeared to be oligo-ubiquitylated under the same experimental conditions (Figure 2D). Mass spectrometry indicated that Lats1 is modified by the addition of a single ubiquitin chain at K830 (Figure 2E, Table S1). K→R substitution of residue 830 did not reduce poly-ubiquitylation of Lats1 (Figure S2D), pointing to compensatory ubiquitylation of additional lysine residues (King et al., 1996). In contrast, Lats2 was ubiquitylated at 4 sites (K383, K527, K633, K968) (Figure 2E, Table S1). Simultaneous K→R substitutions at these residues did not reduce oligo-ubiquitylation of Lats2, again pointing to compensatory ubiquitylation of additional lysine residues (Figure S2E).
CRL4DCAF1 promotes proteasomal degradation of Lats1
To examine if CRL4DCAF1 promotes poly-ubiquitylation of Lats1, we performed in vivo ubiquitylation experiments. Expression of DCAF1 led to a large increase in poly-ubiquitylation of Lats1 in Meso-33 cells, and simultaneous expression of Merlin reversed this effect (Figure 3A). Moreover, whereas treatment with MG132 increased the level of poly-ubiquitylation of Lats1 in control 293T cells, it failed to produce this effect in DCAF1-depleted cells, indicating that CRL4DCAF1 is required for efficient poly-ubiquitylation of Lats1 (Figure S3A). To directly test if CRL4DCAF1 mediates ubiquitylation of Lats1, we assembled recombinant CRL4DCAF1 in vitro and tested its ability to ubiquitylate purified Lats1. We found that reconstituted CRL4 E3 ligase ubiquitylates Lats1 in vitro (Figure 3B). However, since we did not detect robust extension of ubiquitin chains under the conditions of the assay, we infer that additional factors may be required for chain elongation in vivo.
Figure 3.

CRL4DCAF1 ubiquitylates and inhibits Lats. (A) Meso-33 cells were transfected with His-Ubiquitin and the indicated recombinant proteins. Ubiquitylated proteins were nickel purified and immunoblotted with anti-HA. Total lysates were immunoblotted as indicated. (B) CRL4DCAF1 was reconstituted in vitro using purified recombinant Cul4A/Rbx1, DDB1, and DCAF1 and incubated with recombinant FH-Lats1 as indicated. Reaction mixtures were immunoblotted as indicated. (C) Meso-33 cells were transfected with the indicated si-RNAs and subjected to a cycloheximide chase assay. Lysates were immunoblotted as indicated. (D) The results in (C) and an additional replicate experiment were quantified by densitometry. Error bar, +/−SEM. (E) 293T cells expressing FH-DCAF1 in combination with increasing quantities of HA-Merlin were immunoprecipitated with anti-Flag and immunoblotted as indicated. (F) Diagram illustrates a model based on the definition of the sequence requirements for binding of DCAF1 to Lats and of Merlin to DCAF1. The WD40 domain of DCAF1, which is implicated in substrate selection, binds directly to the kinase domain of Lats (right). Merlin binds through its FERM domain to the C-terminal segment of DCAF1, disrupting the association of DCAF1 with Lats (left). See also Figure S3.
It is well established that poly-ubiquitylation serves as a signal to target proteins for proteasome-mediated degradation. Since we had noticed that re-expression of Merlin and silencing of DCAF1 increase the steady-state levels of Lats1 in NF2 mutant cells (Figures S1H and S1I), we examined if expression of DCAF1 causes the opposite effect. Stable expression of moderate levels of DCAF1 decreased the steady state levels of Lats1 in FC-1801 cells (Figure S3B). Furthermore, cycloheximide chase experiments demonstrated that silencing of DCAF1 prolongs the half-life of Lats1 in Meso-33 cells by more than 2 fold, indicating that CRL4DCAF1 promotes degradation of Lats1 (Figure 3C and 3D).
Since K830 lies within the kinase domain, poly-ubiquitylation of Lats1 may inhibit kinase activity by interfering with binding of ATP or recruitment of substrates. In addition, Lats1 and 2 contain an N-terminal ubiquitin-binding domain (UBA), which could bind in cis or in trans to one or more C-terminal ubiquitylated sequences, inducing conformational changes that interfere with kinase activity (Figure 2E). To examine if poly-ubiquitylation inhibits the activity of Lats1, we expressed HA-Lats1 and His-Ubiquitin in 293T cells and isolated total and His-ubiquitylated Lats1 by sequential affinity binding and elution (Figure S3C, top). An in vitro kinase assay indicated that ubiquitylated Lats1 possesses a severely diminished ability to phosphorylate GST-YAP at serine 127 as compared to total Lats1 (Figure S3C, bottom). These results suggest that poly-ubiquitylation inhibits Lats1 by blocking its kinase activity and by promoting its degradation.
CRL4DCAF1 inhibits the kinase activity of Lats2
To investigate if CRL4DCAF1 promotes ubiquitylation of Lats2, we performed in vivo ubiquitylation experiments. Ectopic expression of DCAF1 increased oligo-ubiquitylation of Lats2 to a large extent, and simultaneous expression of Merlin reversed this process (Figure S3D). Conversely, depletion of DCAF1 suppressed oligo-ubiquitylation of Lats2 (Figure S3E). In addition, we tested the ability of in vitro assembled CRL4DCAF1 to promote ubiquitylation of recombinant Lats2. The results indicated that CRL4DCAF1 can ubiquitylate Lats2 in vitro (Figure S3F). Collectively, these results suggest that CRL4DCAF1 mediates oligo-ubiquitylation of Lats2.
In agreement with the model that mono- and oligo-ubiquitylation modify protein function without affecting protein stability (Chen and Sun, 2009), silencing of DCAF1 did not increase the steady-state levels of Lats2 (Figure S3G). Interestingly, Lats2 is ubiquitylated at K968 within the kinase domain, at K633 near the binding site for the co-activator Mob1/Mats, and at K527 near the PPXY motif involved in binding to YAP, suggesting that oligo-ubiquitylated Lats2 may exhibit reduced ability to phosphorylate YAP/TAZ in vivo. To examine this hypothesis, we performed in-lysate kinase assays. Lysates from cells overexpressing HA-Lats2 were treated with the broad-specificity de-ubiquitylase USP8 or vehicle control and incubated with GST-YAP. Immunoblotting with anti-P-YAP revealed that de-ubiquitylated Lats2 possesses a greatly increased ability to phosphorylate GST-YAP at Ser 127 as compared to total Lats2 (Fig. S3H, top). Immunoblotting confirmed that treatment with USP8 drastically diminishes the total levels of ubiquitylation of endogenous proteins (Figure S3H, bottom). Taken together, these findings indicate that ubiquitylation profoundly inhibits the kinase activity of Lats2.
Merlin inhibits the interaction between CRL4DCAF1 and Lats1
Prior studies have indicated that Merlin inhibits CRL4DCAF1 by binding through its FERM domain to the C-terminal segment of DCAF1. Intriguingly, patient-derived mutants of Merlin lacking the coiled-coil and C-terminal segments bind to DCAF1 but fail to suppress CRL4DCAF1, suggesting that these latter segments are also required for inhibition of ligase activity (Li et al., 2010). To test if Merlin inhibits CRL4DCAF1 by interfering with substrate recruitment, we examined if expression of Merlin interferes with the association of Flag-HA-DCAF1 with endogenous Lats1 in 293T cells. Notably, co-transfection of increasing concentrations of HA-Merlin led to a dose-dependent reduction of the association of Flag-HA-DCAF1 with Lats1 (Figure 3E). These results suggest that the coiled-coil and C-terminal segments of Merlin may occlude the binding site for Lats on the WD40 domain of DCAF1 (Figure 3F).
Nuclear Merlin promotes phosphorylation of YAP
Since CRL4DCAF1 accumulates predominantly in the nucleus (Li et al., 2010), we examined if Merlin needs to enter into this compartment to inactivate YAP and induce proliferation arrest. Firstly, we generated a fusion protein consisting of an N-terminal ERT2 domain joined by a short linker to full-length Merlin or, as a control, to dsRed (Figure S4A). The ERT2 domain drives passenger proteins to the nucleus in response to tamoxifen binding (Feil et al., 1997). As anticipated, treatment with tamoxifen induced accumulation of ERT2-Merlin and ERT2-DsRed in the nucleus (Figure S4B). Intriguingly, nuclear accumulation of Merlin, but not DsRed, induced phosphorylation of YAP and inhibited the proliferation of Meso-33 cells (Figures S4C and S4D). These results indicate that nuclear accumulation of Merlin is sufficient to induce phosphorylation of YAP and to inhibit proliferation.
To examine if nuclear translocation of Merlin is required to induce proliferation arrest, we used mutational analysis. Analysis of a series of deletion mutants revealed a potential nuclear localization motif at position 24–27 (VRIV). Whereas alanine permutation of each residue or combinations of two or three residues within this motif inhibited nuclear accumulation of Merlin partially, simultaneous alanine substitution of all four residues (24–27A) blocked this process completely (Figures 4A and 4B). A mutant form of Merlin lacking the VRIV sequence (Δ24–27) did not accumulate in the nucleus even if Crm1-dependent nuclear export was blocked with Leptomycin B, confirming that this motif acts as a non-canonical nuclear localization sequence (Figure 4C). Functional analysis revealed that re-expression of Merlin Δ24–27 or Merlin 24–27A does not induce phosphorylation of YAP or inhibit cell proliferation and soft agar growth in Meso-33 cells (Figures 4D-F and S4E). These deficiencies do not arise from defective folding, as these mutations are within the short and flexible N-terminal extension, which distinguishes Merlin from ERM proteins. Furthermore, both Merlin Δ24–27 and Merlin 24–27A associated efficiently with Angiomotin (Figure S4F), a recently identified Merlin-binding protein primarily localized at tight junctions (Yi et al., 2011). In fact, possibly resulting from their increased accumulation in the cytoplasm, these Merlin mutants combined with Angiomotin to a larger extent as compared to wild-type Merlin (Figure S4F). These results indicate that Merlin needs to translocate into the nucleus to inactivate YAP and to suppress cell proliferation, suggesting that the two events are linked.
Figure 4.

Nuclear Merlin promotes YAP phosphorylation and inhibits proliferation. (A) Summary of biochemical subcellular fractionation experiments. Cytoplasmic/membrane (CM) and soluble nuclear fractions (N) from Meso-33 cells expressing Merlin or indicated mutants were immunoblotted for Merlin. (B) Subcellular fractions from Meso-33 cells expressing wild-type Merlin or the indicated mutants were immunoblotted as indicated. (C) Meso-33 cells expressing wild-type Merlin or Merlin Δ24–27 were treated or not with leptomycin B to block nuclear export. Fixed cells were immunostained as indicated. Scale bar, 20μm. Meso-33 cells expressing Merlin or its mutants were subjected to immunoblotting as indicated (D) or BrdU assay (E). (F) FC-1801 cells transduced with wild-type Merlin or its mutants were subjected to soft agar assay. Error bar, +/−SEM. See also Figure S4.
CRL4DCAF1 inhibits Lats in the nucleus
Based on immunofluorescence experiments on cells overexpressing tagged constructs or subcellular fractionation experiments excluding the nuclear fraction, it has been proposed that Lats1 and 2 execute their function at the cell cortex or in the cytosol (Toji et al., 2004; Yang et al., 2004; Yin et al., 2013). Since de-repressed CRL4DCAF1 exerts its pro-oncogenic function in the nucleus (Li et al., 2010), we considered the possibility that its substrates Lats1 and 2 localize in this compartment. Subcellular fractionation experiments indicated that endogenous LATS1 and 2 accumulate in the nuclear fraction, whereas MST1 and Sav1 partition preferentially in the non-nuclear fraction in normal mesothelial Met-5A cells (Figure 5A). In fact, similar proportions of all of these signaling components were found in the non-nuclear and the nuclear fraction under sparse or confluent conditions (Figure 5A). Thus, although all of these components may shuttle in and out of the nucleus, MST1 and Sav1 are present predominantly in the non-nuclear fraction and Lats1 and 2 in the nuclear fraction at steady state. Immunofluorescent staining with a rabbit monoclonal antibody against Lats1 confirmed that Lats1 accumulates in the nucleus in the large majority of Met-5A cells. Intriguingly, however, it also detected a fraction of the kinase at or near cell-to-cell junctions (Figure 5B). In agreement with the observation that Meso-33 cells do not express E-cadherin nor assemble adherens junctions, we did not detect endogenous Lats1 at cell junctions or at the cell cortex in these cells (Figure 5C), suggesting that de-regulated CRL4DCAF1 does not target Lats1 at the cell cortex in NF2 mutant cells.
Figure 5.

CRL4DCAF1 inhibits Lats in the nucleus. (A) Total lysates, cytosolic and crude membrane fractions (CM), and nuclear fractions (N) from sparse (Sp) or confluent (Con) Met-5A cells were immunoblotted as indicated. (B) and (C) Sparse Met-5A cells and Meso-33 cells were stained with an anti-Lats1 antibody and DAPI. Scale bar, 20 μm. (D) Endogenous Lats1 was immunoprecipitated with an anti-Lats1 antibody from cytosolic and crude membrane fractions and nuclear fractions from sparse or confluent Met5-A cells and were immunoblotted as indicated. (E) 293T cells were transfected with empty vector (HA Ctrl) or Flag-HA-tagged Merlin (wild type or its mutants) and were lysed 24 hours later in RIPA buffer without SDS. Flag immunoprecipitates were washed using RIPA buffer without SDS. Flag immunoprecipitates and input were immunoblotted as indicated. (F) Fold enrichment of immunoprecipitated DCAF1 and Lats1 was estimated by densitometry of blots in (E), where enrichment is expressed as the total density of the immunoprecipitated bands normalized to their respective inputs. (G) Meso-33 cells expressing Merlin or its mutants were subjected to immunoblotting as indicated. See also Figure S5.
In the core hippo kinase cassette, MST1 and 2 activate Lats1 by phosphorylating Thr 1079. Activated Lats1 then undergoes autophosphorylation at Ser 909 (Chan et al., 2005). Immunoblotting indicated that Lats1 becomes activated as YAP is phosphorylated and partially extruded from the nucleus in contact-inhibited Met-5A cells, confirming the specificity of these antibodies (Figures S5A and S5B). Notably, subcellular fractionation revealed that phosphorylated, active Lats1 accumulates almost exclusively in the nucleus in these cells (Figure 5D). Similar results were obtained with human liver epithelial HepG2 cells (Figure S5C) and primary mouse fetal liver progenitor cells (Figure S5D). These observations raise the possibility that MST or another upstream kinase moves into the nucleus to activate Lats. Irrespective of the mechanism involved, the predominant accumulation of CRL4DCAF1 and activated Lats in the nucleus is consistent with the hypothesis that CRL4DCAF1 binds to Lats and inhibits it in the nucleus.
Merlin does not suppress tumorigenesis from the cell cortex
Having noted that Lats1 partially localizes to the cell cortex in normal but not NF2 mutant cells (Figure 5B, C), we asked if re-expression of Merlin modifies the subcellular localization of Lats1 in NF2 mutant cells. Upon transient transfection and ensuing overexpression, wild type Merlin localized predominantly at lamellipodia and membrane ruffles in Meso-33 cells, presumably as a consequence of saturation of the nuclear import machinery, and it enhanced the recruitment of endogenous Lats1 to these locales (Figure S5E). Overexpression experiments indicated that HA-tagged Lats1 combines with Flag-HA-tagged Merlin in 293T cells (Figure S5F), in agreement with the recently proposed hypothesis that Merlin recruits Lats to the plasma membrane to promote its activation (Yin et al., 2013). Mutagenesis experiments indicated that the kinase domain of Lats1 interacts with the C-terminal coiled-coil segment of Merlin (Merlin 341–595), but not with its FERM domain (Merlin 341X) (Figures S5F and S5G). Together with the results of a recently published mutational analysis (Yin et al., 2013), these results suggest that Merlin can recruit Lats at the plasma membrane by binding through the N-terminal segment of its coiled-coil domain to the kinase domain of Lats.
To investigate the functional relevance of the interaction of Merlin with Lats1, we tested four patient-derived missense mutants, which fail to accumulate in the nucleus (L46R, L64P, L141P, and A211D), and one that enters into the nucleus but does not bind to DCAF1 (E270G) (Li et al., 2010). In addition, we examined the two synthetic nuclear-defective mutants, Merlin 24–27A and Merlin Δ24–27 (Figures 4A-C). Co-immunoprecipitation analysis indicated that recombinant wild type Merlin combines with endogenous Lats1 much less efficiently than with CRL4DCAF1 in an extraction buffer containing 0.5% Triton X100 but no ionic detergent (Figures 5E and 5F). In addition, whereas all nuclear-defective and patient-derived mutants of Merlin failed to associate with DCAF1 in this assay, none of them exhibited reduced ability to interact with Lats1 (Figures 5E and 5F). Similar results were obtained by using RIPA buffer containing 0.1% SDS (Figure S5H), although we note that this buffer may unfold the FERM domain of Merlin exposing hydrophobic segments (Mani et al., 2011). In agreement with these results, none of the patient-derived mutants exhibited reduced ability to recruit Lats1 to lamellipodia and membrane ruffles upon transient overexpression in Meso-33 cells (Figure S5E). In contrast, they all failed to induce phosphorylation of YAP at Ser 127 (Figure 5G). These findings support the conclusion that Merlin suppresses tumorigenesis by inhibiting CRL4DCAF1 and hence stabilizing activated Lats in the nucleus, rather than by recruiting Lats to the plasma membrane.
CRL4DCAF1 promotes tumorigenesis by inhibiting Lats
To examine if CRL4DCAF1 promotes oncogenesis by activating YAP, we conducted genetic epistasis experiments. As previously shown (Li et al., 2010), depletion of DCAF1 caused a profound proliferation arrest in Meso-33 cells (Figure 6A). Notably, expression of wild type YAP and of YAP-5SA, which cannot be inactivated by Lats1/2 (Zhao et al., 2010b), rescued the DCAF1-silenced tumor cells from proliferation arrest in a manner proportional to their anticipated biological activity (Figure 6A). In addition, YAP-5SA enabled the DCAF1-silenced FC-1801 cells to grow in soft agar and to form tumors upon subcutaneous injection in nude mice (Figures 6B and 6C).
Figure 6.

CRL4DCAF1-mediated inhibition of Lats and de-regulated YAP signaling sustain the oncogenic properties of Merlin-deficient tumor cells. (A) Meso-33 cells expressing GFP, YAP, or YAP-5SA were transfected with a SMART pool control si-RNA or one targeting DCAF1 and subjected to BrdU incorporation assay. (B) FC-1801 cells expressing empty vector or YAP-5SA were transduced with the indicated sh-RNAs and subjected to soft agar assay. The graph illustrates the results (±SEM) normalized to relative control in the vector or YAP-5SA group. (C) FC-1801 cells treated as in (B) were injected subcutaneously in nude mice. (D) Meso-33 cells transduced with a control sh-RNA or sh-RNAs targeting Lats1/2 were transduced with a control sh-RNA or two different sh-RNAs targeting DCAF1 and subjected to BrdU incorporation assay. (E) Meso-33 cells treated as in (D) were subjected to soft agar assay. Typical cell colonies in individual culture wells in the 24-well plate are shown (F). (G) FC-1801 cells transduced with a control sh-RNA (top) or sh-RNAs targeting Lats1/2 (bottom) were subsequently transduced with a control sh-RNA or a sh-RNA targeting DCAF1 and were injected subcutaneously in nude mice. Error bar, +/− SEM. See also Figure S6.
Next, we tested if simultaneous depletion of Lats1 and 2 rescues the tumorigenic potential of DCAF1-silenced NF2 mutant cells. Simultaneous silencing of Lats1 and 2 largely reversed the proliferation arrest induced by depletion of DCAF1 in Meso-33 cells (Figure 6D). In support of the specificity of this effect, silencing of Lats1/2 did not enhance the ability of DCAF1-competent cells to proliferate in this assay (Figure 6D). Furthermore, silencing of Lats1/2 enabled DCAF1-depleted Meso-33 cells to grow in soft agar as efficiently as those expressing DCAF1 (Figures 6E and 6F). Similar results were obtained with FC-1801 cells (Figure S6A).
Finally, we examined if silencing of Lats1/2 rescues the tumorigenic ability of DCAF1-depleted FC-1801 cells. Intriguingly, silencing of Lats1/2 rescued the ability of DCAF1-depleted Schwannoma cells to form tumors to a very large extent (Figures 6G and S6B). We consider this effect specific, because silencing of Lats1/2 alone did not increase tumor growth (Figures 6G and S6B). Taken together, these findings indicate that de-repression of CRL4DCAF1 promotes tumorigenesis of NF2 mutant cells by inactivating Lats1/2 and, hence, activating YAP.
CRL4DCAF1 promotes YAP-dependent oncogenic gene expression in NF2 mutant tumors
To explore the clinical relevance of the signaling mechanism we had identified, we firstly examined a panel of 11 human malignant mesothelioma cell lines carrying or not inactivating mutations at the NF2 locus. Immunoblotting revealed that the NF2 mutant lines exhibited severely diminished levels of Lats1, but not Lats2, as compared to NF2 wild type lines (Figures 7A and S7A). In addition, all except one of the NF2 mutant cell lines displayed decreased phosphorylation of YAP, and 4 out of 7 lines displayed increased expression of the YAP target gene CTGF (Figures 7A and S7A). These results are consistent with the conclusion that CRL4DCAF1 promotes activation of YAP in mesothelioma at least in part by increasing degradation of Lats1.
Figure 7.
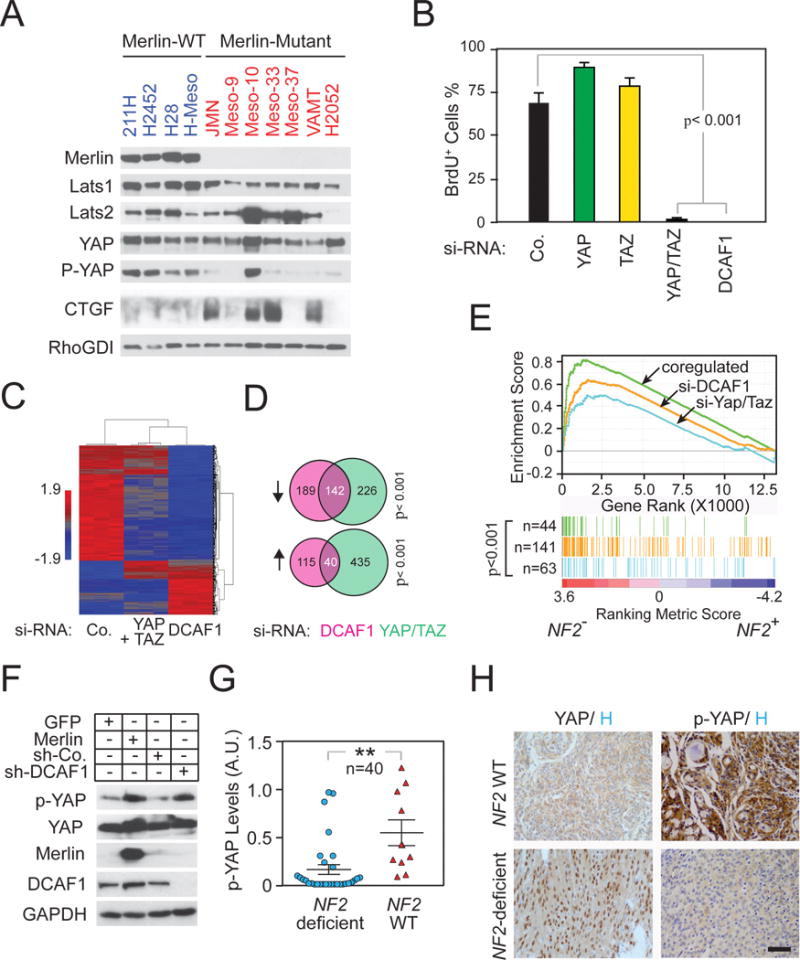
CRL4DCAF1 controls YAP/TAZ-dependent gene expression in NF2 mutant tumors. (A) A panel of human mesothelioma cell lines, including Merlin-wild-type (WT) and Merlin-mutant, were cultured under the same condition. Lysates were immunoblotted as indicated. (B) Meso-33 cells transfected with the indicated SMART pool si-RNAs were subjected to BrdU incorporation assay. (C) Meso-33 cells transfected with the indicated SMART pool si-RNAs were subjected to DNA microarray analysis and unsupervised hierarchical clustering. (D) Venn diagram analysis of the genes downregulated or upregulated following depletion of DCAF1 or both YAP and TAZ. P-values were obtained by Fisher Exact Test. (E) Genes whose expression depend on DCAF1 (orange), on both DCAF1 and YAP/TAZ (green), or on YAP/TAZ alone (blue), were subjected to GSEA in the gene expression profiles of human malignant mesothelioma classified by NF2 mutation status (NF2−, mutant; NF2+, wild-type). (F) Merlin-deficient primary human Schwannoma cells were transduced with a vector encoding GFP, Merlin, and control or DCAF1 sh-RNAs and subjected to immunoblotting. (G) Human meningioma and schwannoma samples classified by NF2 mutation status were analyzed for YAP phosphorylation (arbitrary unit, A.U.), which was normalized against total YAP expression. (H) Representative formalin-fixed and paraffin-embedded NF2 wild-type or NF2-deficient meningioma sections immunostained for YAP/TAZ and phospho-YAP and counterstained with hematoxylin. Scale bar, 100 μm. Error bar, +/−SEM. See also Figure S7 and Table S2–6.
To examine if loss of NF2 induces YAP-dependent oncogenic gene expression in clinical samples, we generated a gene expression signature reflective of CRL4DCAF1-dependent YAP activity. Since only simultaneous depletion of YAP and TAZ induced Meso-33 cells to undergo a proliferation arrest as profound as that induced by depletion of DCAF1 (Figures 7B and S7B), we used DNA microarray analysis to compare the effect of inactivation of CRL4DCAF1 and simultaneous inactivation of YAP and TAZ on gene expression. Whereas depletion of YAP or TAZ did not lead to profound changes in gene expression in Meso-33 cells, simultaneous depletion of both co-activators caused a significant decline in gene expression, confirming that YAP and TAZ function redundantly to control gene expression in mesothelioma cells (Figure 7C). Notably, silencing of DCAF1 suppressed the expression of a large fraction of YAP/TAZ target genes (~39%) (Figures 7C, 7D, and S7C; Table S2). Ingenuity pathway analysis indicated that these genes comprise a large number of cell cycle genes controlled by the RB-E2F network (Figure S7D; Tables S3 and S4). In addition, silencing of DCAF1 enhanced the expression of a somewhat smaller fraction of the genes repressed by YAP/TAZ (~8%), including some TGF-β target genes (Figures 7C, 7D, and S7D; Table S3 and S5). These results indicate that CRL4DCAF1 controls a large fraction of the transcriptional output of YAP in NF2 mutant cells.
We next examined the gene expression profiles of 53 human mesotheliomas, 39 of which harbored mutation or genomic loss at the NF2 locus (Bott et al., 2011). Unsupervised hierarchical clustering strongly suggested that the NF2 mutant mesotheliomas were endowed with a largely distinct gene expression program (Figure S7E). Gene set enrichment analysis (GSEA) revealed that CRL4DCAF1-activated genes were highly enriched in NF2 mutant mesotheliomas as compared to the remaining mesotheliomas (Figure 7E). Intriguingly, among DCAF1-activated genes, those regulated via YAP and TAZ were enriched to an even higher extent, whereas those genes activated by Yap or TAZ alone were less enriched (Figure 7E). These results indicate that CRL4DCAF1 controls YAP/TAZ-dependent gene expression in NF2 mutant mesotheliomas.
To further examine the clinical relevance of our findings, we studied additional NF2 mutant tumor types. Firstly, we analyzed primary human schwannoma cells freshly isolated from a patient affected by NF2. Prior studies had shown that re-expression of Merlin or silencing of DCAF1 suppresses the ability of these cells to proliferate in vitro (Li et al., 2010; Schulze et al., 2002). Interestingly, re-expression of Merlin and silencing of DCAF1 induced a similar level of phosphorylation of YAP in these cells (Figure 7F), suggesting that CRL4DCAF1 controls the output of the Hippo pathway also in freshly explanted, patient-derived NF2 mutant Schwannoma cells. We then examined activation of YAP in 40 human meningioma and vestibular schwannoma samples classified as NF2 mutant or not by using targeted genomic sequencing and immunoblotting with anti-Merlin (Figure S7 and Table S6). Immunoblotting tumor lysates with anti-p- YAP antibodies indicated that the level of phosphorylation of YAP was significantly lower in NF2 mutant tumors as compared to that of other tumors (Figures 7G and S7F). Finally, we examined the expression of YAP and phosphorylation of YAP on whole tissue sections of 31 meningiomas with a sufficient quality and quantity of formalin-fixed paraffin-embedded tissue available. We observed strong nuclear staining of tumor cells for YAP in 16/22 (73%) and no staining in 6/22 (27%) of samples. In addition, we noted weak to absent cytoplasmic staining of tumor cells for P-YAP in 12/23 (52%) and moderate to strong staining in 11/23 (48%) samples (Figure 7H and Table S6). There was a strong positive correlation of nuclear YAP staining and weak or absent cytoplasmic P-YAP staining in tumors with low Merlin expression (p=0.001 for YAP and p=0.001 for phospho-YAP, Mann-Whitney U test), indicating that loss of Merlin causes activation of YAP via reduced phosphorylation and increased accumulation in the nucleus. These clinical findings corroborate the hypothesis that CRL4DCAF1 inhibits Lats and hence activates YAP in NF2 mutant tumors.
Discussion
Our results reveal that de-repressed CRL4DCAF1 functions in the nucleus of NF2 mutant cells to promote activation of YAP. Mechanistically, CRL4DCAF1 binds directly to Lats1 and 2 and directs their conjugation to ubiquitin. Whereas Lats1 is poly-ubiquitylated and targeted for proteasomal-dependent degradation, Lats2 is oligo-ubiquitylated at multiple sites, resulting in loss of kinase activity. As a consequence, active YAP and TAZ accumulate in the nucleus and function redundantly to support the oncogenic potential of NF2 mutant cells. These results indicate that de-repressed CRL4DCAF1 promotes tumorigenesis by inhibiting Lats1 and 2 and thus promoting YAP/TAZ and TEAD-dependent transcription (Figure 8).
Figure 8.

The model illustrates the mechanism by which CRL4DCAF1 promotes oncogenesis. In Merlin-deficient cells, CRL4DCAF1 promotes ubiquitylation of Lats1/2, and suppresses phosphorylation and inactivation of YAP. YAP promotes TEAD-dependent expression of pro-proliferative genes. It is likely that CRL4DCAF1 contributes to oncogenesis by ubiquitylating additional targets. In normal cells, anti-mitogenic signals promote the accumulation of the de-phosphorylated, active form of Merlin. Upon translocation in the nucleus, this form of Merlin binds to DCAF1 and suppresses CRL4DCAF1 activity. This model does not exclude that Merlin activates the core Hippo kinase cascade by a distinct mechanism.
Genetic epistasis experiments in Drosophila have suggested that Merlin cooperates with Expanded and Kibra to activate the core Hippo kinase cassette (Halder and Johnson, 2011). Although Merlin activates Hippo signaling and thereby restricts activation of YAP also in mammalian cells (Zhao et al., 2007), the molecular underpinnings of the connection of Merlin to the Hippo pathway have remained unclear. However, recent studies have suggested that Merlin recruits Lats to the plasma membrane, facilitating its activation by an upstream kinase distinct from MST (Yin et al., 2013). Our results do not preclude the possibility that Merlin limits the proliferation of normal cells - for example during contact inhibition - by multiple, possibly redundant, mechanisms, including those proposed above. However, they indicate that Merlin suppresses tumorigenesis largely, if not exclusively, by repressing CRL4DCAF1-dependent ubiquitylation of Lats in the nucleus.
We observed that re-expression of Merlin induces phosphorylation and inactivation of YAP in NF2 mutant cells without increasing the levels of activation of MST1/2 or Lats1. In addition, Merlin-mediated inactivation of YAP proceeded unabated in the absence of Sav1, which functions as an essential adaptor linking MST1/2 to Lats1/2. These results suggest that Merlin does not restrict activation of YAP by engaging the core kinase cassette, but rather by regulating Lats1 and 2. These observations are consistent with prior evidence suggesting that Lats-mediated phosphorylation and inactivation of Yap can occur independently of MST1/2 in certain mammalian tissues (Yu et al., 2012; Zhou et al., 2009) and with a more recent study suggesting that Merlin facilitates Lats activation by an unidentified upstream kinase distinct from MST (Yin et al., 2013).
The results of our biochemical studies suggest that CRL4DCAF1 employs distinct mechanisms to inhibit Lats1 and 2. Upon DCAF1-mediated recruitment to the ligase complex, Lats1 is poly-ubiquitylated and, hence, targeted for proteasome-mediated degradation. In contrast, Lats 2 is oligo-ubiquitylated at multiple sites, including at lysines in the kinase domain, near the Mob1-binding site, and near the PPXY motif required for interaction with YAP. Presumably as a result of these multiple modifications, Lats2 becomes inactive. Future studies will be required to determine the impact of each one of these modifications on the kinase activity of Lats2 and its interaction with substrates or necessary cofactors. Notably, both Lats1 and Lats2 possess an N-terminal UBA domain, suggesting the possibility that an intramolecular association of this domain with a C-terminal ubiquitylated sequence motif may contribute to maintain the two kinases in an inactive state in NF2 mutant tumor cells.
To examine the relevance of CRL4DCAF1-mediated inhibition of Lats1/2 in NF2 loss-driven oncogenesis, we reconstituted NF2 mutant cells with wild type and mutant forms of Merlin. Deletion or alanine substitution of a short nuclear localization sequence prevented Merlin from interacting with CRL4DCAF1 and from suppressing activation of YAP and cell proliferation. Conversely, tamoxifen-induced translocation of ERT2-Merlin in the nucleus and binding to CRL4DCAF1 resulted in inhibition of YAP and cell proliferation. Furthermore, analysis of a large panel of patient-derived missense mutants of Merlin, which do not combine with CRL4DCAF1 as a result of their inability to enter into the nucleus and/or to bind to the Merlin-binding segment of DCAF1 (Li et al., 2010), revealed that all of them exhibit a severely reduced capacity to inhibit activation of YAP and cell proliferation. Finally, in vitro and in vivo genetic epistasis experiments demonstrated that YAP and TAZ are necessary to maintain the oncogenicity of NF2 mutant tumor cells, and that simultaneous inactivation of Lats1 and 2 rescues the oncogenicity of DCAF1-depleted NF2 mutant tumor cells. In our view, these results provide strong genetic evidence that Merlin suppresses tumorigenesis by inhibiting CRL4DCAF1 and, hence, increasing the levels of active Lats in the nucleus.
Based on the apparently prevalent localization of Hippo pathway components at the cell cortex or in the cytoplasm in Drosophila, it has been proposed that activated Lats phosphorylates YAP in the cytoplasm, preventing its accumulation in the nucleus through redundant mechanisms, such as binding to 14–3–3 or β-TRCP-mediated ubiquitylation (Dong et al., 2007; Zhao et al., 2010b; Zhao et al., 2007). Consistently, overexpressed tagged forms of Lats 1 and 2 accumulate predominantly in the cytosol or at centrosomes, respectively (Toji et al., 2004; Yang et al., 2004), whereas a mutant form of YAP that cannot be phosphorylated by Lats accumulates in the nucleus and promotes TEAD-dependent transcription and oncogenesis (Zhao et al., 2007). Since overexpression can saturate the nuclear import machinery and exogenous tags can interfere with transport into the nucleus of recombinant proteins, we examined the localization of endogenous Lats in mammalian cells by using immunofluorescence staining and subcellular fractionation. We found that endogenous Lats accumulates in the nucleus independently of growth conditions. Importantly, phosphorylated and activated Lats was almost exclusively found in the nucleus. These observations suggest that MST1/2 or another unidentified upstream kinase phosphorylates and activate Lats in the nucleus. Activated Lats in turn phosphorylates YAP, promoting its extrusion from the nucleus.
Our results do not exclude the possibility that Merlin may have additional functions at the cell cortex and, in fact, may also regulate the Hippo pathway from this location. Of note, we observed that Merlin can bind, albeit weakly, to Lats1 and Lats2 and we detected a fraction of Lats1 near the plasma membrane in normal mesothelial cells undergoing contact-mediated inhibition of proliferation. However, in contrast to the results of a more limited but similar analysis (Yin et al., 2013), we found that mutations that abolish the tumor suppressor function of Merlin do not interfere with its ability to bind to Lats1 and recruit it to the cell cortex. However, they consistently fail to bind to CRL4DCAF1 and to inactivate YAP. These results confirm that Merlin’s function at the cell cortex is insufficient to suppress tumorigenesis.
We speculate that the signaling pathway we have delineated may have evolved after the separation of chordates from arthropods to provide additional control over the function of YAP. In fact, Drosophila Merlin contains a Glycine residue at the position corresponding to E270 in human Merlin, a substitution that is predicted to abolish interaction with DCAF1 (Li et al., 2010). Moreover, the Drosophila ortholog of DCAF1, Mahjong (Tamori et al., 2010), does not contain a C-terminal Merlin-binding segment. These considerations suggest that the interaction of Merlin with CRL4DCAF1 has evolved in chordates to function as a clamp to restrain Hippo signaling to YAP. In this model, Merlin and CRL4DCAF1 act as a negative and a positive component of a switch that fine-tunes the activation of YAP in mammalian cells. Inactivation of NF2 eliminates the clamp, causing deregulated YAP/TEAD dependent transcription of pro-survival and mitogenic genes. It remains to be examined if this clamp on the activation of YAP operates also during development to limit stem cell self-renewal, organ size, and cell fate specification, as it is anticipated from the known physiological function of YAP and its deregulation following inactivation of NF2 during tumorigenesis.
We validated the relevance of our findings for human tumorigenesis by using a variety of approaches. First, genetic manipulation indicated that re-expression of Merlin and silencing of DCAF1 inhibit activation of YAP and cell proliferation in primary schwannoma cells from NF2 patients. Second, immunoblotting and immunostaining of meningioma and vestibular schwannoma samples revealed a striking correlation between the loss of Merlin and the activation of YAP. Third, analysis of a panel of human mesothelioma lines indicated that deletion of NF2 correlates with reduced levels of Lats1 protein and increased activation of YAP. Finally, GSEA demonstrated that the gene expression program regulated by CRL4DCAF1 through activation of YAP/TAZ is highly enriched in NF2 mutant mesotheliomas as compared to those lacking NF2 alterations.
We note that simultaneous depletion of Lats1 and 2 did not completely rescue the ability of DCAF1-silenced NF2 mutant cells to overproliferate in vitro and in vivo, suggesting that CRL4DCAF1 may have additional substrates that contribute to its pro-oncogenic function. RORα, which has been recently identified as a CRL4DCAF1 substrate and exhibits severely reduced expression in human breast cancers (Lee et al., 2012), is a potential candidate, but other still unidentified substrates may contribute as well. These considerations suggest that entry of Merlin into the nucleus and inhibition of CRL4DCAF1 may have evolved to limit not only the activation of YAP but also that of additional pro-oncogenic signals. In addition to their biological implications, these findings indicate that pharmacological targeting of CRL4DCAF1 is a rational approach for the treatment of Neurofibromatosis type 2 and malignant pleural mesothelioma. We suggest that, since CRL4DCAF1 may promote oncogenesis through multiple mechanisms, CRL4DCAF1 inhibitors may display enhanced efficacy in NF2 mutant tumors as compared to drugs that interfere with the interaction of YAP with TEAD (Jiao et al., 2014; Liu-Chittenden et al., 2012).
Experimental Procedures
Patients and tumor samples
Human studies were approved by the Institutional Review Boards of MSKCC, NYU-Langone Medical Center and the Plymouth University Peninsula School of Medicine and Dentistry. See details in Supplemental Experimental Procedures.
Subcellular Fractionation
Nuclear and non-nuclear fractions were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents following manufacturer’s instructions (Thermo Fisher Scientific).
Immunofluorescent Staining
Cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.3% sodium deoxycholate and 0.3% Triton X-100 for 30 min on ice. After blocking with 5% BSA, samples were incubated overnight at 4°C with primary antibodies diluted in PBS/2.5% BSA/0.05% Triton X-100. After washing with PBS/0.1% Triton X-100, samples were incubated with secondary antibodies diluted as described above for 2 hours at 4°C. Samples were finally washed with PBS/0.1% Triton X-100, rinsed with PBS, and mounted in Gelvatol.
In vitro Ubiquitylation Assay
Affinity-purified recombinant proteins, including E1 (Calbiochem), E2 (UBCH5c), GST-Cul4A, His-Rbx1, GST-DDB1, His-ubiquitin, and FH-DCAF1 and FH-Lats1 (purified from 293T cells) were incubated in 20 μl reaction buffer (50 mM Tris-HCl pH 7.5, 0.2 M NaCl, 10 mM MgCl2, 4 mM ATP, 1 mM DTT) at 25 °C for 2 hours. Reactions were terminated by boiling in SDS loading buffer.
Soft Agar and Tumorigenicity Assay
For soft agar assay, cells were trypsinized, resuspended in complete medium, and plated in 0.34% low melting temperature agarose (FMC Bioproducts) in complete medium at 2×104 (Meso-33 cells) or 1.2×104 (FC-1801 cells) per well in 24-well Ultra Low Cluster Plates (Costar). For tumorigenicity assay, 1×106 FC-1801 cells were suspended in Ca/Mg-free PBS and injected subcutaneously into the right flank of nude mice. Tumor volumes were determined by caliper measurement. Animal studies were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of MSKCC.
Statistical Methods
Statistical significance was determined by Student’s t test unless indicated otherwise.
Supplementary Material
Highlights.
De-regulated CRL4DCAF1 induces activation of YAP in NF2 mutant tumor cells
CRL4DCAF1 promotes ubiquitin-mediated proteasomal degradation of Lats1
CRL4DCAF1-mediated oligo-ubiquitylation inhibits the kinase activity of Lats2
CRL4DCAF1 activates Yap-dependent oncogenic gene expression
Significance.
In spite of significant advances, the mechanism by which Merlin/NF2 suppresses tumorigenesis has remained incompletely understood. We have found that Merlin translocates into the nucleus, where it inhibits the pro-mitogenic E3 ubiquitin ligase CRL4DCAF1. Here, we provide evidence that CRL4DCAF1 promotes YAP-dependent transcription and oncogenesis by ubiquitylating and thereby inhibiting the Hippo pathway components Lats 1 and 2. Analysis of clinical samples indicates that this signaling mechanism operates in NF2 mutant tumors. These results reveal the mechanism by which Merlin activates the Hippo pathway and suppresses tumorigenesis.
Acknowledgments
We thank I. Farrance, M. Giovannini, K. Guan, S. Jhanwar and D. Lim, M. Pagano and D. Pan for reagents, R. Mukherjee and N. Antao for experimental assistance, the Core Facilities of MSKCC and NYU-Langone Medical Center for sample processing and analysis, and members of the Giancotti laboratory for discussions. This work was supported by the CTF Young Investigator Award 2009-01-013 (to W.L.), the NIH Grants R01 CA152975 (to F.G.G.), S10 OD010591 (to the Microchemistry and Proteomics Core Laboratory of MSKCC), UL1 TR000038 (to the BioRepository and Immunohistochemistry Core Laboratory of NYU-Langone Medical Center) and P30 CA08748 (to MSKCC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- Bossuyt W, Chen CL, Chen Q, Sudol M, McNeill H, Pan D, Kopp A, Halder G. An evolutionary shift in the regulation of the Hippo pathway between mice and flies. Oncogene. 2014;33:1218–1228. doi: 10.1038/onc.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, Creaney J, Lake RA, Zakowski MF, Reva B, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24:2076–2086. doi: 10.1038/sj.onc.1208445. [DOI] [PubMed] [Google Scholar]
- Chen ZJJ, Sun LJJ. Nonproteolytic Functions of Ubiquitin in Cell Signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Deng Y, Pang A, Wang JH. Regulation of mammalian STE20-like kinase 2 (MST2) by protein phosphorylation/dephosphorylation and proteolysis. J Biol Chem. 2003;278:11760–11767. doi: 10.1074/jbc.M211085200. [DOI] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed M, Anders RA, Maitra A, Pan D. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre Recombinase Activity by Mutated Estrogen Receptor Ligand-Binding Domains. Biochem Biophys Res Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- Genevet A, Wehr MC, Brain R, Thompson BJ, Tapon N. Kibra is a regulator of the salvador/warts/hippo signaling network. Dev Cell. 2010;18:300–308. doi: 10.1016/j.devcel.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantschnig H, Rodan GA, Reszka AA. Mapping of MST1 Kinase Sites of Phosphorylation. J Biol Chem. 2002;277:42987–42996. doi: 10.1074/jbc.M208538200. [DOI] [PubMed] [Google Scholar]
- Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development. 2011;138:9–22. doi: 10.1242/dev.045500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8:27–36. doi: 10.1038/ncb1339. [DOI] [PubMed] [Google Scholar]
- Hariharan IK, Bilder D. Regulation of imaginal disc growth by tumor-suppressor genes in Drosophila. Annu Rev Genet. 2006;40:335–361. doi: 10.1146/annurev.genet.39.073003.100738. [DOI] [PubMed] [Google Scholar]
- Harvey K, Tapon N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat Rev Cancer. 2007;7:182–191. doi: 10.1038/nrc2070. [DOI] [PubMed] [Google Scholar]
- Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, et al. A Peptide Mimicking VGLL4 Function Acts as a YAP Antagonist Therapy against Gastric Cancer. Cancer cell. 2014;25:166–180. doi: 10.1016/j.ccr.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A Family of Diverse Cul4-Ddb1-Interacting Proteins Includes Cdt2, which Is Required for S Phase Destruction of the Replication Factor Cdt1. Mol Cell. 2006;23:709–721. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- King RW, Glotzer M, Kirschner MW. Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Molecular biology of the cell. 1996;7:1343–1357. doi: 10.1091/mbc.7.9.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand D, Manent J, Couvelard A, Watilliaux A, Siena M, Chareyre F, Lampin A, Niwa-Kawakita M, Kalamarides M, Giovannini M. Merlin regulates transmembrane receptor accumulation and signaling at the plasma membrane in primary mouse Schwann cells and in human schwannomas. Oncogene. 2009;28:854–865. doi: 10.1038/onc.2008.427. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim TS, Yang TH, Koo BK, Oh SP, Lee KP, Oh HJ, Lee SH, Kong YY, Kim JM, et al. A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J. 2008;27:1231–1242. doi: 10.1038/emboj.2008.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Lee JS, Kim H, Kim K, Park H, Kim JY, Lee SH, Kim IS, Kim J, Lee M, et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell. 2012;48:572–586. doi: 10.1016/j.molcel.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Li W, Cooper J, Karajannis MA, Giancotti FG. Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO reports. 2012;13:204–215. doi: 10.1038/embor.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140:477–490. doi: 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani T, Hennigan RF, Foster LA, Conrady DG, Herr AB, Ip W. FERM domain phosphoinositide binding targets merlin to the membrane and is essential for its growth-suppressive function. Mol Cell Biol. 2011;31:1983–1996. doi: 10.1128/MCB.00609-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103:12405–12410. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The Hippo Signaling Pathway in Development and Cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze KM, Hanemann CO, Muller HW, Hanenberg H. Transduction of wild-type merlin into human schwannoma cells decreases schwannoma cell growth and induces apoptosis. Hum Mol Genet. 2002;11:69–76. doi: 10.1093/hmg/11.1.69. [DOI] [PubMed] [Google Scholar]
- St John MAR, Tao W, Fei X, Fukumoto R, Carcangiu ML, Brownstein DG, Parlow AF, McGrath J, Xu T. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat Genet. 1999;21:182–186. doi: 10.1038/5965. [DOI] [PubMed] [Google Scholar]
- Tamori Y, Bialucha CU, Tian AG, Kajita M, Huang YC, Norman M, Harrison N, Poulton J, Ivanovitch K, Disch L, et al. Involvement of Lgl and Mahjong/VprBP in cell competition. PLoS Biol. 2010;8:e1000422. doi: 10.1371/journal.pbio.1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Harvey KF, Bell DW, Wahrer DCR, Schiripo TA, Haber DA, Hariharan IK. salvador Promotes Both Cell Cycle Exit and Apoptosis in Drosophila and Is Mutated in Human Cancer Cell Lines. Cell. 2002;110:467–478. doi: 10.1016/s0092-8674(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Toji S, Yabuta N, Hosomi T, Nishihara S, Kobayashi T, Suzuki S, Tamai K, Nojima H. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes to Cells. 2004;9:383–397. doi: 10.1111/j.1356-9597.2004.00732.x. [DOI] [PubMed] [Google Scholar]
- Xiao L, Chen Y, Ji M, Volle DJ, Lewis RE, Tsai M-Y, Dong J. KIBRA phosphorylation is regulated by the mitotic kinase aurora and protein phosphatase 1. J Biol Chem. 2011 doi: 10.1074/jbc.M111.246850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Yu K, Hao Y, Li D-m, Stewart R, Insogna KL, Xu T. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol. 2004;6:609–617. doi: 10.1038/ncb1140. [DOI] [PubMed] [Google Scholar]
- Yi C, Troutman S, Fera D, Stemmer-Rachamimov A, Avila J, Christian N, Persson N, Shimono A, Speicher D, Marmorstein R, et al. A Tight Junction-Associated Merlin-Angiomotin Complex Mediates Merlin’s Regulation of Mitogenic Signaling and Tumor Suppressive Functions. Cancer Cell. 2011;19:527–540. doi: 10.1016/j.ccr.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial Organization of Hippo Signaling at the Plasma Membrane Mediated by the Tumor Suppressor Merlin/NF2. Cell. 2013;154:1342–1355. doi: 10.1016/j.cell.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–791. doi: 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D. The Merlin/NF2 Tumor Suppressor Functions through the YAP Oncoprotein to Regulate Tissue Homeostasis in Mammals. Dev Cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010a;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2010b;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. Mst1 and Mst2 Maintain Hepatocyte Quiescence and suppress Hepatocellular Carcinoma Development through Inactivation of the Yap1 Oncogene. Cancer Cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.