

Abstract
The diaryl ethers are a novel class of antituberculosis drug candidates that inhibit InhA, the enoyl-ACP reductase involved in the fatty acid biosynthesis (FASII) pathway, and have antibacterial activity against both drug-sensitive and drug-resistant strains of Mycobacterium tuberculosis. In the present work we demonstrate that two time-dependent B-ring modified diaryl ether InhA inhibitors have antibacterial activity in a mouse model of TB infection when delivered by intraperitoneal injection. We propose that the efficacy of these compounds is related to their residence time on the enzyme, and to identify structural features that modulate drug-target residence time in this system, we have explored the inhibition of InhA by a series of B-ring modified analogues. Seven ortho substituted compounds were found to be time dependent inhibitors of InhA where the slow step leading to the final EI* complex is thought to correlate with closure and ordering of the InhA substrate binding loop. A detailed mechanistic understanding of the molecular basis for residence time in this system will facilitate the development of InhA inhibitors with improved in vivo activity.
Keywords: InhA inhibitors, diaryl ethers, time-dependent inhibition, structure-activity relationship, fatty acid biosynthesis
Introduction
Tuberculosis (TB) is a global infectious disease that is a serious threat to human health due to emergence of multidrug-resistant (MDR-TB) and extensively drug-resistant (XDR-TB) strains of Mycobacterium tuberculosis.[1] In 2008, 22% of new TB cases were reported to be MDR-TB,[2] however there is a current lack of new TB-specific chemotherapeutics to combat the spread of these resistant organisms. In the search for novel TB lead candidates, we and others have developed inhibitors of InhA, the enoyl-ACP reductase involved in the M. tuberculosis fatty acid biosynthesis (FASII) pathway.[3] InhA plays an essential role in cell viability and is a target for the TB drug isoniazid (INH).[4] Since resistance to INH results primarily from defects in drug activation and not from mutations in InhA,[5] compounds that directly inhibit InhA should be active against INH-resistant strains. Based on this premise, we developed a series of diaryl ethers (Figure 1) that are potent inhibitors of InhA and that have antimicrobial activity against both INH-sensitive and resistant strains of M. tuberculosis.[3a] Unfortunately, a previous study demonstrated that PT004 (Figure 1 b) had efficacy in a macrophage TB assay but did not show efficacy in the rapid murine model of TB infection.[6] Our studies on the enoyl-ACP reductase enzymes in other pathogens revealed that the in vivo efficacy of diaryl ether inhibitors correlated with their residence time on the enzyme target (tR = 1/koff) but not with their Ki values for enzyme inhibition or their in vitro antibacterial activity (MIC).[7] Since Ki and MIC values are determined at constant drug concentration, the studies support the importance of drug-target residence time in determining in vivo drug activity given that the in vivo drug concentration is not constant.[8] Based on this hypothesis, we developed long residence time inhibitors of InhA and discovered PT070,[9] the first slow-onset diaryl ether inhibitor which showed a 430-fold increase in binding affinity to InhA compared to PT004 (Figure 1 b). PT070 has a residence time of 24 min on InhA and binds to the enzyme through a two-step induced-fit mechanism, in which the rapid formation of the initial EI complex is followed by the slow formation of the final EI* complex (Figure 2).
Figure 1. The diaryl ether scaffold.

The diaryl ethers share a scaffold consisting of an alkyl phenol A-ring and a B-ring with various substituents. a) The general structure of the diaryl ether scaffold. b) Lead compounds for SAR studies in this paper. c) Newly synthesized derivatives including ortho-substituted derivatives, di-substituted derivatives, and derivatives with heterocyclic B-rings (2’-pyridyl or 4’-pyridyl).
Figure 2. Kinetic scheme for time-dependent inhibition.

The time-dependent diaryl ether inhibitors bind through a two-step induced-fit mechanism in which the rapid formation of the initial enzyme-inhibitor complex (EI) is followed by a slow step leading to the final enzyme-inhibitor complex (EI*).
In order to further rationally modulate residence time in this system, we need to understand the molecular basis that governs the interconversion of EI and EI*. The structural change that occurs between EI and EI* has been linked to motions of the substrate binding loop (residues 198-208) which closes over the active site when the inhibitor is bound.[9] Based on the premise that substituents on the B-ring of the diaryl ether play a critical role in the energetics of the EI to EI* transition, we now report SAR studies on a series of B-ring modified diaryl ethers that provide insight into the role of specific interactions between the inhibitor and the substrate binding loop in the time-dependent inhibition of InhA. In addition, to further substantiate the link between residence time and in vivo antibacterial activity, we have determined the efficacy of PT070 in a mouse model of TB infection together with that of an additional time dependent InhA inhibitor, PT091.
Chemistry
Previous approaches to the preparation of diaryl ethers by us involved a Buchwald-Hartwig cross-coupling to link the A-ring and B-ring, and the attachment of an alkyl group using Negishi coupling (Scheme 1).[3a, 10] This synthesis route employed air- and moisture-sensitive catalysts, and resulted in low (10-20%) overall yields. To stream-line this process, we established a new scheme in which intermediate 3 was prepared from vanillin (Scheme 2). This involved protection of the vanillin hydroxyl with a benzyl group, attachment of a pentenyl group using a Wittig reaction, hydrogenation to reduce the double bond and subsequent cleavage of the protecting group. Once compound 3 was obtained, a variety of aromatic groups were coupled to the phenol to provide intermediates for the target molecules (Schemes 3–8). Although additional steps were employed in this new approach, the overall yield of PT092, which has a similar structure to previously reported diaryl ethers, was 37% (Scheme 4). For simple aryl halides, the reported CuI catalyzed coupling reactions[11] are generally useful for the synthesis of aryl ethers, such as compound 4. However, this method is limited by reactivity and steric hindrance of the aryl halide, whereas the recently published air-stable copper (I) bipyridyl complex[12] showed higher catalytic efficiency and afforded more complex biaryl coupled products, such as compound 28. We also modified the Ullmann protocol to use the nucleophilic aromatic substitution reaction with fluoronitrobenzenes,[10, 13] benzonitriles, picolinonitrile, pyridine N-oxide,[14] or isonicotinonitrile to afford the aryl ethers 5, 15–18, 27, 29, 31, 33-34, 36, and 38. The ether intermediates were transformed into the approprtiately substituted diaryl ethers 6, 19–26, 30, 32, 35, 37, and 39 using group conversions such as reductions, deaminations,[13, 15] diazotizations, hydrolysis, and fluoration. Final compounds were obtained after the subsequent demethylation reaction with boron tribromide.[13, 15a, 15c]
Scheme 1.

Previously reported synthesis of diaryl ethers.3a Reagents and conditions: a) (CuOTf)2•PhH, Cs2CO3, EtOAc, [ArCO2H], toluene, 110 °C; b) RZnCl, Pd(P(tBu)3)2,THF/NMP, 130 °C; c) BBr3, CH2Cl2, −78 °C.
Scheme 2.

Synthesis of intermediate 3. Reagents and conditions: a) BnBr, KOH aq, MeOH, reflux, 2 h, 91%; b) n-C5H11PPh3Br, nBuLi, THF, −78 °C to rt, 2.5 h, 82%; c) H2, Pd/C, EtOH, 6 h, 89%.
Scheme 3.

Synthesis of PT095. Reagents and conditions: a) 1-iodo-2-(trifluoromethyl)benzene, (CuOTf)2•PhH, Cs2CO3, 1-naphthoic acid, EtOAc, toluene, 110 °C, 24 h, 75%; b) BBr3, CH2Cl2, −78 °C to rt, 3 h, 88%.
Scheme 8.

Derivatives with 4’-N pyridyl B-rings. Reagents and conditions: a) NaOH, MeCN, 80 °C, 2 h, 60%; b) Fe, AcOH, H2O, 80 °C, 2 h, 57%; c) BBr3, CH2Cl2, −78 °C to rt, 5 h; d) K2CO3, DMAc, 160 °C, 7 h, 69%; e) KOH, MeOH, 80 °C, 2 h, 82%.
Scheme 4.

Derivatives with mono-substituted B-rings. Reagents and conditions: a) K2CO3, 1-fluoro-2-nitrobenzene, 18-crown-6, DMF, 110°C, 3 h, 66%; b) H2, Pd/C, EtOH, 6 h, 91%; c) NaNO2, AcOH, H2O, CuX, 0 °C, 30 min; d) BBr3, CH2Cl2, −78 °C to rt, 5 h.
The synthesis of PT134 is challenging. It was first attempted by using several metal catalyzed coupling conditions[11-12] to link compound 3 with Boc protected 5-bromopyrimidin-4-yl amine, however none of these conditions afforded the desired product. To address this hurdle, we constructed the pyrimidine ring using a 5 step synthesis that employed relatively simple reaction conditions (Scheme 9). The alkylation of 3 with ethyl bromoacetate provided 40, which was then subjected to formylation followed by condensation to give 41. Conversion of the hydroxyl to a chloro group using POCl3 and nucleophilic substitution by ammonia at 130°C provided 43, which was subsequently demethylated using boron tribromide to give the final product PT134.
Scheme 9.

Derivatives with a pyrimidyl B-ring. Reagents and conditions: a) Ethyl bromoacetate, NaOEt, EtOH, 80 °C, 16 h, 35%; b) Ethyl formate, NaH, THF, 65 °C, 4 h; Formamidine acetate, EtOH/MeOH, 80 °C, 4 h, 47%; c) POCl3, 70°C, 3 h, 46%; d) NH4OH, 130 °C, 18 h, 87%; e) BBr3, CH2Cl2, −78°C to rt, 3 h, 68%.
Results and discussion
We previously described the synthesis of a series of diphenyl ether inhibitors of InhA, the most potent of which had hexyl or octyl substituents on the inhibitor A-ring (Ki’ 9.4 and 1.1 nM, respectively).[3a] We evaluated the pharmacodynamic properties of the hexyl analog (PT004) in a mouse model of TB infection, but failed to observe a significant reduction in bacterial load.[6] Pharmacokinetic analysis of PT004 suggested that improvements in ClogP might result in improved in vivo activity, and we subsequently synthesized a series of B-ring substituted PT004 analogs.[10] These studies, coupled with additional SAR data on the inhibition of the enoyl-ACP reductase in other organisms,[16] indicated that modification to the B-ring might also further improve the affinity of this inhibitor series for InhA, leading to the synthesis of an ortho methyl-substituted analog with significantly improved affinity for InhA (PT070).[9] PT070 was found to be slow-onset inhibitor of InhA with a residence time of 24 min on the enzyme. Based on the knowledge that drug-target residence time could have a dramatic impact on in vivo drug activity,[8a, 8b, 8f] we set out to explore the effect of B-ring substituents on the time-dependent inhibition of InhA and on in vivo activity. We show here that time-dependent inhibition is sensitive to the substitution pattern. We also show that PT004, together with an analog bearing an ortho chloro group (PT091) reduce bacterial load in the spleens of mice infected with M. tuberculosis.
Ortho substituted diaryl ethers
Compounds PT004,[3a] PT010,[10] PT013,[10] and PT070[9] have been reported in previous SAR studies. The improvement in binding affinity of PT070 for InhA compared to PT004 is thought to result from reduced freedom of rotation about the ether bond, together with increased hydrophobic contacts between the B-ring and Ala198, Met199, Ile202, and Val203 in the substrate binding loop based on the structural data.[9] Introduction of an ortho methyl group on the B-ring also resulted in an additional interaction between the inhibitor and Ala198. These increased contacts are thought to be critical for the formation of the EI* complex in which helix-6 of the substrate binding loop has closed over the active site.[9, 17] Replacement of the methyl group with an amino group resulted in an analog with similar IC50 and MIC values but also impacted the ability to detect slow-onset inhibition, supporting the importance of ortho B-ring substitution for time-dependent inhibition. Consequently, to better understand the mechanism of the time-dependent kinetics and further modulate the residence time, analogs with various ortho substituents were designed and synthesized (Table 1).
Table 1.
Derivatives with ortho-substituent groups.

| |||||
|---|---|---|---|---|---|
| Compound | IC50[a] (nM) | MIC[b] (μg/mL) | Ki (nM) | Slow-onset | Residence Time (min) |
| R | |||||
| PT004 -H | 11 ± 1[c] | 2.1 | 9.4 ± 0.5 | No | |
| PT010 -NO2 | 182 ± 2.0[d] | 12.50 | N.D.[f] | Yes | 27 ± 6 |
| PT013 -NH2 | 61.9 ±4.5[d] | 3.13 | N.D.[f] | No | |
| PT070 -CH3 | 50.7 ± 4[d] | 3.125 | 0.044 ± 0.005 | Yes | 24 ± 2 |
| PT091 -Cl | 49.5 ± 2.2[d] | 1.56 | 0.96 ± 0.14 | Yes | 21 ± 3 |
| PT092 -Br | 10.0 ± 0.8[e] | 3.125 | 0.20 ± 0.05 | Yes | 30 ± 3 |
| PT095 -CF3 | 29.7 ± 1.2[e] | 50.00 | N.D.[f] | No | |
| PT096 -I | 44.6 ± 7.5[d] | 25 | 3.72 ± 5.13 | No | |
| PT113 -F | 12.1 ± 4.8[e] | 1.56 | 0.09 ± 0.02 | Yes | 9 ± 3 |
| PT114 -OH | 48 ± 3[d] | 12.5 | 15.9 ± 3.7 | No | |
| PT119 -CN | 235.6 ± 10.0[d] | 2.5 | 2.14 ± 0.35 | Yes | 80 ± 12 |
The half maximal inhibitory concentration (IC50) is the concentration of an inhibitor that is required for inhibition of 50% of the enzyme activity.
The minimum inhibitory concentration (MIC) is the lowest concentration of an inhibitor that is adequate to inhibit visible growth of bacteria.
IC50 values were determined at an enzyme concentration of 1 nM.
IC50 values were determined at an enzyme concentration of 100 nM.
IC50 values were determined at an enzyme concentration of 20 nM.
N.D. = Not determined.
Values of IC50, Ki, and residence time are average values from 3 independent experiments with standard deviation.
Compared to PT070, compounds PT113 (F), PT091 (Cl), and PT092 (Br) all have sub-nM binding affinities (Ki) with similar or shorter residence times. In contrast, under the assay conditions employed, the only analogue with a longer residence time, PT119 (tR 80 min), had a binding affinity that is reduced ~50-fold compared to PT070.
In an attempt to rationalize these observations we determined the crystal structures of InhA bound to PT092[17] and PT119 (Figure 3 and 4). These compounds form a ternary complex with enzyme and the oxidized cofactor (NAD+), and in both ternary complexes the substrate binding loop of InhA forms an α-helix that closely interacts with the B ring, similar to the enzyme-inhibitor complexes formed by other time-dependent inhibitors.[9, 17-18] This is in contrast to the structures of complexes formed with rapid reversible inhibitors in which the substrate binding loop is either disordered[3a] or forms an α-helix in a much more open conformation.[17] The results support the correlation between slow-onset inhibition and ordering coupled to closure of the substrate binding loop previously suggested for the FabI class of enoyl-ACP reductases.[19]
Figure 3. Superimposed structures of InhA-complexes involving PT070, PT092 and PT119.


a) Overlay of PT070 (magenta, 2×23.pdb) and PT092 (lime, 4ohu.pdb) bound to InhA. b) Overlay of PT070 (magenta, 2×23.pdb) and PT119 (cyan, 4oim.pdb) bound to InhA. The figures were generated using PyMol.[34]
Figure 4. Interactions in the PT070 and PT119 enzyme-inhibitor complexes.


a) Hydrophobic interactions between the PT070 B-ring and the substrate binding loop (magenta, 2×23.pdb). b) Hydrophobic and hydrogen bonding interactions of the PT119 B-ring with the substrate binding loop and cofactor (cyan, 4oim.pdb). The figures were generated using PyMol.[34]
The substrate binding loop in the PT092 complex exhibits a very similar conformation to that observed in the PT070 complex (Figure 3 a), which is expected from their similar residence times and binding affinities. However, in one of four subunits in the asymmetric unit, a different binding mode of PT092 relative to helix-6 is observed: A201 and I202, instead of I202 and V203, make van der Waals contacts with the inhibitor, which is accompanied by a twist of helix-6 backbone and displacement of the adjacent helix-7. Interestingly, the altered position of the substrate binding loop in this InhA-PT092 subunit is found exclusively in the structure of PT119 bound to InhA (Figure 3 b, 4 b). We speculate that this altered helix-6 conformation rationalizes the increased in residence time of PT119 (tR 80 min) relative to PT070 (tR 24 min) and propose that replacement of the B-ring methyl group with a cyano group raises the barrier between EI and EI* on the binding reaction coordinate.
Owing to the crystallization conditions and the potential impact from crystal packing, the observed structure for the PT119 complex could represent a snapshot along the binding coordinate from EI to EI*. Nevertheless, the reduced overall binding affinity of PT119 compared to PT070 indicates that EI* for PT119 is destabilized relative to PT070. Since the overall energy barrier between EI and EI* has increased for PT119, it follows that the transition state between EI and EI* is even more destabilized (ΔΔGTS > ΔΔGEI*) (Figure 5). In the crystal structure, PT119 appears to make reduced van der Waals contacts with helix-6 residues (Figure 4). In addition, the bulkier cyano substituent approaches the adjacent NAD and InhA backbone within an unfavorably distance of 3.4 Å. These less-than-optimal interactions provide a plausible explanation for the proposed destabilization.
Figure 5. Free energy diagram for the interaction of PT070 and PT119 with InhA.

These inhibitors bind through a two-step mechanism in which the barrier between EI and EI* is assumed to control the rate of formation and breakdown of EI*. Solid line: PT070. Dashed line: PT119.
The results of the binding studies are consistent with a previous proposal that B-ring substituents should generally be small in order to be accommodated in the InhA active site.[10] However, binding affinity is even more sensitive to substituent size than we originally recognized. In particular, introduction of –CF3 or –CN groups into the ortho position of the B-ring resulted in 50-100 fold weaker binding affinities compared to PT070, while the introduction of polar groups such as –OH (PT114) also had a significant effect on binding kinetics, which indicates that hydrophobic interactions between the ortho group and the protein are critical. Further studies with compounds containing ortho halogens showed no correlation between the binding affinity and electronegativity. These results confirm that a small (containing one or two heavy atoms) ortho substituent is necessary for high affinity binding, and that this group should not be a hydrogen bond donor.
Derivatives with di-substituted B-rings
One hypothesis for the improved binding affinity of PT070 compared to PT004 is that the additional methyl group restricts rotations around the ether linkage and stabilizes the conformation observed in the structure of the diphenyl ether bound to the enzyme.[9] Consequently, a series of di-ortho substituted B-ring analogs were designed with two groups ortho to the ether bond (Table 2). If hindered rotation about the ether bond is important for binding, these compounds are expected to have increased affinity compared to the analogs with a single ortho substituent (Table 1). The two ortho groups were chosen from the most potent compounds identified in Table 1, and included methyl, halogen, and cyano groups. Compound PT107, an analog of PT070 was also synthesized to examine the tolerance for an additional para group.
Table 2.
Derivatives with di-substituted B-ring.

| ||||||
|---|---|---|---|---|---|---|
| Compound |
IC50[a] (nM) | MIC[b] (μg/mL) | Ki (nM) | |||
| R1 | R2 | R3 | ||||
| PT004 | -H | -H | -H | 11 ± 1[c] | 2.1 ± 0.9 | 9.4 ± 0.5 |
| PT070 | -CH3 | -H | -H | 50.7 ± 4 | 3.125 | 0.044 ± 0.005 |
| PT107 | -CH3 | -H | -NO2 | 50±5 | 6.25 | 0.13 ± 0.03 |
| PT108 | -CH3 | -CH3 | -H | 1570 ± 200 | 100 | N.D.[e] |
| PT109 | -Cl | -Cl | -H | 86 ± 6 | 25 | N.D.[e] |
| PT110 | -CH3 | -NH2 | -H | N.I.[d] | >100 | N.D.[e] |
| PT111 | -F | -CN | -H | 100±9 | 25 | N.D.[e] |
| PT131 | -C(NH)NH2 | -F | -H | N.I.[d] | N.D.[e] | N.D.[e] |
| PT133 | -F | -Cl | -H | 79.7 ± 24.4 | 25 | N.D.[e] |
The half maximal inhibitory concentration (IC50) is the concentration of an inhibitor that is required for inhibition of 50% of the enzyme activity.
The minimum inhibitory concentration (MIC) is the lowest concentration of an inhibitor that is adequate to inhibit visible growth of bacteria.
IC50 values were determined at an enzyme concentration of 1 nm.
N.I. = No inhibition observed at 2000 nm.
N.D. = Not determined.
IC50 and Ki values are average results from 3 independent experiments with standard deviation.
Unfortunately, none of these derivatives had improved activity compared to their parent compounds (Table 2). To rationalize these results the docked structure of the dichloro analogue PT109 was compared with the corresponding structure of PT091 (monochloro) bound to InhA (Figure 6). This analysis demonstrated that PT109 has a higher Grid score, van der Waals, and internal energy than PT091, which explains the reduced binding affinity of this analog for the enzyme (Table S1): the extra steric hindrance has resulted in unfavorable van der Waals interactions and thus lowered the binding affinity of the inhibitor.
Figure 6. Superimposed structures of InhA-complexes involving PT091 and PT109.

PT091 (yellow) and PT109 (magenta) were docked into the InhA active site (lime) as described in the text. Also shown is the NAD+ cofactor (gray). The figure was generated with PyMol.[34]
Enzyme inhibition and antibacterial growth assays revealed that the compounds have similar SAR to compounds with ortho substituted B-rings: protic or large groups are not tolerated at the ortho position of the B-ring. Replacing a methyl group in PT108 with an amino group (PT110) resulted in a total loss of binding affinity and inhibition of bacterial growth. Introduction of the large carbamimidoyl pharmacophore (PT131) also led to a total loss of inhibitory activity, compared to smaller groups such as cyano or chloro (PT111 and PT133, respectively). Thus, substituents that include halogen or cyano groups are preferred. Although MIC values for the di-ortho analogs are raised 4-15 fold compared to PT070, the ortho, para-di substituted B-ring analog PT107 has similar IC50 and MIC values, implying that small para groups on the B-ring are well tolerated.
Derivatives with heterocyclic B-rings
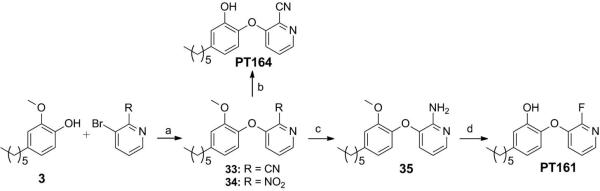
In order to improve hydrophilicity of the diaryl ethers we also explored analogues with pyridyl B-rings. Previous studies showed that although introduction of an ortho nitrogen atom into the B-ring reduced activity significantly, para and meta nitrogen atoms were well tolerated.[10] However, the position of the meta nitrogen is ambiguous on the unsubstituted B-ring. To differentiate 2’- and 4’-N (Figure 1 c), 1’-substituted pyridyl derivatives were synthesized and evaluated (Table 3). The substituents included fluorine and cyano groups, which were found to be preferred in studies with the phenyl B-ring analogs, together with an amino group. In addition, a carboxylate group was also introduced to explore the effect of an ionizable group on activity. The pyridyl B-rings were found not to alter the preference for specific ortho substituents: the fluorine substituent resulted in the lowest IC50 value (PT161) within the 2’-pyridyl derivatives, as did the cyano group (PT115) within the 4’-pyridyl derivatives. Importantly, introduction of fluorine or cyano groups led to slow-onset inhibition (PT164, PT161), in contrast to the parent compound PT077, suggesting that these substituents have an important effect on time-dependent kinetics. Experiments with the carboxylic group (PT116) indicated that an ionizable group, presumably bearing a formal negative charge is not tolerated at the ortho position.
Table 3.
Derivatives with heterocyclic B-rings.
| Compound | IC50 (nM)[a][c] | MIC[b] (μg/mL) | Slow-onset | Residence time (min) | ||
|---|---|---|---|---|---|---|
| R | ||||||
| PT077 |

|
H | 190 ± 12 | 3.13 | No | |
| PT164 | -CN | 308 ± 4.2 | <0.39 | Yes | N.D.[d] | |
| PT161 | -F | 40 ± 20 | 0.3125 | Yes | N.D.[d] | |
| PT112 |

|
-NH2 | 69000 ± 700 | 50 | N.D.[d] | |
| PT115 | -CN | 890.6 ± 105.8 | 12.5 | Yes | >20 | |
| PT116 | -COOH | >12000 | 25 | N.D.[d] | ||
| PT134 |

|
238.8 ± 46.8 | 3.125 | N.D.[d] | ||
The half maximal inhibitory concentration (IC50) is the concentration of an inhibitor that is required for inhibition of 50% of the enzyme activity.
The minimum inhibitory concentration (MIC) is the lowest concentration of an inhibitor that is adequate to inhibit visible growth of bacteria.
IC50 values were determined at an enzyme concentration of 100 nM.
N.D. = Not determined.
IC50 and Ki values are average results from 3 independent experiments with standard deviation.
We found that in 1’-substituted B-rings, the 2’-N analogues generally had lower MIC's and IC50 values compared to the 4’-N compounds (PT164 vs PT115). Although the amino group previously resulted a 5-fold increase in the IC50 value compared to the fluorine group (PT013 vs. PT113), this difference increased to 1700-fold (PT112 vs. PT161), very likely caused by the different scaffolds. The benefit of the 2’-N atom was also seen in PT134 which has both 2’- and 4’-N and improved IC50 and MIC values compared to the corresponding 4’-N analogue PT112 which has a 300-fold lower IC50 value. The X-ray structure of PT070 bound to InhA was used to identify potential differences in the interaction of 2’- and 4’-N analogues with the enzyme. This analysis revealed that although the 2’- and 4’-N atoms have similar hydrophobic contacts with Ile202 and Met161, the 2’-N position is able to form additional hydrogen bonding interactions with the backbone amide of Gly96 (Figure 7). The poor MIC values for the 4’-pyridyl compounds is consistent with the relatively weak affinity of these inhibitors for InhA, while the 2’-pyridine compounds had MIC values that were generally better than compounds with phenyl B rings (Table 1) suggesting that the pyridine B ring may aid uptake into the bacterium.
Figure 7. Interactions between the PT070 B ring and InhA.

Possible hydrophobic and hydrogen bonding interactions are shown for the 2’- and 4’- positions of the PT070 (green) B-ring. The figure was generated from 2×23.pdb using PyMol.[34]
Rapid model of in vivo efficacy
PT004 reduced the bacterial load in the rapid macrophage model of TB infection,[6] demonstrating that it was able to enter the macrophage and kill M. tuberculosis bacteria under altered growth conditions. However, PT004 did not significantly reduce the bacterial load in the lung or spleen in the rapid murine model of TB.[6] In the present study, the time-dependent inhibitors PT070 and PT091 were evaluated for efficacy when delivered IP in the same mouse model. Compared to untreated controls, PT070 reduced the bacterial counts in the spleen 0.57 ± 0.26 log10 with a p value = 0.007 while PT091 reduced the bacterial counts in the spleen 0.69 ± 0.22 log10 with a p value = 0.0002 (Figure 8 a). Importantly, the spleen is a secondary site of TB infection and if a compound can reduce the bacterial burden in the spleen it is indicative of controlling dissemination, and disease progression. Notably, in many of the mice there was a greater reduction of bacterial burden than others (Figure 8 b) which suggests that these candidates may have better efficacy with optimized dosing and alternative delivery regimens.
Figure 8. a) Histogram and b) scatter plot of CFU data from the lung and spleen of animals infected with M. tuberculosis (Erdman) and either untreated (Control) or drug treated.
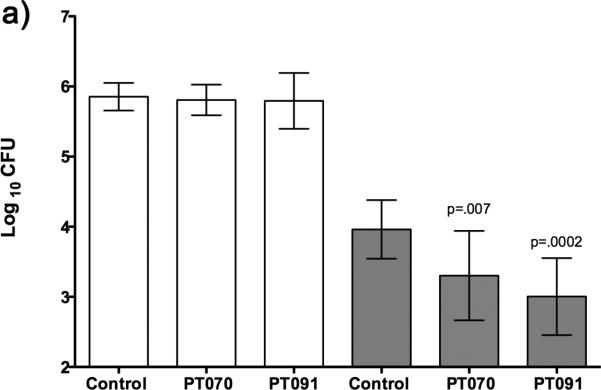

Open bars or symbols represent CFU data from the lung whereas shaded bars and symbols represent CFU data from the spleen. All compounds were delivered IP in 15% ethanol, 20% propylene glycol, 40% polyethylene glycol 400, PBS. Animals were treated with PT070, 50 mg/kg BID Day 2, 25 mg/kg BID Day 3-10, or with PT091, 25mg/kg BID Day 1-4, 12.5 mg/kg BID Day 5-6, 12.5 mg/kg SID and 25 mg/kg SID Day 7-10. The significance between control and treatment groups was determined via an unpaired, one tailed t-test (t-test1, 2) with 95% confidence intervals from mean CFU's for the organs (GraphPad Software ver. 5, San Diego CA USA, www.graphpad.com)
In contrast, PT004 is a rapid reversible InhA inhibitor that does not demonstrate efficacy in the murine model of TB. Importantly, PT070 and PT091 are slow onset inhibitors having residence times of 24 and 21, respectively. The improved efficacy of diaryl ethers that have increased residence time is consistent with observations in the F. tularensis infection model,[7] and highlights the potential importance of drug-target residence time for modulating in vivo drug efficacy.[8e, 8f, 20]
Conclusion
Twenty compounds were synthesized based on the diaryl ether scaffold, with a hexyl-substituted A-ring and various modifications to the B-ring. All the compounds were tested for enzyme inhibition and cell based antibacterial activity. Crystallographic data demonstrated that the slow-onset inhibitors PT092 and PT119 resulted in a more ordered and closed InhA substrate binding loop, in agreement with our hypothesis that motions of this loop are related to the structural change that accompanies the EI to EI* transition for slow-onset inhibitors. The relatively large cyano group in PT119 creates steric hindrance, introduces conformational change of helix-6, and preferentially destabilizes the transition state between EI and EI* compared to the EI* ground state , thus resulting in a longer residence time. The activity of the diaryl ether compounds is highly sensitive to the size of ortho substituents on the B-ring. Once the group has more than two heavy atoms, the activity is significantly reduced. In addition, within the acceptable size limits for this group, non-hydrogen bonding donors are preferred. Introduction of groups on both ortho positions resulted in a loss of activity which, according to docking studies, leads to a conformation in which there are unfavorable van der Waals interactions with the enzyme. The ortho, para di-substituted compound PT107 showed similar activity to that of the parent compound, suggesting that para groups on the B-ring are well tolerated. We were able to distinguish 2’-N and 4’-N pyridyl B-ring analogs by introducing ortho substituents into the B-ring. The 2’-N pyridyl B-rings had significantly better activity than the 4’-N analogues which may reflect the improvement in cell penetration of these compounds and suggests that the pyridyl B-ring is a better scaffold than the phenyl B-ring. For all the diaryl ether compounds, slow-onset inhibitors always have higher binding affinity than rapid reversible inhibitors. In contrast to the previously reported lack of efficacy for the rapid onset inhibitor PT004, the slow-onset inhibitors PT070 and PT091 demonstrated efficacy in a rapid animal model of TB infection, causing a reduction in bacterial CFUs in the spleen. Together, these results support the importance of slow-onset kinetics for in vivo drug efficacy.16
Experimental Section
Chemistry
All commercially available chemicals and solvents were used without further purification. All new compounds gave satisfactory spectroscopic and/or analytical data. 1H and 13C NMR spectra were recorded at 300 or 400 MHz (Varian INOVA), and chemical shifts are reported in parts per million (δ) downfield from the internal standard tetramethylsilane (TMS). Mass spectra were obtained using electrospray (ES) ionization techniques (Agilent Technologies, 1100 Series LC/MSD). The purity of all target compounds is > 98% as determined by 1H NMR or HPLC. General procedures of diazotization, aromatic nucleophilic substitution, reduction, deamination, and demethylation are described below.
General procedure of diazotization (for 7, 8, 9, 10)
The reactant aniline (1 mmol) was dissolved in AcOH (10 mL) and H2O (100 mL), and cooled to −10°C. NaNO2 (103.9 mg, 1.5 mmol) was added slowly, followed by the appropriate halide (1.5 mmol) after 30 min. The ice bath was removed, and the reaction was stirred for approximately 2 h. The reaction mixture was then extracted with CH2Cl2 (250 mL) and H2O (150 mL). The organic layer was dried over MgSO4 and dried in vacuo to give the crude products.
General procedure for the aromatic substitution reaction (for 5, 15, 16, 17, 178, 27, 29, 31, 38)
The nitrobenzene (1.24 mmol), K2CO3 (1.03 mmol), and a catalytic amount of 18-crown-6 ether were added to a stirred solution of the corresponding phenol (1.03 mmol) in DMF (3 mL) at rt, and the solution was then heated to 110°C for 3 h. After completion of the reaction, as shown by TLC, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (2 × 20 mL). The organic layer was washed with brine (30 mL), dried over anhydrous Na2SO4, and dried in vacuo. The crude product was purified by column chromatography to afford the substituted nitrobenzene.
General procedure for reduction of nitrobenzene to aniline (for 6, 19, 20, 21, 22, 30)
Concentrated HCl (1.3 mL) was added dropwise to a stirred solution of the nitrobenzene (0.78 mmol) in ethanol (15 mL) at 0°C and stirred for 5 min. Zn powder (17.3 mmol) was then added slowly and the reaction mixture was allowed to come to rt. The stirred reaction mixture was monitored by TLC, and after completion (~ 1 h), the reaction was quenched with triethylamine and filtered. The filtrate was dried in vacuo and the crude product was purified by column chromatography to afford the corresponding anilines.
General procedure for deamination of anilines (for 23, 24, 25, 26)
Concentrated H2SO4 (5 mL) was added dropwise to a stirred solution of the aniline (4.3 mmol) in ethanol (25 mL) at 0 °C. A saturated aqueous solution of NaNO2 (8.6 mmol) in H2O (5 mL) was added very slowly, and the reaction mixture was stirred at 0 °C for 30 min. The solution was allowed to come to rt and Zn powder (43 mmol) was added. After refluxing for 0.5 h, the remaining Zn powder was added and the mixture refluxed for an additional 2.5 h. After completion of the reaction, the mixture was filtered and neutralized with saturated NaHCO3 (pH 7-8). The organic solvent was removed by evaporation and the aqueous solution was then extracted with EtOAc (2 × 50 mL). The organic layer was washed with water (50 mL), dried over Na2SO4, and dried in vacuo. The crude product was subsequently purified by column chromatography to afford the appropriate deaminated product.
General procedure of demethylation (for PT091, PT092, PT095, PT096, PT107-PT116, PT19, PT131, PT133, PT134, PT161, PT164)
The methoxybenzene (0.627 mmol) was dissolved in dry CH2Cl2 (50 mL), and BBr3 (0.94 mL, 2M in CH2Cl2, 1.88 mmol) was added dropwise at −40°C. The reaction was gradually warmed to rt and stirred for 3 h. When TLC showed completion, the reaction was cooled to −40°C and quenched with MeOH. The solution was dried in vacuo to give the crude product.
4-(Benzyloxy)-3-methoxybenzaldehyde (1)
Vanillin (10 g, 65.7 mmol), benzyl bromide (12.4 g, 72.3 mmol) and KOH (4.1 g, 72.3 mmol) were dissolved in 50% MeOH/H2O (150 mL) and heated to reflux for 2 h. After TLC showed completion, the reaction was extracted with H2O (300 mL) and CH2Cl2 (2 × 100 mL). The organic layer was dried with MgSO4 and dried in vacuo to give the crude product. Purification with flash chromatography (EtOAc/petroleum ether = 8%) gave 1 as white crystals (14.5 g, 91%). 1H NMR (400 MHz, CDCl3): δ 9.81 (s, 1H), 7.42-7.29 (m, 7H), 6.96 (d, J = 6.3 Hz, 1H), 5.22 (s, 2H), 3.92 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 191.1, 153.8, 150.3, 136.2, 130.5, 128.9, 128.4, 127.4, 126.8, 112.6, 109.6, 71.09, 56.3 ppm. HRMS-ES+: m/z [M+H]+ calcd for C15H15O3: 243.1021, found: 243.1022.
1-(Benzyloxy)-4-(hex-1-en-1-yl)-2-methoxybenzene (2)
n-BuLi (2.45 mL, 4.9 mmol, 2 M in cyclohexane) was added dropwise to a solution of n-pentyl-triphenylphosphonium bromide (2 g, 4.9 mmol) in dry THF (100 mL) at −78°C. After 30 min, a solution of 1 (1.0 g, 4.1 mmol) in dry THF (50 mL) was added dropwise. After an additional 30 min the cooling bath was removed, and the reaction was stirred for another 1.5 h. When TLC showed completion, the reaction was quenched with HCl (5 mL 1 m) and extracted with H2O (100 mL). The aqueous layer was washed with CH2Cl2 (3 × 25 mL). The organic layers were combined, dried with MgSO4, and dried in vacuo to give the crude product, which was subsequently purified by flash chromatography (EtOAc/petroleum ether = 5%) to give 2 as white crystals (1.0 g, 82%). 1H NMR (400MHz, CDCl3): δ 7.27-7.43 (m, 5H), 6.91 (s, 1H), 6.79 (s, 1H), 6.29 (d, J = 11.7 Hz, 2H), 6.08 (td, J = 11.7, 5.1 Hz, 1H), 5.13 (s, 2H), 3.89 (s, 3H), 2.18 (dd, J = 5.1, 4.8 Hz, 2H), 1.46-1.29 (m, 4H), 0.91 (t, J = 5.4 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 150.0, 147.6, 137.5, 131.98, 129.7, 129.5, 28.7, 128.0, 127.5, 118.9, 114.5, 109.5, 71.4, 57.2, 32.9, 31.9, 22.5, 14.2 ppm. HRMS-ES+: m/z [M+H]+ calcd for C20H24O2: 297.1855, found: 297.1855.
4-Hexyl-2-methoxyphenol (3)
Activated palladium on charcoal (40 mg) was added to a solution of 2 (800 mg, 2.7 mmol) in ethanol (100 mL). The reaction was stirred under hydrogen for 6 h and then filtered. The solvent was dried in vacuo to give the crude product. Flash chromatography (EtOAc/petroleum ether = 5%) gave 3 as a colorless liquid (500 mg, 89%). 1H NMR (400 MHz, CDCl3): δ 6.80 (d, J = 6.0 Hz, 1H), 6.65-6.63 (m, 2H), 5.42 (s, 1H), 3.85 (s, 3H), 2.50 (t, J = 5.7 Hz, 2H), 1.56-1.54 (m, 2H), 1.30-1.27 (m, 6H), 0.86 (t, J = 5.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) ppm: δ 146.5, 143.7, 135.2, 121.1, 114.3, 111.1, 56.1, 35.9, 32.0, 32.0, 29.2, 22.8, 14.3 ppm. MS (ESI+, 70 eV): m/z (%): 209.0 (100) [M+H]+; MS (ESI-, 70 eV): m/z (%): 207 (100) [M-H]−.
4-Hexyl-2-methoxy-1-(2-(trifluoromethyl)phenoxy)benzene (4)
Reagents 1-iodo-2-(trifluoromethyl)benzene (2.0 g, 7.35 mmol), 3 (1.10 g, 7.35 mmol), Cs2CO3 (10.5 g, 32.3 mmol), (CuOTf)2•PhH (185.0 mg, 0.37 mmol, 5.0 mol % Cu), and 1-naphthoic acid (5.56 g, 32.3 mmol) were dissolved in EtOAc (1 mL) and toluene (75 mL) in an oven-dried 150 mL two-necked round-bottomed flask. Molecular sieves 4 Å (1.8 g) were added into the flask while nitrogen gas is flushing, and then the flask was sealed with a septum and heated to 110 °C under nitrogen. After 24 h stirring, upon cooling to rt, CH2Cl2 was added and the organic phase was obtained by filtration. This solution was washed with 5% NaOH (20 mL). The aqueous layer was then extracted with CH2Cl2 (2 × 25 mL) and the combined organic layers were washed with brine (50 mL), dried over Mg2SO4, and dried in vacuo to give the crude product, which was then purified by flash chromatography on silica gel (4% EtOAc/hexane) to give pure 4 as a light yellow liquid (1.94 g, 75%). MS (ESI+, 70 eV): m/z (%): 353.0 (100), 354.2 (21) M+H]+.
4-Hexyl-2-methoxy-1-(2-nitrophenoxy)benzene (5)
A solution of 3 (800 mg, 3.8 mmol), 1-fluoro-2-nitrobenzene (536.2 mg, 3.8 mmol), and 18-crown-6 (50.2 mg, 0.19 mmol) in 40 mL dry DMF was heated to 110°C under nitrogen and stirred for 3 h. The reaction was cooled to rt and extracted with H2O (200 mL) and CH2Cl2 (200 mL). The organic layer was dried with MgSO4 and evaporated to give crude product. Purification with flash chromatography (EtOAc/petroleum ether = 5%) gave 5, as a light yellow oil (826 mg, 66%). 1H NMR (300 MHz, CDCl3): δ 7.94 (dd, J = 8.4, 1.5 Hz, ,1H), 7.40 (td, J = 7.5, 1.8 Hz, 1H), 7.08 (td, J = 7.5, 1.5Hz, 1H), 6.99 (d, J = 7.8 Hz, 1H), 6.81 (dd, J =8.4, 1.2 Hz, 1H), 6.80 (dd, J = 8.1, 1.8 Hz, 1H), 6.76 (d, J = 2.1 Hz, 1H), 3.77 (s, 3H), 2.61 (t, J = 7.8 Hz, 2H), 1.62 (m, 2H), 1.32 (m, 2H), 0.89 (t, J = 6.9 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 152.3, 151.3, 141.8, 141.3, 134.1, 125.8, 121.9, 121.2, 118.1, 113.6, 56.2, 36.1, 31.9, 31.8, 29.2, 22.8, 14.3 ppm; HRMSES+: m/z [M+H]+ calcd for C19H23NO4: 330.1705, found: 330.1703.
2-(Hexyl-2-methoxyphenoxy)aniline (6)
Activated palladium on charcoal (25 mg) was added to a solution of 5 (500 mg, 1.5 mmol) in ethanol (100 mL). The reaction was stirred under hydrogen for 6 h and then filtered. The crude product was obtained by evaporation and used in the next step without further purification (413 mg, 91%). 1H NMR (400 MHz, CDCl3): δ 6.88 (td, J = 5.7, 1.2 Hz, 1H), 6.78-6.75 (m, 3H), 6.72 (dd, J = 6.0 Hz, 1.2 Hz, 1H), 6.65 (dd, J = 5.1, 1.5 Hz, 1H), 6.62 (dd, J = 6.0, 1.2 Hz, 1H), 3.86 (s, 2H), 3.84 (s, 3H), 2.55 (t, J = 6.0 Hz, 2H), 1.59-1.56 (m, 2H), 1.32-1.28 (m, 6H), 0.87 (t, J = 5.4 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 150.5, 144.9, 143.5, 139.2, 138.1, 124,0, 120.8, 119.1, 118.7, 118.4, 116.4, 113.2, 56.9, 36.0, 31.9, 31.5, 29.2, 22.8, 14.3 ppm; HRMS-ES+: m/z [M+H]+ calcd for C19H25NO2: 300.1964, found: 300.1963.
1-(2-Chlorophenoxy)-4-hexyl-2-methoxybenzene (7)
Compound 7 was obtained from compound 6 (300 mg, 1.0 mmol) and CuCl using the general procedure for diazotization, as a light yellow solid (188 mg, 59%). The crude product 7 was used in the next step without purification.
1-(2-Bromophenoxy)-4-hexyl-2-methoxybenzene (8)
Compound 8 was obtained using the general procedure for diazotization from 6 (300 mg, 1.0 mmol) and CuBr, as a yellow solid (233 mg, 64%). Purification using flash chromatography (EtOAc/petroleum ether = 5%) gave pure 8. Yield 63%. 1H NMR (300 MHz, CDCl3): δ 7.26 (dd, J = 6.9, 1.5 Hz, 1H), 7.18 (dd, J = 4.8, 1.5 Hz, 1H), 6.96-6.64 (m, 5H), 3.82 (s, 3H), 2.60 (t, J = 7.5 Hz, 2H), 1.65-1.60 (m, 2H), 1.33-1.22 (m, 6H), 0.90 (t, J = 6.3 Hz, 3H); MS (ESI+, 70 eV): m/z (%): 363.1 (100), 364.0 (21), 365.4 (100) [M+H]+.
4-Hexyl-2-methoxy-1-(2-methoxy-4-nitrophenoxy)benzene (15)
Compound 15 was obtained, as a yellow solid (1.7 g, 98%), from 3 (1.0 g, 4.8 mmol) and 11 (1.0 g, 5.8 mmol) using the general procedure for the aromatic substitution reaction. 1H NMR (300 MHz, CDCl3): δ 7.83 (d, J = 2.4 Hz, 1H), 7.74 (dd, J = 2.7, 6.0 Hz, 1H), 6.98 (d, J = 7.8 Hz, 1H), 6.80 (td, J = 7.8, 1.8 Hz, 2H), 6.60 (d, J = 9.0 Hz, 1H), 4.02 (s, 3H), 3.76 (s, 3H), 2.62 (t, J = 7.8 Hz, 2H), 1.66-1.57 (m, 2H), 1.38-1.28 (m, 6H), 0.89 (t, J = 6.9 Hz, 3H) ppm; MS (ESI+, 70 eV): m/z (%): 360.3 (100), 361.1 (22) [M+H]+.
1,3-Dichloro-2-(hexyl-2-methoxyphenoxy)-5-nitrobenzene (16)
Compound 16 was obtained, as a yellow solid (1.7 g, 88%), from 3 (1.0 g, 4.8 mmol) and 12 (1.2 g, 5.8 mmol) using the general procedure for the aromatic substitution reaction. 1H NMR (400 MHz): δ 8.24 (s, 2H), 6.82 (s, 1H), 6.62 (d, J = 9.0 Hz, 1H), 6.42 (d, J = 2.2 Hz, 1H), 3.94 (s, 3H), 2.60 (t, J = 7.6 Hz, 2H), 1.64-1.58 (m, 3H), 1.38-1.26 (m, 5H), 0.87 (t, J = 6.7 Hz, 3H) ppm.
2-Fluoro-1-(hexyl-2-methoxyphenoxy)-4-nitrobenzene (17)
Compound 17 was obtained as a yellow solid (1.0 g, 60%) from 3 (1.0 g, 4.8 mmol) and 13 (0.92 g, 5.8 mmol) using the general procedure for the aromatic substitution reaction. 1H NMR (400 MHz,): δ 8.00 (d, J = 10.2 Hz,1H), 7.82 (d, J = 9.2 Hz, 1H), 6.96 (d, J = 8.2 Hz, 1H), 6.80-6.6.70 (m, 2H), 6.68 (t, J = 8.1 Hz, 1H), 3.74 (s, 3H), 2.58 (t, J = 6.7 Hz, 2H), 1.64-1.56 (m, 3H), 1.38-1.22 (m, 5H), 0.87 (t, J = 6.6 Hz, 3H) ppm.
1-Chloro-3-fluoro-2-(hexyl-2-methoxyphenoxy)-5-nitrobenzene (18)
Compound 18 was obtained as a yellow solid (1.4 g, 77%) from 3, (1.0 g, 4.8 mmol) and 14 (1.1 g, 5.8 mmol) using the general procedure for the aromatic substitution reaction. HPLC: 95%. 1HNMR (400MHz, CDCl3): δ 8.18 (s, 1H), 7.91-7.90 (m, 1H), 6.80 (s, 1H), 6.72-6.64 (m, 2H), 3.82 (s, 3H), 2.58-2.52 (m, 2H), 1.62-1.54 (m, 3H), 1.38-1.24 (m, 5H), 0.94-0.84 (m, 3H) ppm.
4-(Hexyl-2-methoxyphenoxy)-3-methoxyaniline (19)
Compound 15 (800 mg, 83%) was used in the general procedure for reduction of nitrobenzene to give 19, as a yellow oil (609 mg, 1.8 mmol). MS (ESI+, 70 eV): m/z (%): 330.1 (100), 331.2 (22) [M+H]+.
3-Chloro-5-fluoro-4-(hexyl-2-methoxyphenoxy) aniline (22)
Compound 22 was obtained, as a yellow solid (545 mg, 74%), from 18 (800 mg, 2.1 mmol) using the general procedure for reduction of nitrobenzene. HPLC: 96%. 1HNMR (400MHz, CDCl3): δ 6.80 (s, 1H), 6.70 (s, 1H), 6.62-6.56 (m, 2H), 6.44 (d, J = 7.4 Hz, 1H), 3.94 (s, 3H), 2.58 (t, J = 7.4 Hz, 2H), 1.62-1.54 (m, 2H), 1.38-1.22 (m, 6H), 0.94-0.84 (m, 3H) ppm; MS (ESI+, 70 eV): m/z (%): 352.3 (100), 353.1 (21), 354.4 (33) [M+H]+.
4-Hexyl-2-methoxy-1-(2-methoxyphenoxy)benzene (23)
Compound 23 was obtained, as a yellow oil (406 mg, 85%), from 19 (500 mg, 1.5 mmol) using the general procedure for deamination. MS (ESI+, 70 eV): m/z (%): 315.2 (100), 316.1 (22) [M+H]+.
1-(2,6-dichlorophenoxy)-3-hexyl-2-methoxybenzene (24)
Compound 20 was obtained as a yellow solid (577 mg, 78%) from 16 (800 mg, 2.0 mmol) using the general procedure for reduction of nitrobenzene. The crude compound 20 (577 mg, 1.6 mmol) was used without further purification in the general procedure for deamination to give 24, as a yellow oil (486 mg, 86%). HPLC: 96%. 1H NMR (400MHz, CDCl3): δ 7.38 (m, 2H), 7.16 (t, J = 8.5 Hz, 1H) ,6.82 (s, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.32 (d, J = 7.6 Hz, 1H), 3.80 (s, 3H), 2.56 (t, J = 7.5 Hz, 2H), 1.62-1.54 (m, 3H), 1.38-1.24 (m, 5H), 0.94-0.86 (m, 3H) ppm.
1-(2-Fluorophenoxy)-4-hexyl-2-methoxybenzene (25)
Compound 21 was obtained as a yellow solid (599 mg, 82%) from 17 (800 mg, 2.3 mmol) using the general procedure for reduction of nitrobenzene. The crude compound 21 (599 mg, 1.9 mmol) was used without further purification in the general procedure for deamination to give 25, as a yellow oil (448 mg, 78%). HPLC: 98%. 1H NMR (400 MHz, CDCl3): δ 7.17-7.12 (m, 1H), 7.02-6.99 (m, 2H), 6.88-6.81 (m, 3H), 6.72-6.69 (dd, J = 8.2, 2.2 Hz, 2H), 3.84 (s, 3H), 2.58 (t, J = 7.4 Hz, 2H), 1.64-1.58 (m, 2H), 1.37-1.32 (m, 6H), 0.89 (t, J = 6.6 Hz, 3H) ppm; HRMS-ES+: m/z [M+H]+ calcd for C19H29FO2: 303.1760, found: 300.1758.
1-Chloro-3-fluoro-2-(hexyl-2-methoxyphenoxy)benzene (26)
Compound 26 was obtained, as a yellow oil (407 mg, 85%), from 22 (500 mg, 1.4 mmol) using the general procedure for deamination. HPLC: 95.4%. 1HNMR (400MHz, CDCl3): δ 7.26 (d, J = 7.6 Hz, 1H), 7.18-7.04 (m, 2H), 6.82 (s, 1H), 6.62 (d, J = 8.2 Hz, 1H), 6.42 (d, J = 7.9 Hz, 1H), 3.94 (s, 3H), 2.59 (t, J = 6.9 Hz, 2H), 1.68-1.54 (m, 3H), 1.42-1.24 (m, 6H), 1.89 (t, J = 6.6 Hz, 3H) ppm; MS (ESI+, 70 eV): m/z (%): 337.3 (100), 338.0 (21), 339.4 (33) [M+H]+.
4-Hexyl-2-methoxy-1-(2-methyl-4-nitrophenoxy)benzene (27)
Compound 27 was obtained, as a yellow solid (857 mg, 52%), from 3 (1.0 g, 4.8 mmol) and 1-fluoro-2-methyl-4-nitrobenzene (894 mg, 5.8 mmol) using the general procedure for the aromatic substitution reaction. The crude compound was used for the next step without further purification. MS (ESI+, 70 eV): m/z (%): 344.1 (100), 345.0 (22) [M+H]+.
1-(2,6-Dimethylphenoxy)-3-hexyl-2-methoxybenzene (28)
2-Bromo-1, 3-dimethylbenzene (0.667 g, 3.6 mmol) and K3PO4 (1.0 g, 4.8 mmol) were added to a stirred solution of 3 (0.5 g, 2.403 mmol) in DMF (5 mL). The reaction mixture was degassed for 30 min after which catalyst (0.112 g, 0.24 mmol) was added and the mixture was stirred at 120 °C for 48 h. After TLC showed that the reaction was complete, the solution was extracted with EtOAc (40 mL) and water (2 × 40 mL). The organic layer was washed with brine (60 mL), dried over Na2SO4, and dried in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane = 3%) to afford 28 as a yellow solid (0.075 g, 10%). 1HNMR (400MHz, CDCl3): δ 7.10-7.02(m, 2H), 6.80 (s, 1H), 6.54 (d, J = 7.4 Hz, 1H), 6.20 (d, J = 8.0 Hz, 2H), 3.98 (s, 3H), 2.54 (t, J = 7.5 Hz, 2H), 2.18 (s, 3H), 1.64-1.48 (m, 6H), 1.38-1.24 (m, 5H), 0.89 (t, J = 6.7 Hz, 3H) ppm.
1-(2-Methyl-6-nitrophenoxy)-3-hexyl-2-methoxybenzene (29)
Compound 29 was obtained, as a yellow solid (692 mg, 42%), from 3 (1.0 g, 4.8 mmol) and 2-fluoro-1-methyl-3-nitrobenzene (908 mg, 5.8 mmol) using the general procedure for the aromatic substitution reaction. HPLC: 99%. 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 2.1 Hz, 1H), 7.28-7.22 (m, 2H), 6.82 (s, 1H), 6.58 (d, J = 2.1 Hz, 1H), 6.38 (m, 1H), 3.88 (s, 3H), 2.56 (t, J = 7.2 Hz, 2H), 2.22 (s, 3H), 1.62-1.54 (m, 3H), 1.38-1.22 (m, 5H), 0.89 (t, J = 6.7 Hz, 3H) ppm; MS (ESI+, 70 eV): m/z (%): 366.1 (100) [M+Na]+.
2-(Hexyl-2-methoxyphenoxy)-3-methylbenzenamine (30)
Compound 30 was from 29 (500 mg, 1.5 mmol) using the general procedure for reduction of nitrobenzene, as a dark yellow solid (187 mg, 41%). HPLC: 99%. 1HNMR (400MHz, CDCl3): δ 6.99-6.94 (t, J = 8.4 Hz, 1H), 6.78 (d, J = 7.6 Hz, 2H), 6.64 (m, 1H), 6.59 (m, 1H), 6.42 (m, 1H), 3.89 (s, 3H), 2.56 (t, J = 7.5 Hz, 2H), 2.18 (s, 3H), 1.62-1.54 (m, 2H), 1.42-1.24 (m, 5H), 0.89 (t, J = 6.5 Hz, 3H) ppm; MS (ESI+, 70 eV): m/z (%): 314.0 (100), 315.3 (22) [M+H]+.
2-(Hexyl-2-methoxyphenoxy)-3-fluorobenzonitrile (31)
Compound 31 was prepared from 3 (1.0 g, 4.8 mmol) and 2-bromo-3-fluorobenzonitrile (1.2 g, 5.8 mmol) using the general procedure for the aromatic substitution reaction, as a yellow solid (629 mg, 40%). HPLC: 95%. 1HNMR (400MHz, CDCl3): δ 7.38-7.22 (m, 3H), 6.98 (d, J = 8.2 Hz, 1H), 6.82-6.78 (m, 2H), 6.72 (d, J = 7.8 Hz, 1H), 3.78 (s, 3H), 2.63 (t, J = 7.4 Hz, 2H), 1.72-1.64 (m, 3H), 1.42-1.24 (m, 6H), 0.92 (t, J = 6.8 Hz, 3H) ppm.
2-(Hexyl-2-methoxyphenoxy)-3-fluorobenzamidine (32)
Li(TMS)2 (0.18 g, 1.18 mmol) was added to a stirred solution of 31 (0.2 g, 0.61 mmol) in THF (2 mL) at 0 °C and the reaction was stirred at rt for 3 h. After the reaction was shown to be complete by TLC, the mixture was diluted with H2O (15 mL) and extracted with CH2Cl2 (30 mL × 2). The organic layer was washed with brine (15 mL), dried over Na2SO4, and dried in vacuo. The crude product was purified by flash chromatography (MeOH/CH2Cl2 = 1%) to give 32 as a yellow solid (0.096 g, 46%). HPLC: 64%. 1HNMR (400MHz, CDCl3): δ 8.96 (bs, 1H), 7.98 (br, 2H), 7.42 (d, J = 8.0 Hz, 1H), 7.12-7.03 (m, 2H), 6.90-6.81 (m, 2H), 6.74 (d, J = 7.6 Hz, 1H), 3.80 (s, 3H), 2.60 (t, J = 7.2 Hz, 2H), 1.66-1.60 (m, 2H), 1.42-1.38 (m, 6H), 0.98-0.92 (m, 3H) ppm; MS (ESI+, 70 eV): m/z (%): 345.0 (100) [M+H]+.
3-(Hexyl-2-methoxyphenoxy)-2-nitropyridine (34)
3 (0.5 g, 2.4 mmol) was added to an aqueous solution of NaOH (5%, 7.68 g, 9.61 mmol) and stirred for 10 min. The solution was dried in vacuo, CH3CN (5 mL) and 3-bromo-2-nitropyridine (0.455 g, 2.88 mmol) were added to the remaining residue and the reaction mixture was refluxed at 80 °C for 2 h. After the reaction was shown to be complete by TLC, the reaction mixture was filtered and washed with H2O (15 mL) and CH3CN (50 mL). The organic layer was dried in vacuo, and the crude product was purified by flash chromatography (EtOAc/hexane = 10%) to give 34 as a yellow solid (0.13 g, 16%). HPLC: 76%. 1H NMR (400MHz, CDCl3): δ 8.37 (d, J = 8.1 Hz, 1H), 8.31 (d, J = 5.0 Hz, 1H), 7.10-7.09 (m, 2H), 6.82 (d, J = 6.0 Hz, 2H), 3.71 (s, 3H), 2.62 (t, J = 8.0 Hz, 2H), 1.64-1.44 (m, 2H), 1.35-1.32 (m, 6H), 0.90 (t, J = 6.5 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 156.1, 151.8, 150.9, 141.8, 139.3, 135.2, 133.9, 122.4, 120.7, 117.7, 113.1, 55.9, 35.9, 31.6, 31.2, 29.0, 22.5, 14.0 ppm; HRMS-ES+: m/z [M+H]+ calcd for C18H22N2O4: 331.1658, found: 331.1652.
3-(Hexyl-2-methoxyphenoxy)pyridin-2-amine (35)
Fe powder (81 mg, 3.1 mmol) was added to a stirred solution of 34 (128 mg, 0.38 mmol) in AcOH/H2O (4:1, 5 mL) and the mixture was then refluxed at 100 °C for 30 min. After the reaction was shown to be complete by TLC, the mixture was filtered and washed first with water then with dichloromethane (40 mL). The organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give 35 (92 mg, 77%). The crude compound was used for the next step without further purification. MS (ESI+, 70 eV): m/z (%): 301.2 (100), 302.0 (20) [M+H]+.
3-(Hexyl-2-methoxyphenoxy)-4-nitropyridine 1-oxide (36)
Compound 3 (0.5 g, 2.4 mmol) was added to an aqueous solution of NaOH (0.385 g, 9.6 mmol, 1 mL) and stirred for 5 min, and then water was dried in vacuo to afford a white solid. This solid was taken up in CH3CN (7 mL) and 3-bromo-4-nitropyridine 1-oxide (0.63 g, 2.8 mmol) was added. The mixture was refluxed at 80 °C for 2 h and after the reaction was shown to be complete by TLC the reaction mixture was filtered, and the filtrate was dried in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane = 50%) to afford 36 as a dark yellow solid (0.5 g, 60%). 1H NMR (400 MHz,): δ 7.86 (d, J = 2.2 Hz, 1H), 7.84 (d,J = 8.0 Hz, 1H), 7.74 (s, 1H), 7.04 (d, J = 7.8 Hz, 1H), 6.82 (m, 2H), 3.78 (s, 3H), 2.60 (t, J = 7.6 Hz, 2H), 1.64-1.58 (m, 3H), 1.38-1.26 (m, 5H), 0.89 (t, J = 6.7 Hz, 3H) ppm; MS (ESI+, 70 eV): m/z (%): 347.2 (100), 348.1 (20) [M+H]+.
3-(Hexyl-2-methoxyphenoxy)pyridin-4-amine (37)
A stirred solution of 36 (0.5 g, 1.4 mmol) in AcOH/H2O (8:2, 100 mL) was heated to 80 °C, and Fe powder (4 g) was added portion wise at 80 °C and stirred at the same temperature for 2 h. After the reaction was shown to be complete by TLC, the reaction mixture was quenched with water and extracted with CH2Cl2 (2 × 20 mL). The organic layer was washed with brine (15 mL), dried over Na2SO4, and dried in vacuo. The crude product was purified by flash chromatography (MeOH/CH2Cl2 = 5%) to give 37 as a dark yellow oil (0.25 g, 57%). HRMS-ES+: m/z [M+H]+ calcd for C18H24N2O2: 301.1916, found: 301.1906.
3-(Hexyl-2-methoxyphenoxy)isonicotinonitrile (38)
Compound 38 was obtained, as a dark yellow oil (1.0 g, 69%), from 3 (1.0 g, 4.8 mmol) and 3-chloroisonicotinonitrile (0.8 g, 5.8 mmol) using the general procedure for the aromatic substitution reaction. HPLC: 98%. 1H NMR (400 MHz, CDCl3): δ 8.38 (d, J = 1.5 Hz, 1H), 8.14 (s, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 7.8 Hz, 1H), 6.82 (d, J = 7.5 Hz, 2H), 3.78 (s, 3H), 2.62 (t, J = 7.4 Hz, 2H), 1.66-1.62 (m, 2H), 1.42-1.34 (m, 6H), 0.91 (t, J = 6.6 Hz, 3H) ppm.
3-(Hexyl-2-methoxyphenoxy)isonicotinic acid (39)
KOH pellets (0.025 g) were added to a stirred solution of 38 (0.07 g, 0.15 mmol) in MeOH (2 mL) at rt. The reaction mixture was then refluxed at 80 °C for 2 h. After the reaction was shown to be complete by TLC, the reaction mixture was neutralized with 1 n HCl solution (pH 1-2) and extracted with CH2Cl2 (2 × 20 mL). The organic layer was washed with H2O (10 mL), dried over Na2SO4, and dried in vacuo to give the crude compound, which was then purified with HPLC to give pure 39 as a dark yellow oil (61 mg, 82%). 1H NMR (400 MHz, DMSO): δ 13.56 (s, 1H), 8.38 (d, J = 2.1 Hz, 1H), 7.98 (s, 1H), 7.62 (d, J = 2.2 Hz, 1H), 7.00 (s, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 3.78 (s, 3H), 2.60 (t, J = 7.4 Hz, 2H), 1.60 (t, J = 6.8 Hz, 2H), 1.38-1.24 (m, 6H), 0.89 (t, 3H) ppm.
Ethyl 2-(hexyl-2-methoxyphenoxy) acetate (40)
A solution of 3 (2 g, 9.6 mmol) in ethanol (5 mL) was added to a solution of sodium ethoxide (4.6 ml, 14.4 mmol) in ethanol (15 mL) at rt. Ethyl bromo acetate (1.6 mL, 14.4 mmol) was added dropwise and the reaction mixture was refluxed at 80 °C for 16 h. After the reaction was shown to be complete by TLC, the solvent was dried in vacuo, H2O was added, and the mixture was extracted with EtOAc (2 × 50 mL). The organic layer was washed with brine (50 mL), dried over Na2SO4, and dried in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane = 10%) to give 40 as a yellow solid (0.98 g, 35%). HPLC: 97%. 1H NMR (400MHz, CDCl3): δ 6.78 (d, J = 8.2 Hz, 1H), 6.72 (s, 1H), 6.64 (d, J = 7.8 Hz, 1H), 4.70 (s, 2H), 4.26 (t, J = 7.4 Hz, 2H), 3.82 (s, 3H), 2.57 (t, J = 7.2 Hz, 3H), 1.62-1.54 (m, 4H), 1.38-1.24 (m, 6H) ppm; MS (ESI+, 70 eV): m/z (%): 295.4 (100), 296.1 (19) [M+H]+.
5-(Hexyl-2-methoxyphenoxy) pyrimidin-4-ol (41)
40 (1 g, 3.4 mmol) and ethyl formate (2.5 g, 34 mmol) were added to a stirred solution of NaH (0.146 g, 6.1 mmol) in THF (10 mL) at rt. The mixture was then heated to 65 °C for 4 h. After the reaction was shown to be complete by TLC, the solvent was dried in vacuo, and the crude compound was dissolved in MeOH/EtOH = 50% (12 mL). Formamidine acetate (0.353 g, 3.4 mmol) was added and the reaction was stirred at 80 °C for 4 h. After the reaction was shown to be complete by TLC, the solvent was dried in vacuo and the crude product was purified by flash chromatography (MeOH/CH2Cl2 = 8%) to give 41 as a yellow solid (0.47 g, 47%). HPLC: 99%. 1H NMR (400 MHz, CDCl3): δ 7.88 (s, 1H), 7.38 (s, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.82-6.78 (m, 2H), 3.82 (s, 3H), 2.60 (t, J = 7.2 Hz, 2H), 1.68-1.48 (m, 4H), 1.38-1.22 (m, 4H), 0.89 (t, J = 6.8 Hz, 3H) ppm; HRMS-ES+: m/z [M+H]+ calcd for C17H22N2O3: 303.1709, found: 303.1708.
4-Chloro-5-(hexyl-2-methoxyphenoxy) pyrimidine (42)
POCl3 (0.3 mL) was added dropwise to a stirred solution of 41 (25 mg, 0.08 mmol) in CHCl3 (5 mL), and stirred at 70 °C for 3 h. After the reaction was shown to be complete by TLC, the solvent was removed under reduced pressure and extracted with CH2Cl2 (20 mL) and water (2 × 20 mL). The organic layer was washed with water (20 mL), brine (20 mL), dried over Na2SO4, and dried in vacuo. The crude product was purified by flash chromatography (MeOH/CH2Cl2 = 5%) to give 42 as a dark yellow solid (12 mg, 46%). 1HNMR (400MHz, CDCl3): δ 7.93 (s, 1H), 7.38 (s, 1H), 6.96 (d, J = 7.8 Hz, 1H), 6.88-6.81 (m, 2H), 3.80 (s, 3H), 2.58-2.54 (t, J = 7.2 Hz, 2H), 1.62-1.54 (m, 2H), 1.38-1.24 (m, 6H), 0.98-0.92 (m, 3H) ppm.
5-(Hexyl-2-methoxyphenoxy)pyrimidin-4-amine (43)
NH4OH (1.5 mL) was added dropwise to a stirred solution of 42 (55 mg, 0.17 mmol) in CH2Cl2 (10 mL) at rt, and then heated to 130 °C for 18 h. After the reaction was shown to be complete by TLC, the solvent was dried in vacuo, and the crude product purified by column chromatography (MeOH/CH2Cl2 = 8%) to give 43 as a dark yellow oil (45 mg, 87%). HPLC: 96%. 1H NMR (400 MHz, CDCl3): δ 8.30 (s, 1H), 7.70 (s, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.80 (s, 1H), 6.75 (d, J = 7.9 Hz, 1H), 5.20 (bs, 2H), 4.80 (s, 3H), 2.64 (t, J = 7.4 Hz, 2H), 1.70-1.50 (m, 2H), 1.40-1.20 (m, 6H), 0.85 (t, J = 6.7 Hz, 3H) ppm; HRMS-ES+: m/z [M+H]+ calcd for C17H23N3O2: 302.1869, found: 302.1868.
2-(2-Chlorophenoxy)-5-hexylphenol (PT091)
PT091 was obtained from 7 (300 mg, 0.94 mmol) using the general demethylation procedure. Purification by silica gel chromatography (EtOAc/petroleum ether = 5%) gave pure PT091 as a light yellow oil (255 mg, 89%). 1H NMR (400 MHz, CDCl3): δ 7.45 (dd, J = 7.9, 1.6 Hz, 1H), 7.20 (td, J = 8.0, 1.0 Hz, 1H), 7.07 (td, J = 7.9, 1.5 Hz, 1H), 6.96 (dd, J = 8.0, 1.5 Hz, 1H), 6.88 (d, J = 2.0 Hz, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.64 (dd, J = 8.2, 2.0 Hz, 1H), 5.50 (br, 1H), 2.55 (t, J = 7.9 Hz, 2H), 1.61-1.58 (m, 2H), 1.37-1.26 (m, 6H), 0.89 (t, J = 9.0 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ 152.5, 147.7, 141.1, 140.3, 130.8, 128.0, 124.9, 124.6, 120.5, 119.2, 118.0, 116.3, 35.5, 31.7, 31.4, 28.9, 22.6, 14.1 ppm; HRMS-ES+: m/z [M+NH4]+calcd for C18H21ClO2: 322.1230, found: 322.1572.
2-(2-Bromophenoxy)-5-hexylphenol (PT092)
PT092 was obtained from 8 (300 mg, 0.83 mmol) using the general demethylation procedure. Purification with flash chromatography (EtOAc/petroleum ether = 5%) gave pure PT092 (236 mg, 82%) as a light yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.58 (dd, J = 7.8, 1.6 Hz, 1H), 7.36 (td, J = 8.0, 1.2 Hz, 1H), 7.02 (td, J = 7.8, 1.6 Hz, 1H), 6.93 (dd, J = 8.0, 1.2 Hz, 1H), 6.80 (d, J = 2.0 Hz, 1H), 6.75 (d, J = 8.2 Hz, 1H), 6.68 (dd, J = 8.2, 2.0 Hz, 1H), 5.32 (br, 1H), 2.60 (t, J = 8.0 Hz, 2H), 1.62-1.58 (m, 2H), 1.32-1.21 (m, 6H), 0.80 (t, J = 9.0 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ 154.5, 152.1, 143.2, 138.7, 131.9, 126.3, 125.1, 124.6, 123.5, 117.1, 113.0, 110.4, 35.9, 31.8, 31.1, 28.8, 22.5, 14.2 ppm; HRMS-ES+: m/z [M+NH4]+calcd for C18H21BrO2: 348.0725, found: 348.0732.
5-Hexyl-2-(2-(trifluoromethyl)phenoxy)phenol (PT095)
PT095 was obtained from 4 (1.0 g, 2.8 mmol) using the general demethylation procedure. Purification with flash chromatography (EtOAc/petroleum ether 5%) gave pure PT095 as a light yellow oil (844 mg, 88%). 1H NMR (300MHz, CDCl3): δ 7.67 (dd, J = 7.8, 1.5 Hz, 1H), 7.44 (td, J = 7.8, 1.2 Hz, 1H), 7.16 (tt, J = 8.1, 0.9 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.90 (d, J = 2.1 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 6.69 (dd, J = 8.1, 1.8 Hz, 1H), 5.43 (s, 1H), 2.57 (t, J = 7.8 Hz, 2H), 1.64-1.56 (m, 2H), 1.37-1.28 (m, 6H), 0.90 (m, 3H) ppm; HRMS-ES+: m/z [M+Na]+ calcd for C19H21F3O2: 361.1391; found: 361.1396.
5-Hexyl-2-(2-iodophenoxy)phenol (PT096)
Compound 9 was obtained using the general procedure for diazotization from 6 (300 mg, 1.0 mmol) and CuI. Crude 9 was used in the general demethylation procedure to give PT096, as a light yellow oil (334 mg, 84%). Purification by flash chromatography (EtOAc/petroleum ether = 5%) gave pure PT096 as a light yellow oil. HRMS-ES+: m/z [M+Na]+ calcd for C18H21IO2: 419.0484, found: 419.0479.
5-Hexyl-2-(2-methyl-4-nitrophenoxy)phenol (PT107)
PT107 was obtained from 27 (200 mg, 0.59 mmol) using the general demethylation procedure. Purification with flash chromatography (EtOAc/hexane = 15%) gave pure PT107 as a yellow solid (169 mg, 88%). 1H NMR (300 MHz, CDCl3): δ 8.10 (dd, J = 0.6, 1.8 Hz, 1H), 7.94 (ddd, J = 0.6, 3.0, 9.3 Hz, 1H), 6.91 (d, J = 2.1 Hz, 1H), 6.75 (m, 2H), 6.71 (d, J = 1.8 Hz, 1H), 5.41 (s, 1H), 2.58 (t, J = 7.8 Hz, 2H), 2.44 (s, 3H), 1.61 (m, 2H), 1.32 (m, 6H), 0.89 (t, J = 6.9 Hz, 3H) ppm; HRMS-ES+: m/z [M+Na]+ calcd for C19H23NO4: 352.1525, found: 352.1528.
2-(2,6-Dimethylphenoxy)-5-hexylphenol (PT108)
PT108 was obtained from 28 (200 mg, 0.64 mmol) using the general demethylation procedure. Purification with flash chromatography (EtOAc/hexane = 15%) gave pure PT108 as a dark yellow oil (153 mg, 80%). HPLC: 94%. 1H NMR (400 MHz, CDCl3): δ 7.11-7.07 (m, 3H), 6,86 (s, 1H), 6.48 (d, J = 8.2 Hz, 1H), 6.20 (d, J = 7.8 Hz, 1H), 5.78 (bs, 1H), 2.51 (t, J = 7.8 Hz, 2H), 2.14 (s, 6H), 1.58 (t, J = 6.9 Hz, 2H), 1.30-1.26 (m, 6H), 0.88-0.86 (t, 3H) ppm; HRMS-ES+: m/z [M+Na]+ calcd for C20H26O2: 321.1831, found: 321.1832.
2-(2,6-Dichlorophenoxy)-5-hexylphenol (PT109)
PT109 was obtained from 24 (300 mg, 0.85 mmol) using the general demethylation procedure. Purification with flash chromatography (EtOAc/hexane = 10%) gave PT109 as a dark yello oil (274 mg, 95%). HPLC: 99%. 1H NMR (400 MHz, CDCl3): δ 7.40 (d, J = 9.0 Hz, 2H), 7.26-7.20 (t, J = 8.5 Hz, 1H), 6.87 (d, J = 2.2 Hz, 1H), 6.54-6.52 (dd, J = 8.8, 1.8 Hz, 1H), 6.33 (d, J = 8.2 Hz, 2H), 5.72 (bs, 1H), 2.51 (t, J = 7.5 Hz, 2H), 1.59-1.55 (m, 2H), 1.36-1.24 (m, 6H), 0.87 (t, J = 6.6 Hz, 3H) ppm; HRMS-ES+: m/z [M+Na]+ calcd for C18H20Cl2O2: 361.0738, found: 361.0746.
2-(2-Amino-6-methylphenoxy)-5-hexylphenol (PT110)
Compound PT110 was obtained from 30 (200 mg, 0.64 mmol) using the general demethylation procedure, as a dark yellow oil (122 mg, 64%). HPLC: 99%. 1H NMR (400 MHz, DMSO): δ 9.11 (s, 1H), 6.83 (t, J = 8.7 Hz, 1H), 6.69 (s, 1H), 6.62 (d, J = 8.2 Hz, 1H), 6.46-6.41 (m, 2H), 6.22 (d, J = 7.6 Hz, 1H), 4.71 (s, 1H), 2.42 (t, J = 7.8 Hz, 2H), 1.98 (s, 3H), 1.54-1.48 (m, 2H), 1.34-1.22 (m, 6H), 0.94-0.82 (m, 3H) ppm; HRMS-ES+: m/z [M+H]+ calcd for C19H25NO2: 300.1964, found: 300.1959.
3-Fluoro-2-(4-hexyl-2-hydroxyphenoxy) benzonitrile (PT111)
Compound PT111 was obtained from 31 (200 mg, 0.61 mmol) using the general demethylation procedure, as a yellow solid (103 mg, 54%). HPLC: 99%. 1H NMR (400 MHz, CDCl3): δ 7.35-7.34 (d, J = 2.2 Hz, 1H), 7.27-7.23 (m, 1H), 7.00 (d, J = 9.2 Hz, 1H), 6.83 (d, J = 1.8 Hz, 1H), 6.70 (d, J = 8.5 Hz, 1H), 6.63-6.62 (d, J = 8.5, Hz, 2H), 5.24 (bs, 1H), 2.49 (t, J = 7.4 Hz, 2H), 1.53-1.51 (m, 2H), 1.26-1.18 (m, 6H), 0.82-0.80 (m, 3H) ppm.
2-((4-Aminopyridin-3-yl)oxy)-5-hexylphenol (PT112)
Compound PT112 was obtained from 37 (300 mg, 1.0 mmol) using the general demethylation procedure as a brown solid (97 mg, 34%). HPLC: 98%. 1H NMR (400 MHz, DMSO): δ 9.37 (s, 1H), 7.80 (d, J = 1.8 Hz, 1H), 7.60 (s, 1H), 6.75 (d, J = 8.1 Hz, 1H), 6.73 (d, J = 2.2 Hz, 1H), 6.64 (d, J = 8.2 Hz, 1H), 6.58-6.56 (dd, J = 8.1, 2.1 Hz,1H), 5.90 (s, 2H), 2.47 (t, J = 7.8 Hz, 2H), 1.54-1.50 (m, 2H), 1.27-1.22 (m, 6H), 0.86 (t, J = 6.7 Hz, 3H) ppm;HRMS-ES+: m/z [M+H]+ calcd for C17H22N2O2: 287.1760, found: 287.1761.
2-(2-Fluorophenoxy)-5-hexylphenol (PT113)
Compound PT113 was obtained from 25 (300 mg, 1.0 mmol) using the general demethylation procedure, as a yellow oil (126 mg, 44%). HPLC: 98%. 1H NMR (400 MHz, CDCl3): δ 7.20-7.18 (m, 1H), 7.17-6.99 (m, 3H), 6.87 (d, J = 7.8 Hz, 1H), 6.72 (d, J = 2.4 Hz, 1H), 6.64-6.62 (dd, J = 7.8, 2.2 Hz, 1H), 5. 65 (s 1H), 2.54 (t, J = 7.5 Hz, 2H), 1.58 (t, J = 7.8 Hz, 2H), 1.35-1.25 (m, 6H), 0.88 (t, J = 6.5 Hz, 3H) ppm; HRMS-ES+: m/z [M+Na]+ calcd for C18H21FO2: 311.1423, found: 311.1433.
5-Hexyl-2-(2-hydroxyphenoxy)phenol (PT114)
PT114 was obtained from 23 (300 mg, 1.0 mmol) using the general demethylation procedure. Purification with chromatography (EtOAc/hexane = 20%) gave pure PT114 as a yellow oil (152 mmol, 53%). 1H NMR (300MHz, CDCl3): δ 7.03-7.00 (m, 2H), 6.87 (d, J = 2.4 Hz, 1H), 6.82-6.80 (m, 2H), 6.76 (d, J = 8.4 Hz, 1H), 6.65 (dd, J = 2.4, 8.4 Hz, 1H), 6.00 (br, 2H), 2.54 (t, J = 7.8 Hz, 2H), 2.03 (s, 2H), 1.59-1.57 (m, 2H), 1.36-1.27 (m, 6H), 0.91 (t, J = 6.6 Hz, 3H) ppm; HRMS-ES+: m/z [M+Na]+ calcd for C18H22O3: 309.1467, found: 309.1466.
3-(4-Hexyl-2-hydroxyphenoxy)isonicotinonitrile (PT115)
Compound 38 (200 mg, 0.64 mmol) was used in the general demethylation procedure to give PT115 as a dark yellow oil (80 mg, 42%). HPLC: 96%. 1H NMR (400 MHz, CDCl3): δ 8.46 (d, J = 1.8 Hz, 1H), 8.34 (s, 1H), 7.56 (d, J = 2.1 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 5.38 (s, 1H), 2.62-2.58 (m, 2H), 1.64-1.54 (m, 3H), 1.38-1.24 (m, 5H), 0.94-0.84 (m, 3H) ppm; HRMS-ES+: m/z [M+H]+ calcd for C18H20N2O2: 297.1603, found: 297.1599.
3-(4-Hexyl-2-hydroxyphenoxy)isonicotinic acid (PT116)
Compound 39 (200 mg, 0.61 mmol) was used in the general demethylation procedure to give PT116 as a dark yellow oil (144 mg, 75%). HPLC: 93%. 1H NMR (400 MHz, DMSO): δ 8.32 (d, J = 1.8 Hz, 1H), 8.02 (s, 1H), 7.58 (d, J = 2.2 Hz, 1H), 6.98 (d, J = 9.1 Hz, 1H), 6.78 (s, 1H), 6.62 (d, J = 8.8 Hz, 1H), 2.58-2.44 (m, 2H), 1.58-1.46 (m, 2H), 1.36-1.24 (m, 6H), 0.94-0.84 (m, 3H) ppm; HRMS-ES+: m/z [M+H]+ calcd for C18H21NO4: 316.1549, found: 316.1541.
2-(4-Hexyl-2-hydroxyphenoxy)benzonitrile (PT119)
Compound 10 was obtained using the general procedure for diazotization from 6 (300 mg, 1.0 mmol) and CuCN. Crude 10 was used in preparation of PT119 without purification, which employs the general demethylation procedure. Purification with flash chromatography (EtOAc/petroleum ether = 5%) gave pure PT119 as a white solid (244 mg, 86%). 1H NMR (300 MHz, CDCl3): δ 7.64 (br. d, J = 7.8 Hz, 1H), 7.46 (br. t, J = 8.1 Hz, 1H), 7.12 (br. t, J = 7.5 Hz, 1H), 6.90-6.83 (m, 3H), 6.72 (br. d, J = 8.1 Hz, 1H), 5.55 (s, 1H), 2.57 (t, J = 7.5 Hz, 2H), 1.60 (br. t, J = 6.6 Hz, 2H), 1.36-1.24 (m, 6H), 0.88 (t, J = 6.6 Hz, 3H). HRMS-ES+: m/z [M+H]+ calcd for C19H21NO2: 296.1651, found: 296.1647.
3-Fluoro-2-(4-hexyl-2-hydroxyphenoxy)benzimidamide (PT131)
PT131 was obtained from 32 (200 mg, 0.58 mmol) using the general demethylation procedure, as a dark yellow solid (119 mg, 62%). HPLC: 91%. 1H NMR (400 MHz, CDCl3): δ 9.12 (bs, 1H), 8.30 (br, 2H), 7.58-7.52 (m, 1H), 7.32-7.18 (m, 2H), 6.88 (s, 1H), 6.76 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 7.8 Hz, 1H), 2.44 (t, J = 7.8 Hz, 2H), 1.56-1.44 (m, 2H), 1.36-1.22 (m, 6H), 0.86 (t, J = 6.7 Hz, 3H) ppm; HRMS-ES+: m/z [M+H]+ calcd for C19H23FN2O2: 331.1822, found: 331.1825.
2-(2-Chloro-6-fluorophenoxy)-5-hexylphenol (PT133)
Compound 26 (300 mg, 0.89 mmol) was used in the general demethylation method to give PT133 (242 mg, 84%). HPLC: 95%. 1H NMR (400 MHz, CDCl3): δ 7.28 (d, J = 8.1 Hz, 1H), 7.18-7.10 (m, 2H), 6.84 (s, 1H), 6.56 (d, J = 7.8 Hz, 1H), 6.42 (d, J = 8.0 Hz, 1H), 5.68 (s, 1H), 2.54 (t, J = 7.5 Hz, 2H), 1.62-1.54 (m, 2H), 1.36-1.24 (m, 6H), 0.89 (t, J = 6.6 Hz, 3H) ppm; HRMS-ES+: m/z [M+Na]+ calcd for C18H20ClFO2: 345.1034, found: 345.1032.
2-((4-Aminopyrimidin-5-yl)oxy)-5-hexylphenol (PT134)
Compound 43 (200 mg, 0.66 mmol) was used in the general demethylation method to give PT134 as a dark yellow oil (130 mg, 68%). HPLC: 98%. 1H NMR (400 MHz, CDCl3): δ 8.18 (s, 1H), 7.38 (s, 1H), 6.98 (d, J = 8.1 Hz, 1H), 6.82 (s, 1H), 6.72 (d, J = 7.8 Hz, 1H), 2.58 (t, J = 7.6 Hz, 2H), 1.66-1.58 (m, 2H), 1.38-1.24 (m, 6H), 0.94 (t, J = 6.7 Hz, 3H) ppm; HRMS-ES+: m/z [M+H]+ calcd for C16H21N3O2: 288.1712, found: 288.1716.
2-((2-Fluoropyridin-3-yl)oxy)-5-hexylphenol (PT161)
A stirred solution of 35 (607 mg, 2.0 mmol) in HF/pyridine (70%, 1 mL) was cooled to −78°C. After an addition of NaNO2 (158.7 mg, 2.3 mmol), the solution was kept at 0 °C for 30 min and then at 60 °C for 1 h. After the reaction was shown to be complete by TLC, the mixture was poured into cold water (10 mL) and neutralized by sat. NaHCO3. After extraction with CH2Cl2 (10 mL), the organic layer was dried over MgSO4 and dried in vacuo to give the crude compound, which was treated with the general demethylation method and purified with flash chromatography (EtOAc/hexane = 15%) to give PT161 as a light yellow solid (284 mg, 49%). 1H NMR (300MHz, CDCl3): δ 7.86 (td, J = 1.5, 4.8 Hz, 1H), 7.28 (td, J = 9.6, 1.5 Hz, 1H), 7.10 (dd, J = 4.8, 7.8 Hz, 1H), 6.91 (d, J = 2.1 Hz, 1H), 6.78 (d, J = 8.1 Hz, 1H), 6.68 (dd, J = 2.1, 8.1 Hz, 1H), 2.55 (t, J = 7.8 Hz, 2H), 1.59 (m, 2H), 1.30 (m, 6H), 0.89 (t, J = 3.3 Hz, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ 155.9, 152.7, 147.1, 141.4, 140.6, 140.3, 140.3, 140.0, 139.7, 127.5, 127.5, 122.0, 122.0, 120.7, 118.9, 117.0, 35.4, 31.7, 31.3, 28.9, 22.6, 14.1 ppm; HRMS-ES+: m/z [M+H]+ calcd for C17H20FNO2: 290.1494, found: 290.1488.
3-(4-Hexyl-2-hydroxyphenoxy)picolinonitrile (PT164)
Compound 33 was obtained as a light yellow solid with 3 (1.0 g, 4.8 mmol) and 3-bromopicolinonitrile (1.1 g, 5.8 mmol), following the same procedure that was used to prepared 34. MS (ESI+, 70 eV): m/z (%): 311.0 (100), 312.2 (23) [M+H]+. The crude compound 33 was used in the general demethylation method to give PT164. Purification with chromatography (EtOAc/hexane = 10%) gave pure PT164 as a light yellow solid (583 mg, 41%). 1H NMR (400MHz, CDCl3): δ 8.43 (d, J = 4.5 Hz, 1H), 7.47 (dd, J = 8.7, 4.5 Hz, 1H), 7.29-7.28 (m, 2H), 6.94 (d, J = 7.0 Hz, 1H), 6.72 (d, J = 9.0 Hz, 1H), 5.69 (br. s, 1H), 2.61 (t, J = 7.5 Hz, 2H), 1.642-159 (m, 2H), 1.39-1.33 (m, 6H), 0.91 (t, J = 6.2 Hz, 3H)ppm. 13C NMR (100 MHz, CDCl3): δ 156.9, 145.1, 143.7, 143.6,129.0, 128.1, 123.6, 118.8, 118.8, 114.4, 108.0, 107.7, 31.6, 29.9, 22.6, 14.1 ppm; HRMSES+: m/z [M+H]+ calcd for C18H20N2O2: 297.1603, found: 297.1607.
Crystallization and structure determination of the InhA:NAD+: PT119 complex
Crystals of the InhA:NAD+:PT119 ternary complex were obtained by the hanging drop, vapor diffusion method.[9] To co-crystallize PT119, 5 mg/mL of InhA was incubated with 2 mm NAD+ and 800 μm PT119 in 8% DMSO for 2 h at rt before mixing with an equal volume of reservoir solution containing 2.4 M sodium acetate pH 5.0, 200mM NaCl, 14% PEG 3350 and 4% DMSO. The crystals were allowed to grow for twenty days before soaking in a solution containing 2.8 M sodium acetate, 75 mm NaCl, 21% DMSO, 2 mm NAD+ and 2 mm PT119 and cryo-cooling in liquid nitrogen. Datasets were collected at beamline X12C (NSLS, BNL). The reflections were indexed, integrated and scaled using HKL2000[21] and structures solved using MolRep[22] using InhA as a model from the protein data bank. NAD+ and PT119 were built into the difference map calculated from the initial solutions. Structural refinement restraints for NAD+ and PT119 were generated using eLBOW.[23] Coordinates, ADP and TLS refinement, simulated annealing, and water picking were performed using Phenix.[24] Model building and real-space refinement were performed in Coot.[25] Data collection and refinement statistics are given in Table S2.
In vivo Efficacy
Bacterial strain
The M. tuberculosis strain used in the mouse studies was Erdman TMCC 107. Working stocks of bacteria were grown in Proskauer-Beck medium to mid-logarithmic phase growth (OD600 nm 0.3-0.5), dispensed in 1 mL aliquots, and stored frozen at -80°C. The bacteria used in all the in vivo experiments were from these stocks.
Mouse studies
In vivo efficacy against M. tuberculosis Erdman was assessed in the rapid M. tuberculosis animal model of efficacy which utilizes gamma interferon gene-disrupted (GKO) C57BL/6 mice (Jackson Laboratories, ME).[26] Treatment started 5 days post-infection with all compounds being delivered IP in a formulation of 15% ethanol, 20% propylene glycol, 40% polyethylene glycol 400, PBS. Control animals were treated with formulation alone. PT070 was given 50 mg/kg BID day1-4, 25 mg/kg BID day5-10 and PT091 was given 25 mg/kg BID day1-4, 12.5 mg/kg day5-6, 12.5 mg/kg SID and 25 mg/kg SID day 7-10. Animals were sacrificed after 10 days of treatment and the spleen was homogenized in sterile saline (0.85%) and homogenates were diluted in saline and plated on 7H11 agar supplemented with 10% OADC, 0.01% asparagine, and antibiotics (10 mg/L cycloheximide, 50 mg/L carbenicillin). Bacterial burden in the spleen was determined by enumeration of bacterial colonies after 3 weeks of incubation. The significance between control and treatment groups was determined via an unpaired, one tailed t-test (t-test1, 2) with 95% confidence intervals from mean CFU's (GraphPad Software ver. 5, San Diego CA USA, www.graphpad.com).
Steady State Kinetics and Progress Curve Assays
Kinetic assays using trans-2-dodecenoyl-Coenzyme A (DD-CoA) and wild-type InhA were performed as described previously to determine IC values and residence times.[9, 19b] 50 IC50 values were determined by varying the concentration of inhibitor in 150 mm PIPES buffer containing 250 μm NADH, 25 μm DD-CoA, and 20, 50, or 100 nm InhA. Progress curves were obtained in reaction mixtures containing InhA (5 nm), glycerol (8%, v/v), BSA (0.1 mg/ml), DMSO (2%, v/v), DD-CoA (300 μm), NADH (250 μm) and inhibitor (0-1000 μm). Inhibition constants for rapid reversible and slow-onset inhibitors were determined using methods reported previously.[9, 27] Data fitting was performed using Grafit 4.0 (Erithacus Software Ltd.).
Docking Experiments
A model for the complex formed between PT109 and InhA was obtained using the DOCK6 suite of programs.[28] The partial atomic charges of cofactor and ligand were computed based on the semiempirical AM1-BCC[29] method using AMBER10.[30] The partial atomic charges of amino acids in the protein were assigned based on the AMBER ff99SB force field.[31] The binding site was prepared as described previously.[32] Briefly, a molecular surface for InhA was computed using the program DMS.[33] The program SPHGEN in DOCK 6 was used to generate a set of spheres at the regions where inhibitor atoms could potentially interact favorably with the receptor. 39 Spheres were used to guide inhibitor placement during flexible docking. The grid file was computed with a 0.3 Å grid space using the program GRID in DOCK 6, and default parameters were then used for flexible docking.
Supplementary Material
Scheme 5.

Derivatives with various B-rings. Reagents and conditions: a) K2CO3, DMAc, 160°C, 3 h; b) Zn, HCl, EtOH/H2O, 0 °C to rt, 1 h; c) NaNO2, H2SO4, Zn, EtOH/H2O, 0 °C to reflux, 3 h; d) BBr3, CH2Cl2, −78 °C to rt, 5 h.
Scheme 6.

Derivatives with di-substituted B-rings. Reagents and conditions: a) K2CO3, DMAc, 160°C, 4 h; b) BBr3,CH2Cl2, −78 °C to rt, 3 h; c) K3PO4,Cu(bipy)2BF4, 120°C, DMF, 48 h, 10%; d) Zn,HCl, 0 °C to rt, 1 h, 41%; e) Li(TMS)2, THF, 0 °C to rt, 3 h, 46%.
Scheme 7.
Derivatives with 2’-N pyridyl B-rings. Reagents and conditions: a) Cs2CO3, MeCN, 80 °C, 2 h; b) BBr3,DCM, -78 °C to rt, 2 h; c) Fe, AcOH/H2O, 100 °C, 30 min, 77%; d) 1. HF/pyridine, 0-60 °C, 1 h; 2. BBr3, DCM, −78 °C to rt, 2 h; 49%.
Acknowledgement
This work was supported by National Institutes of Health Grant AI070383 and GM102864 to P.J.T. Financial support for NSLS beamlines ×29 and ×12c comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources (P41RR012408) and the National Institute of General Medical Sciences (P41GM103473) of the National Institutes of Health.
References and Notes
- 1.a Donoghue HD, Spigelman M, Greenblatt CL, Lev-Maor G, Kahila Bar-Gal G, Matheson C, Vernon K, Nerlich AG, Zink AR. Lancet Infect Dis. 2004;4(9):584–592. doi: 10.1016/S1473-3099(04)01133-8. [DOI] [PubMed] [Google Scholar]; b Wright A, Bai G, Barrera L, Boulahbal F, Martin-Casabona N, Gilpin C, Drobniewsji F, Havelkova M, Lepe R, Lumb R, Metchock B, Portaels F, Rodrigues M, Rusch-Gerdes S, Van Deun A, Vincent V, Leimane V, Riekstina V, Skenders G, Holtz T, Pratt R, Laserson K, Wells C, Cegielski P, Shah NS. JAMA. 2006;295(20):2349–2351. [Google Scholar]
- 2.WHO WHO Report. 2009 http://www.who.int/tb/publications/global_report/2009/pdf/full_report.pdf.
- 3.a Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li H-J, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. ACS Chem. Biol. 2006;1(1):43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]; b Vilcheze C, Baughn AD, Tufariello J, Leung LW, Kuo M, Basler CF, Alland D, Sacchettini JC, Freundlich JS, Jacobs WR., Jr. Antimicrob Agents Chemother. 2011;55(8):3889–3898. doi: 10.1128/AAC.00266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Freundlich JS, Wang F, Vilcheze C, Gulten G, Langley R, Schiehser GA, Jacobus DP, Jacobs WR, Jr., Sacchettini JC. ChemMedChem. 2009;4(2):241–248. doi: 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Dessen A, Quemard A, Blanchard JS, J. r. Jacobs WR, Sacchettini JC. Science. 1995;267(5204):1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]; b Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR., Jr. Science. 1994;263(5144):227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 5.a Lei B, Wei CJ, Tu SC. J. Biol. Chem. 2000;275(4):2520–2526. doi: 10.1074/jbc.275.4.2520. [DOI] [PubMed] [Google Scholar]; b Timmins GS, Deretic V. Mol Microbiol. 2006;62(5):1220–1227. doi: 10.1111/j.1365-2958.2006.05467.x. [DOI] [PubMed] [Google Scholar]
- 6.Boyne ME, Sullivan TJ, amEnde CW, Lu H, Gruppo V, Heaslip D, Amin AG, Chatterjee D, Lenaerts A, Tonge PJ, Slayden RA. Antimicrob Agents Chemother. 2007;51(10):3562–3567. doi: 10.1128/AAC.00383-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu H, England K, am Ende C, Truglio JJ, Luckner S, Reddy BG, Marlenee NL, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. ACS Chem. Biol. 2009;4(3):221–231. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Copeland RA, Pompliano DL, Meek TD. Nat. Rev. Drug Discov. 2006;5(9):730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]; b Swinney DC. Curr Top Med Chem. 2006;6(5):461–478. doi: 10.2174/156802606776743093. [DOI] [PubMed] [Google Scholar]; c Swinney DC. Lett Drug Des Discov. 2006;3(8):569–574. doi: 10.2174/157018006778194655. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tummino PJ, Copeland RA. Biochemistry. 2008;47(20):5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]; e Copeland RA. Expert Opin. Drug Discov. 2010;5(4):1–6. doi: 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]; f Lu H, Tonge PJ. Curr. Opin. Chem. Biol. 2010;14(4):467–474. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luckner SR, Liu N, Am Ende CW, Tonge PJ, Kisker C. J. Biol. Chem. 2010;285:14330–14337. doi: 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.am Ende CW, Knudson SE, Liu N, Childs J, Sullivan TJ, Boyne M, Xu H, Gegina Y, Knudson DL, Johnson F, Peloquin CA, Slayden RA, Tonge PJ. Bioorg. Med. Chem. Lett. 2008;18(10):3029–3033. doi: 10.1016/j.bmcl.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a Wolter M, Nordmann G, Job GE, Buchwald SL. Org Lett. 2002;4(6):973–976. doi: 10.1021/ol025548k. [DOI] [PubMed] [Google Scholar]; b Zhang H, Ma DW, Cao WG. Synlett. 2007;(2):243–246. [Google Scholar]; c Altman RA, Shafir A, Choi A, Lichtor PA, Buchwald SL. J Org Chem. 2008;73(1):284–286. doi: 10.1021/jo702024p. [DOI] [PubMed] [Google Scholar]
- 12.a Niu JJ, Zhou H, Li ZG, Xu JW, Hu SJ. J. Org. Chem. 2008;73(19):7814–7817. doi: 10.1021/jo801002c. [DOI] [PubMed] [Google Scholar]; b Lang H, Zoufala P, Klaib S, del Villar A, Rheinwald G. J Organomet Chem. 2007;692(19):4168–4176. [Google Scholar]
- 13.Marsh G, Stenutz R, Bergman A. Eur. J. Org. Chem. 2003;(14):2566–2576. [Google Scholar]
- 14.a Julemont F, de Leval X, Michaux C, Damas J, Charlier C, Durant F, Pirotte B, Dogne JM. J. Med. Chem. 2002;45(23):5182–5185. doi: 10.1021/jm020920n. [DOI] [PubMed] [Google Scholar]; b Galanakis D, Davis CA, Ganellin CR, Dunn PM. J. Med. Chem. 1996;39(2):359–370. doi: 10.1021/jm950520i. [DOI] [PubMed] [Google Scholar]
- 15.a Neumeyer JL, Baindur N, Yuan J, Booth G, Seeman P, Niznik HB. J. Med. Chem. 1990;33(2):521–526. doi: 10.1021/jm00164a009. [DOI] [PubMed] [Google Scholar]; b Bigelow LAJ, J. R., Sandborn LT. Organic Syntheses Coll. Vol. 1941;1:133. [Google Scholar]; c Gilman H, Van Ess MW, Hayes DM. J. Am. Chem. Soc. 1939;61:643–648. [Google Scholar]
- 16.Lu H, Tonge PJ. Acc. Chem. Res. 2008;41(1):11–20. doi: 10.1021/ar700156e. [DOI] [PubMed] [Google Scholar]
- 17.Li H-J, Lai C-T, Pan P, Yu W, Liu N, Garcia-Diaz M, Bommineni GR, Simmerling C, Tonge PJ. ACS Chem Biol. 2014 doi: 10.1021/cb400896g. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a Rozwarski DA, Grant GA, Barton DHR, Jacobs WR, Jr., Sacchettini JC. Science. 1998;279(5347):98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]; b Stewart MJ, Parikh S, Xiao GP, Tonge PJ, Kisker C. J. Mol. Biol. 1999;290(4):859–865. doi: 10.1006/jmbi.1999.2907. [DOI] [PubMed] [Google Scholar]
- 19.a Rawat R, Whitty A, Tonge PJ. Proc Natl Acad Sci U S A. 2003;100(24):13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Parikh SL, Xiao G, Tonge PJ. Biochemistry. 2000;39(26):7645–7650. doi: 10.1021/bi0008940. [DOI] [PubMed] [Google Scholar]; c Rozwarski DA, Vilcheze C, Sugantino M, Bittman R, Sacchettini JC. J. Biol. Chem. 1999;274:15582–15589. doi: 10.1074/jbc.274.22.15582. [DOI] [PubMed] [Google Scholar]
- 20.Swinney DC. Curr Opin Drug Discov Devel. 2009;12(1):31–39. [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Meth. Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 22.Vagin A, Teplyakov A. J. Appl. Crystallogra. 1997;30:1022–1025. [Google Scholar]
- 23.Moriarty NW, Grosse-Kunstleve RW, Adams PD. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Acta Crystallogr. D Biol. Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.a Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. J Exp Med. 1993;178(6):2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lenaerts AJ, Gruppo V, Brooks JV, Orme IM. Antimicrob. Agents Chemother. 2003;47(2):783–785. doi: 10.1128/AAC.47.2.783-785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sivaraman S, Sullivan TJ, Johnson F, Novichenok P, Cui G, Simmerling C, Tonge PJ. J. Med. Chem. 2004;47(3):509–518. doi: 10.1021/jm030182i. [DOI] [PubMed] [Google Scholar]
- 28.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. RNA. 2009;15(6):1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a Jakalian A, Bush BL, Jack DB, Bayly CI. J Comput Chem. 2000;21(2):132–146. [Google Scholar]; b Jakalian A, Jack DB, Bayly CI. J Comput Chem. 2002;23(16):1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 30.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukherjee S, Balius TE, Rizzo RC. J Chem Inf Model. 2010;50(11):1986–2000. doi: 10.1021/ci1001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DMS . UCSF Computer Graphics Laboratory. San Francisco, CA.: http://www.cgl.ucsf.edu/Overview/software.html. [Google Scholar]
- 34.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.