

Abstract
C-function MADS-box transcription factors belong to the AGAMOUS (AG) lineage and specify both stamen and carpel identity and floral meristem determinacy. In core eudicots, the AG lineage is further divided into two branches, the euAG and PLE lineages. Functional analyses across flowering plants strongly support the idea that duplicated AG lineage genes have different degrees of subfunctionalization of the C-function. The legume Medicago truncatula contains three C-lineage genes in its genome: two euAG genes (MtAGa and MtAGb) and one PLENA-like gene (MtSHP). This species is therefore a good experimental system to study the effects of gene duplication within the AG subfamily. We have studied the respective functions of each euAG genes in M. truncatula employing expression analyses and reverse genetic approaches. Our results show that the M. truncatula euAG- and PLENA-like genes are an example of subfunctionalization as a result of a change in expression pattern. MtAGa and MtAGb are the only genes showing a full C-function activity, concomitant with their ancestral expression profile, early in the floral meristem, and in the third and fourth floral whorls during floral development. In contrast, MtSHP expression appears late during floral development suggesting it does not contribute significantly to the C-function. Furthermore, the redundant MtAGa and MtAGb paralogs have been retained which provides the overall dosage required to specify the C-function in M. truncatula.
Introduction
Genetic regulation of flower development has been subject of study over the last decades, particularly in the model species Arabidopsis thaliana and Antirrhinum majus. These studies provided a general understanding of floral organ development in higher plants and led to the proposal of the ABCDE model, which postulates that floral organ identity in each whorl is defined by five functions named A, B, C, D and E (for review [1], [2], [3], [4]). In particular, C-function is required to promote stamen and carpel identity, to establish the determinate nature of the floral meristem [5] and also to repress A-function in the third and fourth whorls [6]. Cloning of ABCDE organ identity genes in Arabidopsis showed that most of them encode MADS-box transcription factors. Studies of MADS-box genes in higher eudicotyledoneous flowering plants show that they are key regulators of flower development.
In Arabidopsis and Antirrhinum, the C-function is essentially represented by a single gene respectively, AGAMOUS (AG) [5], [7] and PLENA (PLE) [8]. Additional C-function genes have been identified: SHATTERPROOF1 (SHP1) and SHATTERPROOF2 (SHP2) [9], [10] genes in Arabidopsis and FARINELLI (FAR) [11] in Antirrhinum.
Phylogenetic studies using a large data set of AG-like sequences show that AG and PLE actually represent paralogous lineages derived from a duplication in a common ancestor early in the history of the core eudicots. This duplication gave rise to the euAG lineage which includes AG and FAR, and the PLENA lineage (PLE), where SHP1/SHP2 and PLENA are placed [12], [13], [14], [15]. An even more ancient duplication occurred before the radiation of extant angiosperms, producing the C lineage (AG lineage) and the ovule-specific D lineage (AGL11 lineage) [12], [15].
AG, FAR and PLE all display very similar expression patterns during the developing male and female reproductive organs [5], [8], [11]. Mutations of AG and PLENA in their respective species produce identical phenotypes characterized by developing flowers with petals in whorl 3 instead of stamens, and sepals in whorl 4 instead of carpels. In addition, the floral meristem is not determinate [7], [8]. However, far mutation only affects male reproductive organs, causing partial male sterility [11]. Meanwhile, SHP genes are only expressed in the ovules, in the developing pistil and show almost identical expression patterns in developing Arabidopsis fruit [16], [17], [18]. They function redundantly in style and stigma development, notably in the fusion of the carpel [19], and in seedpod shattering [10]. SHP and FAR genes do not contribute significantly to the C-function although FAR is expressed at early stages of flower development.
These studies showed a random evolutionary trajectory for gene functions after a duplication event. AG and PLE, different members of a duplicated gene pair, retained the primary homeotic functions in different lineages. Their respective orthologs have taken completely new roles in fruit dehiscence, as the case of SHP1/SHP2 genes, or contribute redundantly in male reproductive development, as the case of FAR. Representatives of both euAG and PLE lineages have been identified in different core eudicot species and several functional analyses are available for the paralogs from petunia, tomato, and Nicotiana benthamiana [20], [21], [22], [23].
In addition to the ancient duplication events observed in the AGAMOUS subfamily, more recent duplication events appear to have occurred in the euAG lineage [12], [15], [24]. There have been reported two members of the euAG lineage in some species, as for example Populus trichocarpa [25], Cucumis sativus [26], Gerbera hybrida [27], and in the legume species Lotus japonicus [28], Pisum sativum and Medicago truncatula (this study). However, in most cases, functional analyses are lacking. Both phylogenetic and functional data of gene lineages are very important to understand the evolution of gene families [12], [29].
Medicago truncatula (Mt) has three C-lineage genes: two euAG genes (MtAGa and MtAGb) and one PLENA-like gene (MtSHP; [30]). To gain insight into the specific contribution of the euAG and PLE-like paralogous genes in the control of the C-function, we compared the expression patterns of the three C-lineage genes during flower development. We have also characterized MtAGa and MtAGb loss-of-function mutants and plants where both genes have been simultaneously silenced. The particular capacity of these two genes to promote reproductive organ identity has been tested by ectopically expressing them in Arabidopsis. Our results indicate that the members of euAG and PLE lineages in M. truncatula are subfunctionalized, where the C-function is only promoted by the euAG paralogs. They largely overlap in function but the overall dosage of both gene products is critical to promote complete stamen and carpel identity and floral meristem determinacy in M. truncatula.
Material and Methods
Plant material and growth conditions
Medicago truncatula cv. Jemalong lines A17, SA1335 and R108, and Arabidopsis thaliana cv. Columbia plants were used in this study. Plants were grown in the greenhouse, at 22°C (day) and 18°C (night) with a 16 h light/8 h dark photoperiod, in soil:sand (3∶1) irrigated with Hoagland No.1 solution supplemented with oligoelements [31].
The mtaga mutant allele (previously matag-2) was isolated in a previous screening [32] and homozygous plants were used in this study.
Identification of Tnt1 insertion sites in MtAGb and co-segregation test
The M. truncatula population used for the screening of mutants was described in detail [33], [34], [35], [36] (http://bioinfo4.noble.org/mutant/). The mtagb allele was identified by PCR screening of a segregating population of approximately 10,000 independent lines, using primers annealing to the MtAGb sequence (AGb-F, Table S2) in combination with primers annealing to the LTR borders of the Tnt1 retroelement (Tnt1-F; Table S2 and Figure S3). We identified a line (NF4908) with an insertion of the retroelement located in the first intron at 277 bp of the starting codon (Figure 1A). The R1 plants were genotyped by PCR using the Tnt1-F primer in combination with the gene-specific primers MtAGb-F and MtAGb-Rgenomic (Table S2 and Figure S3B). Approximately 70% of the plants showed a mutant phenotype and co-segregated with the Tnt1 insertion.
Figure 1. Two euAGAMOUS genes in Medicago truncatula.
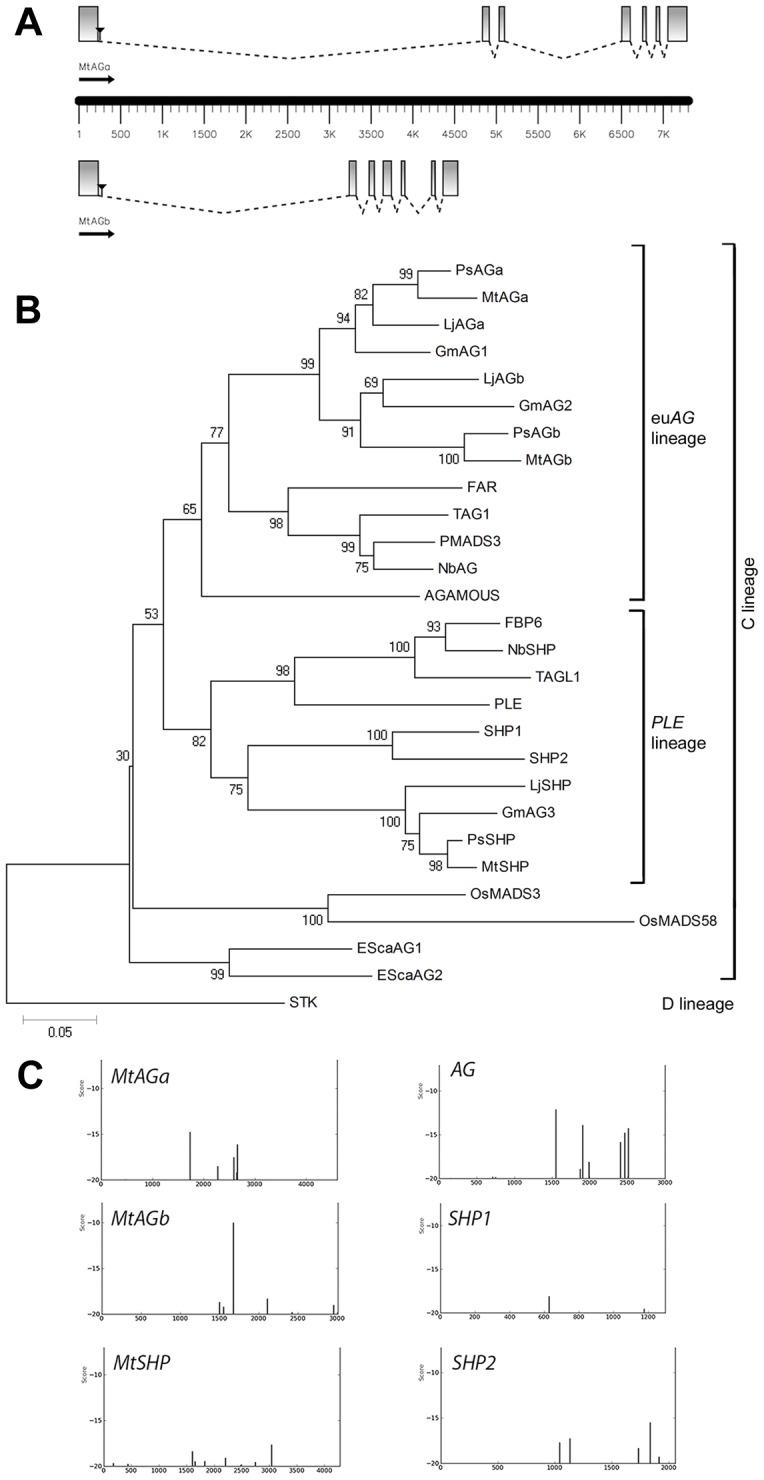
(A) Gene structure of MtAGa and MtAGb. Coding sequences are represented as boxes and introns as dotted lines. Black triangles localize the position of the Tnt1 insertions present in the mutant lines mtaga (NF13380) and mtagb (NF4908) used in this study. (B) Neighbor-Joining Tree of euAG and PLENA homologs from a selection of diverse species. The numbers next to the nodes refer to bootstrap values from 10000 pseudo-replicates. (C) Distribution of putative LFY binding sites in the first intron of MtAGa, MtAGb and MtSHP genes as identified with the use of a position-specific scoring matrix using a cutoff value of −20.
Isolation and sequence analysis
MtAGa and MtAGb cDNAs were isolated from a library of M. truncatula A17 inflorescence apices [37], using the MADS-box fragment of the M. truncatula PISTILLATA gene as a probe. Sequence alignments and similarity comparisons of the inferred proteins were performed using Align and ClustalW tools [38]. The deduced amino acid sequences were aligned using ClustalW and further refined by hand. Genomic sequences search was performed using BLAST [39].
The second intron sequences were obtained by PCR using genomic DNA and the primers MtAGa-intron-F and MtAGa-intron-R for MtAGa and MtSHP-intron- F and MtSHP-intron-R for MtSHP (Table S2). To predict the probability of the presence of LFY binding sites in the intron sequences, we used the Morpheus webpage facility (http://biodev.cea.fr/morpheus/Default.aspx) with the LFY matrix. The score tool permits localization of the best transcription factor binding sites in DNA fragments.
Phylogenetic tree
The phylogenetic tree was inferred by Neighbor-Joining using Poisson-Corrected amino acid distances. Reliability of internal nodes was assessed using bootstrap with 10000 pseudo-replicates. Tree inference was conducted using MEGA version 4 [40]. The data set comprised 28 previously reported C- and D-class genes obtained from GenBank and the two new sequences that we isolated (MtAGa and MtAGb). The tree was rooted using the Arabidopsis D-class gene SEEDSTICK (STK) sequence. All sequences used in this analysis, with their respective species and accession numbers, are included in Table S1.
Southern blot hybridization
Plant genomic DNA was extracted from leaves as described by [41]. Ten micrograms of DNA were digested with restriction enzymes, separated on 0.7% Tris-borate EDTA 1X agarose gels overnight at 1V/cm and transferred to a nylon membrane. Southern blot hybridization was performed by standard methods at 65°C. A 241 bp fragment of MtAGa cDNA (positions 572-813) and a 215 bp fragment of the MtAGb cDNA (positions 558-773) were amplified with the specific primers MtAGadir, MtAGarev, MtAGbdir and MtAGbrev1 (Table S2) and used as probes.
Expression analyses
Total RNA was isolated from frozen plant material using the RNeasy Plant mini Kit (Qiagen, Germany) according to the manufacturer's instructions.
For Northern blot analysis total RNA (15 µg) from frozen leaves, roots, stems, flowers and pollinated ovaries (young fruits) was used. RNA electrophoresis was carried out in formaldehyde-agarose gels, transferred to Hybond N+ membranes (Amersham Biosciences, USA), and hybridized with 32P-labeled probes under standard conditions. The probes were generated from the same gene fragments used for Southern blot analysis.
For Real Time RT-PCR analysis total RNA was treated with rDNaseI of the DNase Treatment and Removal Kit (Ambion, Life Technologies, USA). For first-strand synthesis, total RNA (1 µg) was reverse-transcribed in a 20 µl reaction mixture using the PrimerScript 1st strand cDNA Synthesis Kit (Takara, Japan). One microliter of RT reaction was used for a Real Time RT-PCR analysis with 300 nM of each primer mixed with the Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's instructions. The reaction was carried out into 96 well-optical reaction plates using an ABI PRISM 7500 Sequence Detection System and appropriate software (Applied Biosystems). The relative levels were determined by the 2 –ΔΔCt Method [42]. To normalize the variance among samples, Secret Agent (O-linked N-acetyl glucosamine transferase: TC77416; [43]) was used as an endogenous control. All reactions were performed by triplicate using a biological replicate for each sample. Primers were designed using PRIMER EXPRESS software (Applied Biosystems, USA) using default parameters and are listed in Table S2.
RNA in situ hybridization was performed on 8 µM paraffin sections of M. truncatula inflorescences as described by [16], using digoxigenin-labelled probes. The RNA sense and antisense probes were generated from the same gene fragments used for Southern blot analysis. Both fragments were cloned into the pGEM T-easy vector (Promega, USA) and the probes were synthesized using SP6 or T7 RNA polymerases.
Virus Induced Gene Silencing in Medicago truncatula
pCAPE1 and pCAPE2 derivatives were used as vectors for gene silencing [44]. Two DNA fragments from the 3′ region of the MtAGa (310 bp) and MtAGb (338 bp) genes were obtained by PCR using primers (MtAGaVIGSdir, MtAGbVIGSdir, MtAGaVIGS2rev, MtAGbVIGS2rev) that added restriction sites to both ends of the fragments (Table S2). The amplicons were cloned into pGEM T-easy (Promega, USA), digested using the appropriated restriction enzymes and cloned into a similarly digested pCAPE2 vector [44]. The resulting plasmid (pCAPE2-MtAGab) was confirmed by sequencing before being introduced into Agrobacterium tumefaciens strain C58/pMP90. Agrobacterium inoculation of M. truncatula leaves was performed as described by [44].
Generation of transgenic RNAi plants
Transformation of M. truncatula R108 was performed as described previously [33]. The 35S::RNAi-MtAG construct was performed using a 215 bp fragment from MtAGb (positions 557–772 from the ATG codon), amplified using primers MtAGb-RNAiD and MtAGb-RNAiR (Table S2) that incorporate two restriction sites that are used for cloning into the pHANNIBAL vector [45].
Arabidopsis transformation
MtAGa and MtAGb cDNA fragments were amplified using AGaSBXdir, AGaSBXrev, AGbSBXdir and AGbSBXrev primers (Table S2) and cloned into the pBINJIT60 vector [46], a pBIN19 derivative (Clontech, Palo Alto, CA, USA), which placed gene transcription under the control of a tandem repeat of the CaMV 35S promoter. Arabidopsis plants were transformed by floral dipping according to standard procedures [47] and selected on kanamycin.
Photography, microscopy and cryo-SEM
Light photographs were made with a stereomicroscope Leica MZ16F attached to a DFC300 FX camera (Leica Microsystems, Germany).
After in situ hybridizations, sections were observed and photographed with a Nikon Eclipse E-600 microscope equipped with a digital camera.
For cryo-SEM, samples were frozen in slush nitrogen and attached to the specimen holder of a CryoTrans 1500 Cryo-Preparation System (Oxford Instruments, UK) interfaced with a JEOL JSM-5410 scanning electron microscope. The samples were then transferred from cryostage to the microscope sample stage, where the condensed surface water was sublimed by controlled warming to -85°C. Afterwards, the sample was transferred again to the cryostage in order to gold coat it by sputtering. Finally the sample was put back on the microscope sample stage to be viewed at an accelerating voltage of 15 KeV.
Results
Identification of two euAGAMOUS genes in Medicago truncatula
To identify MADS-box genes involved in flower development, we screened a cDNA library of M. truncatula floral apices using a set of MADS-box fragments from different species as a probe [48], [49]. Among the isolated clones, three corresponded to full-length sequences that present significant similarity to genes of the AGAMOUS subfamily. One of these clones presents high sequence similarity with the Arabidopsis SHP1 and SHP2 genes [17], [18], [50] and with the Antirrhinum PLENA gene (PLE; [8]) and corresponds to the MtSHP gene [30]. The other two clones isolated present high sequence similarity with the C-lineage genes AGAMOUS of Arabidopsis [5] and FARINELLI of Antirrhinum [11] and have been named MtAGa and MtAGb. The MtAGa clone is 1208 bp long, with an ORF of 780 bp and a deduced protein of 260 amino acids. The MtAGb clone is 1099 bp long, with an ORF of 732 bp and the deduced protein has 244 amino acids. MtAGa shows 81% amino acid identity with MtAGb, being more similar in the N-terminal domains (98% in the MADS domain, 76% in the I region and 80% in the K domain) than in the C-terminal region (68% of identity). MtAGa and MtAGb show respectively 71 and 68% amino acid identity with the euAG clade protein FAR and either one 67% amino acid identity with AG (Figure S1).
The MtAGb genomic sequence was found in two BAC clones: mth2-30e7 (GenBank AC137837.4) and mth2-76i7 (GenBank AC153460.24). This gene is organized in seven exons and six introns (GenBank KJ470634; Figure 1A). The genomic sequence of the MtAGa gene was not available in the databases and was obtained in this study (GenBank KJ470633). MtAGa gene is also organized in seven exons and six introns (Figure 1A). Both genes are present as single copy in the genome as confirmed by Southern blot analysis (Figure S2A). Genomic sequences from MtAGa and MtAGb genes were used as BLASTN queries, and displayed using the Chromosome Visualization Tool (CViT, http://www.medicagohapmap.org/tools/blastform). We found that MtAGb gene is located on chromosome 8 (Figure S2B). However, no location was obtained for the MtAGa gene.
Phylogenetic analyses showed that MtAGa and MtAGb are relatively recent paralogs within the euAG subclade (Figure 1B). Other model legumes such as Pisum, Lotus and Glycine used in this analyses, also displayed two paralogs in this subclade. In most of the studied plants, the euAG subclade is represented by one single gene, such as AG for Arabidopsis, FAR for Anthirrinum or TAG1 for tomato.
Genes from the euAG- and PLE-subclades present a well conserved gene structure and characteristic large introns essential for their correct expression pattern [51], [52]. In M. truncatula, both MtAG genes show a large first intron (4607 bp in MtAGa and 3007 bp in MtAGb). We searched in these sequences for the presence of putative LFY binding sites that are characteristic of C-function genes. We included in this study the MtSHP gene and then we have sequenced its first intron of 4281 bp (GenBank KJ470635). We used a bioinformatics tool available at the Morpheus webpage facility (http://biodev.cea.fr/morpheus/Default.aspx) which examines the distribution of individual LFY binding sites. The presence or absence of LFY binding sites identified by this approach appears to be helpful in predicting to what extent a C-clade gene acts as a true C-function gene [53]. Our results show different binding site landscapes for the three genes analysed (Figure 1C). In the case of the MtAGb first intron, there is a main single binding site of very high score similar to the profile found for the PLE gene [53]. This site corresponds to two fully intact CCAAT-boxes. In the first intron of MtAGa, lower affinity sites are present but the binding seems to be compensated through the action of several nearby sites as reported for the sites present in the second intron of AG (Figure 1C). The prediction of binding sites for LFY in the first intron of MtSHP gave only low score values (Figure 1C) indicating a low probability to be regulated by LFY at early stages of floral development.
Globally, these data suggest that the duplicated euAG genes MtAGa and MtAGb might contribute to C-function specification in M. truncatula and play a role during early stages of flower development.
Expression patterns comparison of three M. truncatula C-lineage genes during floral development
The expression patterns of MtAGa, MtAGb and MtSHP were analysed by Northern blot in different plant tissues and the three genes are exclusively expressed in floral and young fruit tissues (Figure 2). We performed detailed in situ hybridization experiments to show the distribution of MtAGa, MtAGb and MtSHP mRNAs during floral development (Figure 3). Both MtAGa and MtAGb transcripts began to accumulate at stage 2 of flower development. At this stage, MtAGb accumulates in the centre of the floral primordia while MtAGa signal was detected throughout the floral meristem (Figure 3F and 3A). At stage 4, MtAGb transcript was located in the region of the common primordia that will give rise to the stamens and also in the central part of the floral apex where the carpel is developing (Figure 3G). However, MtAGa expression is still observed on the whole floral meristem, including petal and sepal primordia (Figure 3B). From stage 5, expression of both paralogs was distributed uniformly in whorls 3 and 4 (Figure 3C-E and 3H-J), although hybridization signal for MtAGb was stronger on the abaxial region of the carpel. In later stages, expression of both transcripts was observed in the developing ovules, in the distal region of the carpel and on the filament of the anthers (Figure 3E and 3J). In contrast, MtSHP mRNA began to accumulate late on flower development and can be detected at stage 6 in the inner cells of the developing carpel (Figure 3M). Since late stage 7, MtSHP expression is exclusively detected in the ovules (Figure 3N).
Figure 2. Expression patterns of MtAGa, MtAGb and MtSHP genes in various plant tissues of M. truncatula.

Northern blot analyses were performed using total RNA prepared from leaves (L), flowers (Fl), young fruits (Fr), stems (S) and roots (R).
Figure 3. Expression pattern of MtAGa, MtAGb and MtSHP genes during early floral development.

In situ localization of MtAGa (A-E), MtAGb (F-J) and MtSHP (K-N) transcripts in M. truncatula wild-type flower buds. Developmental stages were defined according to [74]. MtAGa transcripts are localized in the whole floral meristem at stages 2 (A) and 4 (B). At stage 5 (C) MtAGa mRNA is detected in stamen and carpel primordia. At stage 7 (D-E) expression locates in stamens, carpel and the developing ovules. MtAGb transcripts are localized in the center of the floral meristem at stage 2 (F). At stage 4 (G) expression locates in the carpel primordia and the half of the common primordia that will give rise to the stamens. At stage 5 (H) MtAGb mRNA is detected in stamen and carpel primordia. At stage 7 (I-J) MtAGb mRNA is detected in stamens, carpel and developing ovules. MtSHP transcripts are not detected at stages 2 (K) and 5 (L). At stage 6 MtSHP transcripts are localized in the inner cells of the developing carpel (M). At stage 7 (N) expression can be detected in the developing ovules. F: floral meristem; S: sepal; C: carpel; CP: common primordia; St: stamen; P: petal; Fi: filament; A: anther; Ov: ovule.
We conclude that the expression of the two M. truncatula euAG genes remained largely restricted to the developing male and female organs from early stages and throughout flower development. MtAGa and MtAGb showed nearly identical spatial expression patterns from stage 4 to late developmental stages. Differences were mainly observed at early stages, where MtAGa expression appeared to be wider spread in the floral meristem than MtAGb. Essentially, the duplicated MtAGa and MtAGb genes showed similar spatial and temporal expression patterns to those described for C-function genes from the euAG subclade such as AG and PLE. In contrast, MtSHP was only expressed in the developing carpel and in the ovules, similar to the pattern described for Arabidopsis SHP genes that do not significantly contribute to C-function.
MtAGa and MtAGb loss-of-function analyses
To investigate the specific contribution of MtAGa and MtAGb genes in the M. truncatula floral development, we looked for retrotransposon insertion mutants [32]. These mutants were available for both genes, each containing a single Tnt1 element inserted in the N-terminal part of the gene (Figure 1A). mtagb Tnt1 insertion was identified as described in the experimental procedures and it was located in the first intron at 277 bp from the start codon (Figure 1A). mtaga (previously mtag-2), was isolated as a case study to demonstrate the utility of the reverse genetic platform in the model legume Medicago truncatula [32]. Using quantitative RT-PCR we have confirmed that MtAGa or MtAGb transcripts were nearly undetectable in the corresponding homozygous mutant plants without affecting the expression levels of the corresponding MtAG paralog (Figure S4). mtaga and mtagb mutants exhibit very similar floral phenotypes: flowers are wild-type in appearance, showing only mild developmental defects on the third and fourth-whorl organs (Figure 4). Incomplete fusion of the staminal tube was observed and occasionally, some petaloid prolongation in the anther tip (Figure 4B and 4C). Whorl 4 is frequently composed by multiple carpels (2-3) or by modified carpels with stigmatic protuberances and exposed ovules (Figure 4B and 4C). Flowers were generally sterile, although occasionally a few seeds were produced.
Figure 4. Floral phenotypes of M. truncatula mtagb and mtaga mutants and MtAGab-VIGS silenced plants.

(A) Dissected wild-type M. truncatula flower showing the four floral whorls (W1 to W4). Floral phenotypes are similar in mtaga. (B) and mtagb (C) mutants. Mutant flowers (left) were opened to show the inner whorls. In W3 the staminal tubes are unfused and occasionally petaloid structures appear (arrow) replacing the anthers. Carpels in W4 present stigmatic protuberances (arrow) or multiple unfused carpels and exposed ovules (boxed). (D) MtAGab-VIGS flower with severe homeotic transformation of stamens into petals and carpels into sepaloid structures (arrows). Bars indicate 1 mm.
To investigate the effect of the combined loss-of-function of MtAGa and MtAGb, we performed virus induced gene silencing (VIGS) experiments. We used a VIGS vector based on PEBV (Pea early browning virus) initially developed to induce gene silencing in Pisum [44]. The silencing efficiency of this vector is lower in M. truncatula cultivars than in P. sativum [54]. We generated the pCAPE2-MtAGab construct to simultaneously silence both genes (see Materials and Methods). Twenty one plants were inoculated with the pCAPE2-MtAGab construct and we analysed the phenotype of 150 flowers. 10% of the MtAGab-VIGS flowers showed homeotic transformations in whorls 3 or 4. Only 1% of the MtAGab-VIGS flowers showed a near complete loss-of-C-function phenotype (Figure 4 D). In whorl 3 some anthers are completely converted into petal-like organs, and in the fourth whorl, sepal-like structures arise instead of carpels (Figure 4 D). These flowers also showed indeterminacy of the floral meristem revealed by the presence of multiple petaloid or sepaloid concentric structures in the centre (Figure 4D, arrows). This phenotype suggests that both MtAG genes were strongly down-regulated but, because of floral tissue limitation, we could not determine the expression levels of the targeted genes.
In parallel, we also developed transgenic M. truncatula lines in which MtAG genes were down regulated by RNA interference (RNAi) and three independent lines were obtained. Only one of these lines (line 5.7) showed floral phenotypes similar to those of single mtag mutants (Figure S5). These flowers showed clear morphological aberrations consisting of mild homeotic transformation of stamens to petals and multiple carpels with exposed ovules. We analyzed the expression of both MtAGa and MtAGb genes in the three RNAi lines. These lines showed a different degree of silencing for each targeted gene (Figure S5J). In silenced line 1.3, we found MtAGb levels considerably reduced (∼15% of wild-type level), while MtAGa levels are somewhat decreased (∼90% of wild-type level). Strong silencing of one paralogous gene did not produce homeotic floral defects. However, floral defects in line 5.7 correlate with simultaneous downregulation of both genes (∼40% of wild-type level) (Figure S5J).
MtAGa and MtAGb gain-of-function analyses
Ectopic expression of AG results in the homeotic conversion of sepals and petals into carpels and stamens, respectively [55], [56]. Other reported phenotypes include early flowering and curling of leaves. To investigate whether MtAGa and MtAGb proteins differ in their ability to induce reproductive organ fate we overexpressed these two genes in Arabidopsis. The coding sequence of these genes was located under the control of the CaMV 35S constitutive promoter. We analysed 75 independent primary transgenic (T1) lines for every construct (35S::MtAGa and 35S::MtAGb plants) and found plants with different floral phenotypes that we classified as mild, medium and strong phenotype (Figure 5B-D). Transgenic plants with mild phenotype showed wax accumulations on the surface of the sepals (Figure 5I) that are characteristic of the carpel (Figure 5H). The petals are narrow and asymmetric (Figure 5B and 5J) with partial homeotic conversions to stamens (Figure 5K). Flowers with medium phenotypes showed curved sepals (Figure 5C) with abundant accumulation of wax (Figure 5L). T1 plants with medium and strong phenotypes showed near complete homeotic transformations of sepals to stamens (Figure 5M and 5N). Finally, flowers with strong phenotype showed thickened and curved sepals (Figure 5D) that contain carpelloid structures with ectopic ovules and stigmatic papillae (Figure 5P). Although these three phenotypical classes were observed for both constructs, we notice a higher proportion of plants with medium/strong phenotypes (80.3%) in the MtAGb overexpressing lines (Figure 5Q). In contrast, 59.5% of MtAGa overexpressing lines showed mild homeotic transformations.
Figure 5. Overexpression of MtAGa and MtAGb in Arabidopsis thaliana: floral phenotypes.

Wild-type Arabidopsis flower (A). Floral phenotypes observed in Arabidopsis plants overexpressing MtAGa or MtAGb genes have been classified into mild (B), medium (C) and strong (D). Scanning electron micrographs showing the characteristic cellular types of wild-type sepals (E), petals (F), stamens (G) and carpels (H). Scanning electron micrographs of sepals from overexpression lines with mild phenotype (I) showing wax accumulation (arrow). Narrow petals from overexpressing lines (J) showing staminoid cellular types (K). Wax accumulation in sepals (L) of plants with medium and strong phenotypes. Near complete homeotic conversion of petal into stamen (M) showing characteristic cellular types from anther (N) and filament (O). Flower with strong phenotype showing ectopic ovules and stigmatic tissue (P) in the first whorl. Relative proportion of phenotypes observed in plants overexpressing MtAGa or MtAGb genes (Q). Bars indicate: 1mm in A, B, C, and D.
In summary, constitutive expression of MtAGa and MtAGb in Arabidopsis are able to cause equivalent homeotic alterations in the flowers that are essentially identical to those caused by constitutive expression of other euAG genes [56]. However MtAGb protein seems to be more efficient than MtAGa to induce reproductive organ fate when overexpressed in a heterologous system.
Discussion
The model legume Medicago truncatula harbors two members of the euAG sub-clade (MtAGa and MtAGb) and one member of PLENA (MtSHP) [30]. In this study, we report the functional characterization of the euAG homologs from this species to achieve a more thorough understanding of the evolution of the AGAMOUS subfamily in core eudicots.
Two euAG genes in Medicago truncatula
The finding of two euAG duplicated genes (MtAGa and MtAGb) in M. truncatula, as well as in other legume species (see Figure 1B) suggests that each pair of paralogs arose by a duplication event prior to the speciation of legumes. They could have been originated during the whole-genome duplication (WGD) event that predated speciation of M. truncatula and other legumes around 50-60 Mya [57], [58]. Gene duplications allowed the emergence of critical changes in several MADS-box gene lineages that control floral organ identity. In Medicago, B-function genes are also duplicated: MtAP3-like genes were originated by an ancient duplication concomitant with the base of the core eudicot radiation [49], whereas the two duplicated PI-like genes were probably originated during the WGD event that also originated the two MtAG genes ([48]; Roque et al. unpublished). The changes of these genes after duplication may have played an important role in the evolution of floral morphology and ontogeny in legumes.
The presence of two euAG genes is not only found in legumes. Previous phylogenetic studies have revealed the existence of two members of the euAG-lineage in several core eudicots [12]. This is the case of the paralogous CUM1 and CUM2 from Cucumis sativus, PTAG1 and PTAG2 from Populus trichocarpa, and GAGA1 and GAGA2 from Gerbera hybrida. These studies are mainly focused on the analyses of their expression patterns and limited functional information is available. Recently, phylogenetic analyses using AG protein sequences from several Asteraceae species, have revealed that some of them, harbor two or three members within the euAG-lineage [24].
MtAGa and MtAGb proteins conserve characteristic domains of the C-lineage members, as the presence of an N-terminal extension preceding the MADS domain [59]. The length of this sequence in MtAG proteins falls within the range previously described for other members of this clade (from 13 to 52 amino acids) [59] (Figure S1). However, regarding to DNA sequence, both genes possess six introns rather than the typical eight of the other AG homologs [7], [8], [11], [25], [60], [61]. This genomic organization is observed in both angiosperm and gymnosperm genes suggesting that the presence of eight introns is likely to be primitive in AG subfamily [12]. The genomic organization of M. truncatula euAG genes could have taken place during the WGD event that predates legume speciation followed by gene rearrangement.
Within the highly similar gene structure of AGAMOUS genes, it has been shown that the first and second introns are essential for their correct expression [24]. Among the identified enhancers, the transcription factor LFY is essential for AG activation and binds to cis-elements located in the second intron [52], [62]. The presence of LFY-binding sites has been reported to be critical to establish early expression of C-function genes during flower development [63]. We identified high affinity binding sites for LFY in the first intron of both MtAGa and MtAGb. Although the binding site landscapes observed for each euAG-paralogous gene are different, experimental analyses have demonstrated that both types of distribution of LFY binding sites are functionally relevant [53]. In contrast, in our analyses MtSHP was not found to be a likely LFY target and accordingly, MtSHP expression is exclusively detected in the ovules at later stages of floral development. Similarly, SHP or STK, which play late roles in fruit and ovule development, were not found to be LFY targets in ChIP-seq experiments [53]. Together, our analyses suggest that both MtAG genes have the same probability to be directly activated by LFY, conferring organ identity on floral meristems that arise after the transition to reproductive development.
Two euAG genes control C-function in Medicago truncatula
A typical C-function knock-out phenotype is described as the conversion of stamens to petals, and a flower in place of the fourth whorl, which starts with a whorl of sepals [24]. 1% of MtAGab-VIGS flowers showed a near complete loss-of-C-function phenotype. Stamens were replaced by petals or petal-like organs and the central carpel was replaced by multiple petaloid or sepaloid concentric structures. MtAGab-VIGS plants still showed some reproductive structures probably due to the fact that these lines still produce some MtAGa and MtAGb transcripts. It would be desirable to measure the MtAG mRNAs levels in MtAGab-VIGS flowers to validate this hypothesis, but sufficient plant material was not available. However, using stable MtAG::RNAi lines we were able to correlate the amounts of MtAG gene products with partial loss-of-C-function phenotypes. Only simultaneous down-regulation of both genes (below 40% of the wild-type level) produces mild aberrations in stamen and carpel development. These results support the quantitative model of AG activity, which suggests that the different AG functions vary with respect to the amount of gene product required [47]. The floral determinacy requires the higher levels of AG, while low levels of AG are sufficient for carpel and stamen specification [51]. According to this model, only a strong reduction of both MtAG transcripts could explain the indeterminacy of the floral meristem in MtAGab-VIGS flowers. In each single mtaga and mtagb mutants the amount of the remaining paralog is sufficient to confer almost all the C-function. However, the discrete mutant phenotypes of these plants suggest that a single MtAG gene is not sufficient to maintain the overall dosage of these subcomponents of protein complexes which are needed to specify the C-function in M. truncatula.
The spatial and temporal expression profile of AGAMOUS subfamily genes is closely consistent with their conserved function and subfunctionalization [24]. The expression of AG lineage genes early in the floral meristem, and in the third and fourth floral whorls is probably the ancestral expression profile of these genes [24]. An overlapping conserved expression pattern for both euAG and PLE-like genes is observed in Petunia, N. benthamiana and Antirrhinum, at early floral developmental stages. This expression profile correlates with their functional redundancy to promote the C-function of these genes in the cited species [4], [11], [20]. In A. thaliana, the only C-function factor is AG, because, although the PLE-like proteins (SHP proteins) fully maintain a similar potential activity, they are not physically present in the meristem and primordia cells at the appropriate time. Their functions are rather restricted to regulate specific tissue types after carpel identity has been established. It remains possible that SHP genes redundantly regulate carpel identity with AG because they are expressed in early carpel primordia. The Arabidopsis AG and SHP genes are a clear example of subfunctionalization as a result of a change in expression pattern [64]. M. truncatula could be considered an illustrative example, where the expression pattern of the members of euAG or PLENA lineages would be very well correlated with their function. The two euAG genes (MtAGa and MtAGb) are expressed early during floral development, having a critical role in the specification of reproductive organs and floral determinacy. MtSHP expression appears late during floral development after all organs have been specified, suggesting it does not contribute significantly to the different components of the C-function (stamen and carpel identity, and determinacy). A role for this gene in fruit morphology and valve margin lignification has been suggested [30]. However, we cannot exclude a function for MtSHP in ovule development, associated with its expression in this tissue during floral development.
It seems that changes in coding sequences are not the main cause of the functional divergence between the majority of the euAG and PLENA-like members. In most cases, the ectopic expression of each sub-clade member gave similar results [4], [23], [55], [65], [66], [67]. This demonstrates that both types of proteins have conserved their biochemical interactions. However, an exception to the rule has been observed, for example, in Antirrhinum, where the presence of an additional glutamine in the K3 helix of FAR protein, causes its inability to induce carpel identity in the first whorl [68], [69]. These data suggest that the functional diversification of Antirrhinum C-lineage genes largely relies on changes in the protein ability to interact rather than on changes in their expression patterns. The ectopic expression of M. truncatula euAG- and PLENA-like genes (MtAGa, MtAGb and MtSHP; this work; [20]) in Arabidopsis was able to cause homeotic alterations in the flowers that are essentially identical to those caused by the constitutive expression of other euAG- and PLENA-like genes. However, we noticed that although there are not qualitative differences in the floral phenotypes between 35S::MtAGa and 35S::MtAGb transgenic plants, they differ quantitatively, indicating that MtAGb protein could be more active in Arabidopsis than MtAGa.
Overall, our results show that MtAG genes could have the same probability to be regulated by LFY at early stages of floral development, while the prediction of binding sites for LFY in the first intron of MtSHP indicated a low probability to be regulated by LFY. Both MtAG genes showed nearly identical spatial expression patterns, which are similar to those described for C-function genes from the euAG subclade such as AG and PLE. In contrast, MtSHP is exclusively detected in the ovules at late stages of floral development. It is not physically present in the meristem and primordia cells at the appropriate time to participate in the C-function. Therefore, in M. truncatula, the C-function is redundantly encoded by the two euAG-genes. Finally, the overexpression of MtAGa or MtAGb in Arabidopsis, gave similar results to those observed for the ectopic expression of other euAG-genes. M. truncatula euAG- and PLENA-like paralogs could have subfunctionalized concomitantly with the differential spatial and temporal expression pattern of the ancestral gene lineage. Furthermore, MtAG paralogs are maintained in the genome consistent with the gene balance hypothesis, which predicts that the fate of duplicated genes largely depends on the maintenance of the stoichiometric balance among members of the macromolecular complex [70], [71], [72], [73].
AG subfamily has been shaped by a complex history of gene duplications leading to various degrees of redundancy and/or subfunctionalization of the C-function in individual gene copies. Despite these differences, the control of the development of reproductive organs is strongly conserved across eudicots. We provide insight on the evolution of the AG subfamily in a new core eudicot species, highlighting the dynamic nature of functional evolution following gene duplication in this MADS-box gene subfamily.
Supporting Information
Sequence comparison of MtAGa, MtAGb and related MADS-box proteins. Identical amino acid residues are shaded in black, and similar amino acid residues in grey. The MADS and K domains, and I and C-terminal regions are marked with arrows. The AG motifs I and II are underlined with continuous and discontinuous lines, respectively. A dotted line before the MADS-box indicates the N-terminal sequence characteristic to some C-lineage genes.
(TIF)
Southern blot analysis of MtAGa and MtAGb genes and localization of the MtAGb gene on the M. truncatula physical map. (A) Southern blot analysis of MtAGa and MtAGb. The length of the DNA markers (in Kb) is shown on the left margin. (B) MtAGb sequence is anchored in BAC AC153460, on the chromosome 8. Pseudochromosomes are represented by the succession of BACs (vertical, grey thick lines) separated by gaps (black, horizontal lines). Not all gaps are visible at this resolution. Unmapped BACs are shown on the right as MtChr0. MPM1/2: molecular weight markers.
(TIF)
Molecular characterization of Tnt1 insertion line in the mtagb locus. Genotyping of 20 plants from the Tnt1 insertion population of NF4908 line. (A) PCR results using AGb-F/Tnt1-F primers. (B) PCR results using AGb-F/AGb-R genomic primers. Homozygous mutant plants are indicated with bold. numbers. MPM: molecular weight markers; WT: wild-type control; H2O: distilled water.
(TIF)
qRT-PCR expression analyses of loss-of-function plants. Relative expression of MtAGa or MtAGb genes in flower buds of mtaga and mtagb mutants. The height of the bars for a given gene indicates differences in relative expression levels in floral buds. The wild-type expression value was set to 1.00, and lower values are plotted relative to this value.
(TIF)
Floral phenotype and expression analyses of 35S::RNAi- MtAG plants. (A) Wild-type M. truncatula flower. (B-C) Flowers from 35S::RNAi-MtAG transgenic line 5.7. (D) Wild-type stamens. (E-F) RNAi lines showing unfused staminal tubes and mild homeotic transformations (arrow). (G) Wild-type carpel. (H-I) Multiple unfused carpels showing exposed ovules (arrow). (J) Relative expression of MtAGa or MtAGb genes in flower buds of the three transgenic RNAi lines measured by qRT-PCR. The height of the bars for a given gene indicates differences in relative expression levels in floral buds. The wild-type expression value was set to 1.00, and lower values are plotted relative to this value.
(TIF)
Sequences of the C-lineage MADS-box genes from the different species used in the phylogenetic analysis. The GenBank accession numbers are indicated.
(DOCX)
Primers used in this work.
(DOCX)
Acknowledgments
We are grateful to C. Ferrándiz for helpful comments. Technical assistance of M.C. Rochina in M. truncatula transformation and R. Martínez-Pardo in the greenhouse is gratefully acknowledged.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files. All data concerning gene sequences obtained in this work are freely available in the GenBank public database (http://www.ncbi.nlm.nih.gov/genbank) provided by the National Center for Biotechnology Information (NCBI). The sequences are identified in the manuscript with their respective GenBank accession numbers: MtAGa cDNA: KF159804 MtAGb cDNA: KF159805 MtAGa genomic: KJ470633 MtAGb genomic: KJ470634 MtSHP intron: KJ470635.
Funding Statement
This work was funded by grants BIO2009-08134 and BIO2012-39849-C02-01 from the Spanish Ministry of Economy and Competitiveness and the Ramón y Cajal Program (RYC-2007-00627 to CGM). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Prunet N, Jack TP (2014) Flower development in Arabidopsis: there is more to it than learning your ABCs. Methods Mol Biol 1110: 3–33. [DOI] [PubMed] [Google Scholar]
- 2. Causier B, Schwarz-Sommer Z, Davies B (2010) Floral organ identity: 20 years of ABCs. Semin Cell Dev Biol 21: 73–79. [DOI] [PubMed] [Google Scholar]
- 3. Irish VF (2010) The flowering of Arabidopsis flower development. Plant J 61: 1014–1028. [DOI] [PubMed] [Google Scholar]
- 4. Heijmans K, Morel P, Vandenbussche M (2012) MADS-box genes and floral development: the dark side. J Exp Bot 63: 5397–5404. [DOI] [PubMed] [Google Scholar]
- 5. Bowman JL, Smyth DR, Meyerowitz EM (1989) Genes directing flower development in Arabidopsis. Plant Cell 1: 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowman JL, Smyth DR, Meyerowitz EM (1991) Genetic interactions among floral homeotic genes of Arabidopsis. Development 112: 1–20. [DOI] [PubMed] [Google Scholar]
- 7. Yanofsky MF, Ma H, Bowman JL, Drews GN, Feldmann KA, et al. (1990) The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 346: 35–39. [DOI] [PubMed] [Google Scholar]
- 8. Bradley D, Carpenter R, Sommer H, Hartley N, Coen E (1993) Complementary floral homeotic phenotypes result from opposite orientations of a transposon at the plena locus of Antirrhinum. Cell 72: 85–95. [DOI] [PubMed] [Google Scholar]
- 9. Pinyopich A, Ditta GS, Savidge B, Liljegren SJ, Baumann E, et al. (2003) Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature 424: 85–88. [DOI] [PubMed] [Google Scholar]
- 10. Liljegren SJ, Ditta GS, Eshed Y, Savidge B, Bowman JL, et al. (2000) SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404: 766–770. [DOI] [PubMed] [Google Scholar]
- 11. Davies B, Motte P, Keck E, Saedler H, Sommer H, et al. (1999) PLENA and FARINELLI: redundancy and regulatory interactions between two Antirrhinum MADS-box factors controlling flower development. EMBO J 18: 4023–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kramer EM, Jaramillo MA, Di Stilio VS (2004) Patterns of gene duplication and functional evolution during the diversification of the AGAMOUS subfamily of MADS box genes in angiosperms. Genetics 166: 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Becker A, Theissen G (2003) The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol 29: 464–489. [DOI] [PubMed] [Google Scholar]
- 14. Irish VF (2003) The evolution of floral homeotic gene function. Bioessays 25: 637–646. [DOI] [PubMed] [Google Scholar]
- 15. Zahn LM, Leebens-Mack JH, Arrington JM, Hu Y, Landherr LL, et al. (2006) Conservation and divergence in the AGAMOUS subfamily of MADS-box genes: evidence of independent sub- and neofunctionalization events. Evol Dev 8: 30–45. [DOI] [PubMed] [Google Scholar]
- 16. Ferrandiz C, Liljegren SJ, Yanofsky MF (2000) Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science 289: 436–438. [DOI] [PubMed] [Google Scholar]
- 17. Ma H, Yanofsky MF, Meyerowitz EM (1991) AGL1-AGL6, an Arabidopsis gene family with similarity to floral homeotic and transcription factor genes. Genes Dev 5: 484–495. [DOI] [PubMed] [Google Scholar]
- 18. Savidge B, Rounsley SD, Yanofsky MF (1995) Temporal relationship between the transcription of two Arabidopsis MADS box genes and the floral organ identity genes. Plant Cell 7: 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Colombo M, Brambilla V, Marcheselli R, Caporali E, Kater MM, et al. (2010) A new role for the SHATTERPROOF genes during Arabidopsis gynoecium development. Dev Biol 337: 294–302. [DOI] [PubMed] [Google Scholar]
- 20. Fourquin C, Ferrandiz C (2012) Functional analyses of AGAMOUS family members in Nicotiana benthamiana clarify the evolution of early and late roles of C-function genes in eudicots. Plant J 71: 990–1001. [DOI] [PubMed] [Google Scholar]
- 21. Kapoor M, Tsuda S, Tanaka Y, Mayama T, Okuyama Y, et al. (2002) Role of petunia pMADS3 in determination of floral organ and meristem identity, as revealed by its loss of function. Plant J 32: 115–127. [DOI] [PubMed] [Google Scholar]
- 22. Pan IL, McQuinn R, Giovannoni JJ, Irish VF (2010) Functional diversification of AGAMOUS lineage genes in regulating tomato flower and fruit development. J Exp Bot 61: 1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pnueli L, Hareven D, Rounsley SD, Yanofsky MF, Lifschitz E (1994) Isolation of the tomato AGAMOUS gene TAG1 and analysis of its homeotic role in transgenic plants. Plant Cell 6: 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dreni L, Kater MM (2014) MADS reloaded: evolution of the AGAMOUS subfamily genes. New Phytol 201: 717–732. [DOI] [PubMed] [Google Scholar]
- 25. Brunner AM, Rottmann WH, Sheppard LA, Krutovskii K, DiFazio SP, et al. (2000) Structure and expression of duplicate AGAMOUS orthologues in poplar. Plant Mol Biol 44: 619–634. [DOI] [PubMed] [Google Scholar]
- 26. Perl-Treves R, Kahana A, Rosenman N, Xiang Y, Silberstein L (1998) Expression of multiple AGAMOUS-like genes in male and female flowers of cucumber (Cucumis sativus L.). Plant Cell Physiol 39: 701–710. [DOI] [PubMed] [Google Scholar]
- 27. Yu D, Kotilainen M, Pollanen E, Mehto M, Elomaa P, et al. (1999) Organ identity genes and modified patterns of flower development in Gerbera hybrida (Asteraceae). Plant J 17: 51–62. [DOI] [PubMed] [Google Scholar]
- 28. Dong ZC, Zhao Z, Liu CW, Luo JH, Yang J, et al. (2005) Floral patterning in Lotus japonicus. . Plant Physiol 137: 1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hofer JMI, Noel Ellis TH (2014) Developmental specialisations in the legume family. Current Opinion in Plant Biology 17: 153–158. [DOI] [PubMed] [Google Scholar]
- 30. Fourquin C, del Cerro C, Victoria FC, Vialette-Guiraud A, de Oliveira AC, et al. (2013) A change in SHATTERPROOF protein lies at the origin of a fruit morphological novelty and a new strategy for seed dispersal in Medicago genus. Plant Physiol 162: 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewitt EJ (1966) Sand and Water Culture Methods Used in the Study of Plant Nutrition. Farnham Royal, UK: Commonwealth Agricultural Bureau.
- 32. Cheng X, Wang M, Lee HK, Tadege M, Ratet P, et al. (2014) An efficient reverse genetics platform in the model legume Medicago truncatula . New Phytol 201: 1065–1076. [DOI] [PubMed] [Google Scholar]
- 33. d'Erfurth I, Cosson V, Eschstruth A, Lucas H, Kondorosi A, et al. (2003) Efficient transposition of the Tnt1 tobacco retrotransposon in the model legume Medicago truncatula. . Plant J 34: 95–106. [DOI] [PubMed] [Google Scholar]
- 34. Tadege M, Ratet P, Mysore KS (2005) Insertional mutagenesis: a Swiss Army knife for functional genomics of Medicago truncatula . Trends Plant Sci 10: 229–235. [DOI] [PubMed] [Google Scholar]
- 35. Tadege M, Wen J, He J, Tu H, Kwak Y, et al. (2008) Large-scale insertional mutagenesis using the Tnt1 retrotransposon in the model legume Medicago truncatula . Plant J 54: 335–347. [DOI] [PubMed] [Google Scholar]
- 36. Cheng X, Wen J, Tadege M, Ratet P, Mysore KS (2011) Reverse genetics in Medicago truncatula using Tnt1 insertion mutants. Methods Mol Biol 678: 179–190. [DOI] [PubMed] [Google Scholar]
- 37. Benlloch R, d'Erfurth I, Ferrandiz C, Cosson V, Beltran JP, et al. (2006) Isolation of mtpim proves Tnt1 a useful reverse genetics tool in Medicago truncatula and uncovers new aspects of AP1-like functions in legumes. Plant Physiol 142: 972–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 39. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599. [DOI] [PubMed] [Google Scholar]
- 41. Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA minipreparation: version II. Plant Mol Biol Rep 1: 19–21. [Google Scholar]
- 42. Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- 43. Hartweck LM, Scott CL, Olszewski NE (2002) Two O-linked N-acetylglucosamine transferase genes of Arabidopsis thaliana L. Heynh. have overlapping functions necessary for gamete and seed development. Genetics 161: 1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Constantin GD, Krath BN, MacFarlane SA, Nicolaisen M, Johansen IE, et al. (2004) Virus-induced gene silencing as a tool for functional genomics in a legume species. Plant J 40: 622–631. [DOI] [PubMed] [Google Scholar]
- 45. Wesley SV, Helliwell CA, Smith NA, Wang MB, Rouse DT, et al. (2001) Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J 27: 581–590. [DOI] [PubMed] [Google Scholar]
- 46.Guerineau F, Mullineaux P (1993) Plant transformation and expression vectors. In: Croy R, editor. Plant Molecular Biology. Oxford, UK: Bios Scientific Publishers, Academic Press. pp. 121–147.
- 47. Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana . Plant J 16: 735–743. [DOI] [PubMed] [Google Scholar]
- 48. Benlloch R, Roque E, Ferrandiz C, Cosson V, Caballero T, et al. (2009) Analysis of B function in legumes: PISTILLATA proteins do not require the PI motif for floral organ development in Medicago truncatula . Plant J 60: 102–111. [DOI] [PubMed] [Google Scholar]
- 49. Roque E, Serwatowska J, Cruz Rochina M, Wen J, Mysore KS, et al. (2013) Functional specialization of duplicated AP3-like genes in Medicago truncatula . Plant J 73: 663–675. [DOI] [PubMed] [Google Scholar]
- 50. Flanagan CA, Hu Y, Ma H (1996) Specific expression of the AGL1 MADS-box gene suggests regulatory functions in Arabidopsis gynoecium and ovule development. Plant J 10: 343–353. [DOI] [PubMed] [Google Scholar]
- 51. Sieburth LE, Meyerowitz EM (1997) Molecular dissection of the AGAMOUS control region shows that cis elements for spatial regulation are located intragenically. Plant Cell 9: 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Busch MA, Bomblies K, Weigel D (1999) Activation of a floral homeotic gene in Arabidopsis. Science 285: 585–587. [DOI] [PubMed] [Google Scholar]
- 53. Moyroud E, Minguet EG, Ott F, Yant L, Pose D, et al. (2011) Prediction of regulatory interactions from genome sequences using a biophysical model for the Arabidopsis LEAFY transcription factor. Plant Cell 23: 1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gronlund M, Constantin G, Piednoir E, Kovacev J, Johansen IE, et al. (2008) Virus-induced gene silencing in Medicago truncatula and Lathyrus odorata . Virus Res 135: 345–349. [DOI] [PubMed] [Google Scholar]
- 55. Mandel MA, Bowman JL, Kempin SA, Ma H, Meyerowitz EM, et al. (1992) Manipulation of flower structure in transgenic tobacco. Cell 71: 133–143. [DOI] [PubMed] [Google Scholar]
- 56. Mizukami Y, Ma H (1992) Ectopic expression of the floral homeotic gene AGAMOUS in transgenic Arabidopsis plants alters floral organ identity. Cell 71: 119–131. [DOI] [PubMed] [Google Scholar]
- 57. Cannon SB, Sterck L, Rombauts S, Sato S, Cheung F, et al. (2006) Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc Natl Acad Sci U S A 103: 14959–14964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Young ND, Bharti AK (2012) Genome-enabled insights into legume biology. Annu Rev Plant Biol 63: 283–305. [DOI] [PubMed] [Google Scholar]
- 59. Jager M, Hassanin A, Manuel M, Le Guyader H, Deutsch J (2003) MADS-box genes in Ginkgo biloba and the evolution of the AGAMOUS family. Mol Biol Evol 20: 842–854. [DOI] [PubMed] [Google Scholar]
- 60. Johansen B, Pedersen LB, Skipper M, Frederiksen S (2002) MADS-box gene evolution-structure and transcription patterns. Mol Phylogenet Evol 23: 458–480. [DOI] [PubMed] [Google Scholar]
- 61. Rutledge R, Regan S, Nicolas O, Fobert P, Cote C, et al. (1998) Characterization of an AGAMOUS homologue from the conifer black spruce (Picea mariana) that produces floral homeotic conversions when expressed in Arabidopsis. Plant J 15: 625–634. [DOI] [PubMed] [Google Scholar]
- 62. Parcy F, Nilsson O, Busch MA, Lee I, Weigel D (1998) A genetic framework for floral patterning. Nature 395: 561–566. [DOI] [PubMed] [Google Scholar]
- 63. Causier B, Bradley D, Cook H, Davies B (2009) Conserved intragenic elements were critical for the evolution of the floral C-function. The Plant Journal 58: 41–52. [DOI] [PubMed] [Google Scholar]
- 64. Airoldi CA, Davies B (2012) Gene duplication and the evolution of plant MADS-box transcription factors. J Genet Genomics 39: 157–165. [DOI] [PubMed] [Google Scholar]
- 65. Gimenez E, Pineda B, Capel J, Anton MT, Atares A, et al. (2010) Functional analysis of the Arlequin mutant corroborates the essential role of the Arlequin/TAGL1 gene during reproductive development of tomato. PLoS One 5: e14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kater MM, Colombo L, Franken J, Busscher M, Masiero S, et al. (1998) Multiple AGAMOUS homologs from cucumber and petunia differ in their ability to induce reproductive organ fate. Plant Cell 10: 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsuchimoto S, van der Krol AR, Chua NH (1993) Ectopic expression of pMADS3 in transgenic petunia phenocopies the petunia blind mutant. Plant Cell 5: 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Airoldi CA, Bergonzi S, Davies B (2010) Single amino acid change alters the ability to specify male or female organ identity. Proc Natl Acad Sci U S A 107: 18898–18902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Causier B, Castillo R, Zhou J, Ingram R, Xue Y, et al. (2005) Evolution in action: following function in duplicated floral homeotic genes. Curr Biol 15: 1508–1512. [DOI] [PubMed] [Google Scholar]
- 70. Birchler JA, Veitia RA (2007) The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Birchler JA, Veitia RA (2010) The gene balance hypothesis: implications for gene regulation, quantitative traits and evolution. New Phytol 186: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Edger PP, Pires JC (2009) Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res 17: 699–717. [DOI] [PubMed] [Google Scholar]
- 73. Freeling M, Thomas BC (2006) Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res 16: 805–814. [DOI] [PubMed] [Google Scholar]
- 74. Benlloch R, Navarro C, Beltrán J, Cañas LA (2003) Floral development of the model legume Medicago truncatula: ontogeny studies as a tool to better characterize homeotic mutations. Sexual Plant Reproduction 15: 231–241. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence comparison of MtAGa, MtAGb and related MADS-box proteins. Identical amino acid residues are shaded in black, and similar amino acid residues in grey. The MADS and K domains, and I and C-terminal regions are marked with arrows. The AG motifs I and II are underlined with continuous and discontinuous lines, respectively. A dotted line before the MADS-box indicates the N-terminal sequence characteristic to some C-lineage genes.
(TIF)
Southern blot analysis of MtAGa and MtAGb genes and localization of the MtAGb gene on the M. truncatula physical map. (A) Southern blot analysis of MtAGa and MtAGb. The length of the DNA markers (in Kb) is shown on the left margin. (B) MtAGb sequence is anchored in BAC AC153460, on the chromosome 8. Pseudochromosomes are represented by the succession of BACs (vertical, grey thick lines) separated by gaps (black, horizontal lines). Not all gaps are visible at this resolution. Unmapped BACs are shown on the right as MtChr0. MPM1/2: molecular weight markers.
(TIF)
Molecular characterization of Tnt1 insertion line in the mtagb locus. Genotyping of 20 plants from the Tnt1 insertion population of NF4908 line. (A) PCR results using AGb-F/Tnt1-F primers. (B) PCR results using AGb-F/AGb-R genomic primers. Homozygous mutant plants are indicated with bold. numbers. MPM: molecular weight markers; WT: wild-type control; H2O: distilled water.
(TIF)
qRT-PCR expression analyses of loss-of-function plants. Relative expression of MtAGa or MtAGb genes in flower buds of mtaga and mtagb mutants. The height of the bars for a given gene indicates differences in relative expression levels in floral buds. The wild-type expression value was set to 1.00, and lower values are plotted relative to this value.
(TIF)
Floral phenotype and expression analyses of 35S::RNAi- MtAG plants. (A) Wild-type M. truncatula flower. (B-C) Flowers from 35S::RNAi-MtAG transgenic line 5.7. (D) Wild-type stamens. (E-F) RNAi lines showing unfused staminal tubes and mild homeotic transformations (arrow). (G) Wild-type carpel. (H-I) Multiple unfused carpels showing exposed ovules (arrow). (J) Relative expression of MtAGa or MtAGb genes in flower buds of the three transgenic RNAi lines measured by qRT-PCR. The height of the bars for a given gene indicates differences in relative expression levels in floral buds. The wild-type expression value was set to 1.00, and lower values are plotted relative to this value.
(TIF)
Sequences of the C-lineage MADS-box genes from the different species used in the phylogenetic analysis. The GenBank accession numbers are indicated.
(DOCX)
Primers used in this work.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files. All data concerning gene sequences obtained in this work are freely available in the GenBank public database (http://www.ncbi.nlm.nih.gov/genbank) provided by the National Center for Biotechnology Information (NCBI). The sequences are identified in the manuscript with their respective GenBank accession numbers: MtAGa cDNA: KF159804 MtAGb cDNA: KF159805 MtAGa genomic: KJ470633 MtAGb genomic: KJ470634 MtSHP intron: KJ470635.