

Abstract
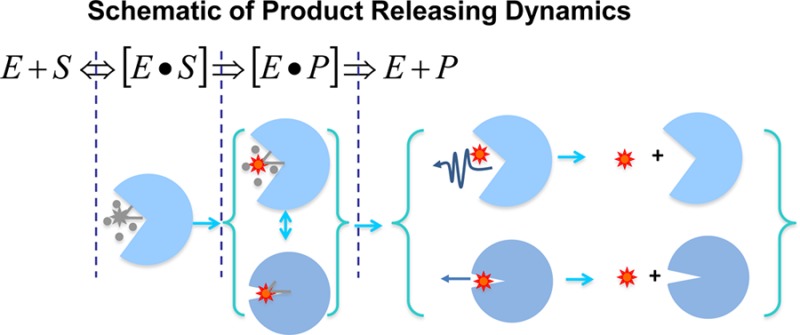
Product releasing is an essential step of an enzymatic reaction, and a mechanistic understanding primarily depends on the active-site conformational changes and molecular interactions that are involved in this step of the enzymatic reaction. Here we report our work on the enzymatic product releasing dynamics and mechanism of an enzyme, horseradish peroxidase (HRP), using combined single-molecule time-resolved fluorescence intensity, anisotropy, and lifetime measurements. Our results have shown a wide distribution of the multiple conformational states involved in active-site interacting with the product molecules during the product releasing. We have identified that there is a significant pathway in which the product molecules are spilled out from the enzymatic active site, driven by a squeezing effect from a tight active-site conformational state, although the conventional pathway of releasing a product molecule from an open active-site conformational state is still a primary pathway. Our study provides new insight into the enzymatic reaction dynamics and mechanism, and the information is uniquely obtainable from our combined time-resolved single-molecule spectroscopic measurements and analyses.
Introduction
Conformational motions of enzymes often play critical roles in enzymatic reactions. A fundamental understanding of enzymatic reaction dynamics and mechanisms relies on the experimental characterization of the specific steps of the enzymatic reactions. Beyond the well-known Michaelis–Menten mechanism, there are a number of fundamental questions that are critical but have not been experimentally answered: for example, how is a product molecule released from an enzymatic active site? Typically, an enzymatic reaction involves multiple kinetic steps, such as substrate binding to form an enzyme–substrate complex, [E•S], nascent product generation, [E•P], and products releasing from the enzyme active site to complete an enzymatic productive turnover, as illustrated in the Michaelis–Menten mechanism, E + S ⇄ [E•S] → [E•P]→ E + P, where E, S, and P represent enzyme, substrate, and product, respectively.1−3 The intrinsically dynamic and inhomogeneous processes of enzymatic reactions involve conformational fluctuations ranging over 10–15 s to >1 s and 10–2 Å to >10 Å, in time and space, respectively.4,5 These spatial and temporal dynamics play critical roles in defining the enzymatic free energy potential surfaces, reactive coordinates, and rate processes.6,7 Particularly, enzymatic conformational dynamics is essential for enzyme specificity and selectivity. Multiple reaction coordinates of the conformational dynamics in an enzyme complicate the understanding of their structure and function.8,9 Therefore, a large variety of experimental and theoretical techniques have been developed to probe internal dynamics of enzymes, particularly at the picosecond to second time scale.10−12
Single-molecule fluorescence spectroscopy and imaging have been proved as a powerful technique in characterizing enzymatic conformational dynamics and mechanisms.4,6,13 Photophysical properties of fluorophores, such as fluorescence intensities, fluorescence quantum yield, and fluorescence decay rate constants, are sensitive to the properties of the local environment.14,15 Fluorescence anisotropy provides information about the motions of the protein fragments to which the fluorescent probe is attached.16,17 For example, time-resolved anisotropy is capable of analyzing the conformational change fluctuations and the protein flexibility.16 Furthermore, single-molecule fluorescence lifetime fluctuation measurements are sensitive in characterizing the electrostatic and hydrophobic interactions between the probe molecules and their fluctuating local environment.18−21 The intrinsic ability to sense the molecular surrounding of the probe molecule makes the single-molecule spectroscopy an ideal tool to study conformational dynamics of enzymes. Complementary to conventional ensemble-averaged measurements that average over a population, single-molecule measurements are able to detect conformational heterogeneities, identify transient or specific conformations, follow conformational changes, and reveal parallel reaction pathways.2,6,22 Generally, three categories of fluorophores have been used in single-molecule studies of enzymatic reaction systems, which include intrinsic fluorescence from tryptophan or tyrosine residues, site-specific fluorescent labeling, and fluorescent products.13 Comparing with others, fluorescent product molecules are generated constantly during the course of an enzymatic reaction, which eliminates the photobleaching problem and provides a chance to record and analyze a long time trajectory of the enzymatic reaction from a single enzyme. In a fluorogenic enzymatic assay measurement, the recorded trajectories are sufficiently long to represent a statistically relevant sampling of all possible conformational states.
Horseradish peroxidase (HRP), a monomeric enzyme 44K Dalton, containing a heme prosthetic group, catalyzes the oxidation of a broad range of substrates such as aromatic amines, indoles, phenols, and sulfonates in the presence of hydrogen peroxide as an oxidizing agent (H2O2).23 The crystal structure and the catalytic mechanism of HRP were described previously.24−26 The HRP catalytic dynamics of turning over the fluorogenic substrate has been investigated by single-molecule fluorescence spectroscopy techniques.9,27−30 However, previously reported results are primarily limited to the reaction rate and the correlation analysis of the dynamic disorder. Specifically, there is still a lack of experimental understanding of the molecular mechanism and dynamics of the interactions between the enzymatic active site and the nascent product and the conformational dynamics associated with the releasing of the product from the enzyme. Nevertheless, various fluorescence characteristics, such as fluorescence intensity, spectrum, lifetime, and anisotropy, deserve to be exploited as they are potentially capable of probing enzyme–substrate and enzyme–product molecular interactions under complex local environments.31−35 Our unique technical approach of multiparameter single-molecule spectroscopic investigation of enzymatic reactions provides information beyond the reaction rates and correlation analysis of dynamic disorder.36,37
In this work, we use HRP-catalyzed oxidation of nonfluorescent substrate Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) to fluorescent resorufin as a model system to study the conformational dynamics of the enzyme during the fluorescent products releasing from the enzymatic active site into the solution, i.e., the last step of the enzymatic reaction turnover cycle. We have used a home-built total internal imaging guided confocal single molecular spectroscopy technique to probe the consecutive fluorescent product releasing from the enzymatic active site. Conformational dynamics of the single-molecule HRP is characterized by measuring real-time photophysical properties of the nascent fluorescent enzymatic products. We have identified a wide distribution of the multiple conformational states involved in active-site interacting with the nascent product molecule during the product releasing, and there is a significant conformational fluctuation between tight states and loose states of the active site of the enzyme involved in the product releasing process. Furthermore, we have experimentally revealed a new pathway in which the product molecules are spilled out from the enzymatic active site, possibly from the squeezing effect, besides the typical pathway of product releasing from an open active-site conformational state through diffusion.
Experimental Methods
1. Chemicals and Sample Preparation
Horseradish peroxidase immobilized on the cover glass was used in our experiments. The cover glass (Gold seal, 3419) was first washed in fresh prepared sulfuric acid dichromate cleaning solution for 1 h to eliminate grease and possible fluorescent spots. After washing with water and drying with nitrogen gas, the cover glass was treated overnight with a mixture solution of 3-mercaptopropyl-trimethoxysilane (Fluka, 09324), isobutyltrimethoxysilane (Sigma, 444065), and dimethyl sulfoxide (Sigma, D4540) with a volume ratio of 1:300:6000. After baking at 110 °C for 10 min, the silanated cover glass was washed with methanol and water. Then the coverslips were incubated in 50 mM PBS buffer (pH 8.0) with about 1 nM maleimide-activated HRP (Thermo scientific, 31485) for 2 h and followed by rinsing with water and PBS buffer. Phosphate buffer (PBS) was prepared with potassium phosphate monobasic solution (Sigma-Aldrich, P8709) and potassium phosphate dibasic solution (Sigma-Aldrich, P8584). Maleimide-activated HRP was linked to the sulfhydryl (−SH) group of 3-mercaptopropyl-trimethoxysilane on the cover glass as shown in Figure 1A. Substrate Amplex Red (Invitrogen, A12222) was dissolved in dimethyl sulfoxide (DMSO) at 5 mg/mL and stored at −20 °C in the dark before use. The reaction solution was prepared just prior to experimentation, with 200 nM Amplex Red and 2 mM H2O2 in PBS buffer (pH 7.4). All chemicals were used without further purification. In our experiment, about 0.5 mL of reaction solution was filled in a home-built magnetic chamber that is composed with the coverglass tethered with HRP at the bottom and a lid on the top of the chamber to eliminate the evaporation.
Figure 1.

Single-molecule fluorescence experimental scheme. (A) Schematic representation of the enzymatic reaction on (3-aminopropyl) trimethoxysilane modified cover glass. Maleimide-activated HRP is linked to the sulfhydryl (−SH) group of 3-mercaptopropyl-trimethoxysilane. Nonfluorescent substrate Amplex red in PBS buffer is converted to fluorescent resorufin product by a single HRP molecule in the presence of hydrogen peroxide initiator, the single-molecule fluorogenic assay. (B) Schematic representation of the total internal reflection fluorescence microscopy imaging-guided confocal fluorescence spectroscopy (TIRFM-CFS). M1: reflection mirror. DM1–DM2: dichroic mirror beam splitters. SPP: side port prism for left/vis obs. TL: tube lens. EF1–EF2: emission filters. L1–L2: lens. PBS: Polarization beam splitter. SPAD1–SPAD2: single photon avalanche photodiode. (C), (D) The typical raw data of single-molecule photon time-stamping spectroscopy of each detector channel with the perpendicular and parallel polarization components, respectively. Each data point represents a detected photon plotted by its arrival time (t) and delay time (Δt).
2. Experimental Setup
The single-molecule enzymatic reaction assay was performed using a total internal reflection fluorescence microscopy imaging-guided confocal fluorescence spectroscopy (TIRFM-CFS) system built in our lab.38 The system is based on an inverted microscope (Axiovert 200M, Carl Zeiss) in an epi-illumination configuration combining the TIRFM mode and the confocal mode, facilitated by a home-developed software for recording images from TIRFM mode and moving the pinpoints of interest from TIRFM imaging to confocal single-molecule spectroscopy measurement.38 The TIRFM mode is used to locate spatially random distributed enzymes from the stochastic on–off bursts of fluorescent signals, and the confocal mode is used for the time-resolved dynamic measurements of the targeted single enzyme.38 Figure 1B shows the experimental setup schematically. Briefly, in the TIRFM mode, a cw laser (GCL-050-L, CrystaLaser) is aligned to the side port by a dichroic mirror beam splitter (DM2) then reflected by the side port prism (SPP) and focused by the tube lens (TL) on the back focal plane of the objective (Plan-Fluar, 1.45 NA, 100×, Carl Zeiss). By slightly deviating DM2 from 45° relative to the incident direction of the beam, the incident angle at the cover glass/solution interface can be adjusted to exceed the critical angle of total internal reflection. The fluorescence emission is collected by the same microscope objective, passing SSP, reflected by M1 to the EMCCD (EMCCD; Photomax 512B, Princeton Instruments), and further the emission signal is purified by an emission filter (EF2). In the confocal mode, a femtosecond pulse laser (Ti:sapphire Mira 900F/P, Coherent Inc.) is used. After the optical parametric oscillator (OPO BASIC, Coherent Inc.) and frequency doubling by a BBO crystal, the linear-polarized pulse laser is aligned to the back port of the microscope stand, reflected by the dichroic mirror beam splitter (DM1) in the filter cube, and focused on the cover glass–solution interface by the objective. The magnetic chamber filled with reaction solution is fixed on the piezoelectric scanning stage (Nano-H100, MCL). Fluorescence emission from the excitation focal volume is collected by the same objective. After transmitting DM1, the signal is reflected by the side port prism (SPP) and transmitting DM2 and refocused by the remagnification lens (L1) to the SPAD1 and SPAD2 (PDM50ct, MPD) entrances. The polarization beam splitter (PBS) is used to separate the signal into parallel and perpendicular polarization components. Here the sensor target diameters of SPADs are 50 μm, which also serve as pinholes to reject stray and ambient light noise. The signals from SPADs are routed by the HRT-82 module and then connected with the single-photon counting module (SPC-830, Becker & Hickl GmbH) to record both photon arrival time (t) and delay time (Δt) between the excitation pulse and excited state emission with high time resolution for each single-photon event. The typical raw data of single-molecule photon time-stamping spectroscopy of each detector channel with the perpendicular and parallel components have been shown as Figure 1 C and D. The arrival times of the photons provide the information about the photon flux, and the histograms of arrival times yield fluorescence intensity trajectories with a given time-bin resolution. Also, the histograms of the delay times give the fluorescence lifetime.39,40 The control experiment has been carried out with the same conditions except no enzyme in the focus volume of laser illumination.
The efficiencies of two detection channels are typically different, primarily because of the difference of the quantum efficiencies of the two SPAD detectors and the bias of the dichroic beam splitter, mirrors, and polarizing beam splitter. To correct the efficiency imbalance of the two detection channels, the overall weighting factor, so-called G factor, is determined by fluorescent intensity response of the two channels with different excitation polarization. In our experiments, the measured weighting factor G is 1.48.
3. Florescence Intensity and Anisotropy
The single-molecule fluorescence intensity decays of the perpendicular and parallel polarized components are typically distinct: The fluorophores that have their transition moment aligned parallel to the electric vector of the excitation laser field preferentially absorb and emit photons. Using the polarization beam splitter, the emission intensity signal is divided into perpendicular and parallel components relative to the polarization of the excitation light. After the signal compensation by the G factor, these intensity trajectories of perpendicular (I⊥(t)) and parallel (I∥(t)) components are used to calculate total fluorescence intensity IT(t) and anisotropy r(t) from the nascent single-molecule product at the enzymatic reaction active site, following eq 1 and eq 2.
| 1 |
| 2 |
4. Florescence Lifetime
Taking convolution with the instrument response function into consideration, the time-resolved polarization intensities I⊥(t) and I∥(t) are determined using eqs 3 and 4, respectively:41
| 3 |
| 4 |
Here, IRF is the instrument response function; τr is the nascent single-molecule product rotational correlation time; τf is fluorescence lifetime; and ∑ sums over all possible terms with different lifetime or rotational correlation time. To simplify the data analysis, the depolarization due to the curvature of the spherical wavefront of the focused field is ignored, due to the low influence even at high apertures.42 We assumed the excited-state population decay as single-exponential functions with lifetime.39 So the total fluorescence intensity could be described by the following equations
| 5 |
Although different methods have been described and applied to extract the lifetime from low count rates,43−47 we use the least-squares (LS) method to get the monoexponential lifetime of the nascent single-molecule enzymatic product since this method is widely applied and converges to maximum likelihood estimation (MLE) at high count rates. The IRF and the time offset are determined from a bulk florescence measurement of the resorufin with the same time binning. Therefore, we have three free parameters to be extracted from the fluorescence data analysis: the fluorescence lifetime, fluorescence amplitude, and constant noise term.
5. Rotational Correlation Time
The rotational correlation time of a fluorescent molecule characterizes the molecule rotation diffusion, being dependent on the size, shape, and hydrodynamics of the molecule, as well as the bulk physical characteristics of the solvent. Therefore, for a fluorescent molecule buried in an enzyme active site, the variation of the rotation correlation time is directly related to the molecular confinement status and its local environment in the enzymatic active site. The rotation correlation time of the rotation fluctuation can be derived through the Perrin equation
| 6 |
Here, r is steady-state anisotropy; r0 is the fundamental anisotropy in the absence of molecular rotation; τf is the fluorescence lifetime; and τr is rotational correlation time. In the current experiment, the fundamental anisotropy of the resorufin is 0.318, which was measured in 80% glycerol solution.48,49
Results and Discussion
Fluorescence intensity is the most straightforward parameter to characterize fluorescence signals of fluorogenic enzymatic reactions. Fluorescence from a single-molecule product can only be detected and identified while it is still confined in the enzyme. After being released from the enzymatic active site, the fluorescent product molecule diffuses out of the laser excitation confocal volume within submilliseconds.50 By the single-molecule photon time stamping spectroscopy technique, we record the intensity trajectories of different polarization channels. Figures 2 A1 and 2 B1 show perpendicular channel and parallel channel fluorescence intensity trajectories, respectively, with a binning time of 50 ms, of the fluorescent products from a single HRP enzyme tethered to the coverglass in PBS buffer solution at pH 7.4. The fluctuations of fluorescence bursts recorded from both parallel and perpendicular polarization channels show similar behaviors and photon count distributions. On average, the intensity of the parallel component is higher than that of the perpendicular component even after the G-factor compensation. The corresponding intensity distributions of different polarizations are shown in Figures 2 A2 and 2 B2, respectively. The black curve is from the intensity distribution of the background, which was taken at the same conditions but without enzyme in the focus volume. This curve clearly shows the profile of the photon time stamping measurement without fluorescence turnovers. The insets show the intensity distributions above the edge of the background. The intensity distribution of the background shows a narrow band below 120 photons, whereas the distributions of intensity beyond the background are asymmetric and elongated toward the higher intensity side. For the distribution portion of intensity beyond the background, it is evident that the distribution likely contains more than one component since the distribution is significantly wider than the highest intensity shot noise.
Figure 2.

Single-molecule fluorescence intensity trajectories and the intensity distributions of HRP-catalyzed oxidation of Amplex Red, binning with 50 ms. (A1) Intensity trajectory of the perpendicular polarization component relative to the excitation polarization after the compensation by the G factor of photon detection. (B1) Intensity trajectory of the parallel polarization component relative to the excitation polarization. (A2) and (B2) The distributions of florescence intensity of the trajectories in A1 and B1, respectively. The black curve denotes the background intensity distribution from control experiment, and the inset is the zoom-in near the threshold of the background.
Generally, the fluorescence intensity of a single fluorophore is proportional to the photon absorption and quantum yield, which is the ratio of emitted photons to the number of absorbed photons. Besides the absorption coefficient, the angle between the excitation polarization and the electron transition dipole also determines the photon absorption probability, which is revealed by the anisotropy measurement. Quantum yields are generally more sensitive to the environment than photon absorption.51 Typical local environmental factors that influence the quantum yield include local refractive index, solvent polarity, proximity and concentrations of quenching species,14 ultimately related to the nonradiative relaxation of the dye molecules, which could be revealed by fluorescence lifetime measurement. Single-molecule fluorescence lifetime analysis is often the choice of probing and characterizing the local environment interactions and fluctuations.18,52,53 Therefore, to identify the relationship between the local environment of a single enzyme’s active site and the fluorescence properties of the fluorescence single-molecule product, it is necessary to perform the anisotropy and lifetime analyses.
Anisotropy provides a critical analysis of the rotational dynamics of a fluorescent molecule; furthermore, specific physical parameters of the local environment influencing the probe molecule rotational motions can also be either qualitatively or semiquantitatively characterized, such as the free space, force field, electric field, hydrophobicity, hydrodynamics, solvation effect, and hydrodynamic volume changes. Figure 3A shows the anisotropy fluctuation trajectory and the corresponding polarization intensity trajectory of a portion of typical single-molecule photon stamping trajectories (Figure 2). Here the intensity is calculated from 300 photons divided by the time intervals for accumulating the consecutive 300 photons to control the shot noise at the same level. The anisotropy shows as constant around zero between the fluorescence photon bursting. During the bursting, the anisotropy shows a dramatic fluctuation. Figure 3B shows the corresponding anisotropy distribution with two distinct peaks. The anisotropy distribution around the peak near zero has a narrow Gaussian-like shape and is dominated by the background, as compared to the control background with mean at −0.02 and with standard deviation as 0.054, as shown in the black curve. The anisotropy distribution at higher anisotropy beyond the background is asymmetric and elongated toward higher values. This broad distribution of the high anisotropy spans from 0.1 and 0.36, which is larger than the standard deviation of the background. To identify the specific interactions of the enzyme–substrate and enzyme–product at the enzymatic active site, it is reasonable to assume that the substrate amplex red and the nascent fluorescent product bind to the active site in a similar configuration as a typical aromatic substrate does, such as a reported binding structure of a HRP–acetate complex.23,25 Aromatic substrates form stable, reversible 1:1 complexes with HRP through both hydrogen-bonded and hydrophobic interactions at the distal side of the heme plane. The amino acid residues Arg38 and His42 play the roles in binding and stabilization of aromatic substrates (see Supporting Information). At the enzymatic active site of the HRP–resorufin complex, the rigid hydrogen bonding of resorufin to the HRP enzyme ensures that the HRP product active site rotates relatively confined, which in turn makes the measured fluorescence anisotropy trajectories likely to yield information about the rotational and conformational dynamics of not only the fluorescent product but also the HRP protein, particularly of the solvent exposed heme prosthetic group at the active site, where the oxidations of aromatic substrates happen. The broad distribution of the anisotropy in Figure 3B also indicates that likely multiple conformational states are involved in active-site interaction with the product molecules during the product releasing, which is consistent with the binding site being a relatively flexible structural region.
Figure 3.
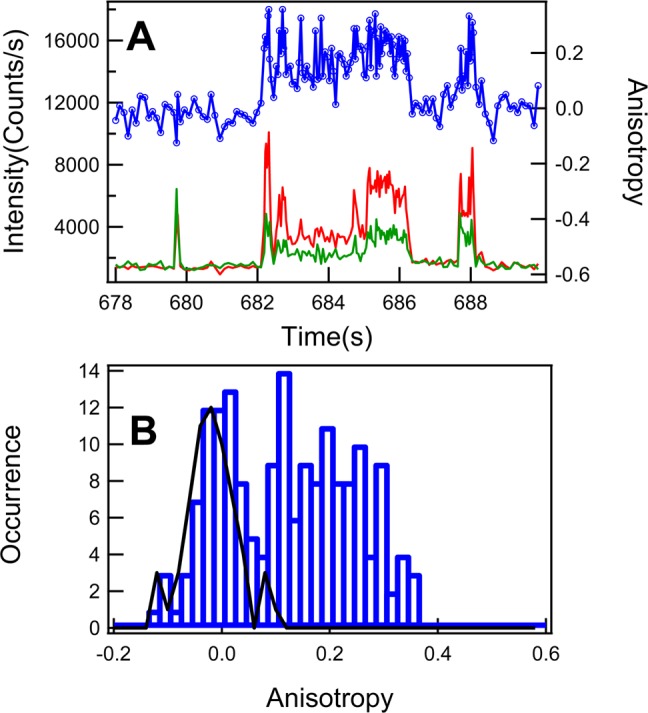
Single-molecule fluorescence anisotropy fluctuation and distribution of single HRP-catalyzed oxidation of Amplex Red fluorogenic assay. (A) Single-molecule fluorescence anisotropy fluctuation. Intensity trajectory from the perpendicular polarization component relative to the polarization of excitation (green) and the simultaneously recorded intensity trajectory from the parallel polarization component relative to the polarization of excitation (red); the calculated anisotropy trajectory (blue) using eq 2 from the pair of polarization components (red and green). (B) Anisotropy distribution from the trajectory (blue) in A. The anisotropy distribution from the background (black) with mean of −0.020 and standard deviation of 0.054. It is evident that the signal anisotropy distribution (blue) is identifiable, beyond the background distribution.
Fluorescence lifetime can be determined independently from molecular rotation through the linear combination of eqs 3 and 4. Figure 4A shows the lifetime fluctuation trajectory and the corresponding polarization intensity trajectories from a portion of the photon stamping trajectories in Figure 2. Each data point in the lifetime trajectory is calculated from the consecutive 300 photons of the two time-resolved photon stamping polarization channels. Figure 4B shows the lifetime distribution of the fluctuation lifetime trajectory in Figure 4A. There are no evident multipeaks in the lifetime distribution as that in the anisotropy distribution. The lifetime background distribution (Figure 4B) shows the mean at 3.0 ± 0.6 ns, which is consistent with the lifetime of free resorufin molecules in pH 7.4 buffer. Compared to the lifetime of the background, the average lifetime of the nascent enzymatic reaction product, resorufin, is much longer than that of free resorufin molecules (Figure 4B). Since the lifetime of the resorufin is sensitive to the pH of the solution,54 the longer average lifetime of the confined resorufin is likely influenced by the local basicity of the active site. This is consistent with the fact that the hydrogen bond from amide oxygen of amino acid residue Asn70 to imidazole NH of amino acid residue His42 contributes to the basicity of the local environment.23 Since the florescence lifetime is sensitive to the fluctuation of the local environment and the movements of macromolecules,52,55−59 the lifetime fluctuations of the confined resorufin most likely reflect the fluctuations of the active-site conformation of the HRP enzyme as well as the enzyme–product molecular interactions. Figure 4 C1 shows the typical fluorescence decay behavior of the free resorufin molecules from point 1 at 678.01 s in Figure 4A. The fluorescence intensity of parallel polarization shows similar decay as that of the perpendicular polarization component, which is consistent with the rotation of the free resorufin molecules being faster than the fluorescence lifetime.60,61 Figures 4 C2 and 4 C3 have shown the fluorescence decay behaviors during the bursting from point 2 at 682.29 s and point 3 at 684.75 s, respectively, in the trajectory 4A. At point 2, the lifetime is shorter than that of the free resorufin molecules in buffer solution, and the shorter lifetime is likely due to the quenching effect from the heme group at the enzymatic active site, since there exists the energy transfer between the enzymatic product, resorufin, and the heme group.56,57,62,63 At point 3, the fluorescence decay shows a longer lifetime than that in point 2. The significant increase of the lifetime suggests a significant change of the enzyme–product molecular interaction at the active site change, presumably making the nascent product molecule more efficiently prevented by the Phe41 residue to access the heme iron,25 which decreases the energy transfer between the product and the heme group. At the enzymatic active site and at the particular time of point 3 the product most likely exists as the resorufin anion (R–) state since it is dramatically different in lifetime compared to that of the protonated form of resorufin (RH).54,64
Figure 4.

Single-molecule fluorescence lifetime fluctuation and distribution of single HRP-catalyzed amplex red fluorogenic assay. (A) Single-molecule fluorescence lifetime fluctuation trajectory. Intensity trajectory from the parallel polarization component relative to the polarization of excitation (red) and the simultaneously recorded intensity trajectory from the perpendicular polarization component relative to the polarization of excitation (green). Lifetime fluctuation trajectory (blue) calculated using eq 5 from the pair of polarization component trajectories (red and green). (B) Single-molecule enzymatic reaction product fluorescence lifetime distribution and the lifetime background distribution (black) from the trajectory (blue) in A. The lifetime background distribution is deduced from the lifetime trajectory from 678 to 681.5 s in (A), and the background distribution gives the mean of 3.01 ns and standard deviation of 0.60 ns. (C1), (C2), and (C3) Fluorescence decays at point 1 at 678.01 s, point 2 at 682.29 s, and point 3 at 684.75 s, respectively. The parallel polarization component decays (red), the perpendicular polarization component decays (green), and the fluorescence decays with fit (blue). The lifetimes at point 1, point 2, and point 3 are 2.9 ± 0.2 ns, 2.7 ± 0.1 ns, and 4.1 ± 0.1 ns, respectively.
Performing correlation analysis of the lifetime, anisotropy, and intensity on the single-molecule photon-by-photon trajectories allows us to characterize the complexity and heterogeneity of enzymatic conformational dynamics and mechanism.37,65,66 Figures 5 A1 and 5 B1 show the correlated distributions of intensity vs anisotropy and fluorescence lifetime vs anisotropy deduced from the trajectories shown in Figure 3 and Figure 4, respectively. Figures 5 A2 and 5 B2 show the correlated counter plots of Figures 5 A1 and 5 B1. There are distinctly three domains in the intensity–anisotropy 2D plot (Figure 5A2), while in the lifetime–anisotropy 2D plot (Figure 5 B2), there are two domains 3′ and 3″ separated at the high anisotropy value, except the same domains 1 and 2 as in Figure 5 A2. These domains reflect the different local environments of the nascent fluorescent enzymatic reaction product, resorufin, at the enzymatic active site. To assign the origin of each domain in the correlation distribution 2D plots, the rotational correlation times (τr) are needed as shown in eq 6.
Figure 5.

Correlation plots of the lifetime, anisotropy, and intensity on the single HRP-catalyzed amplex red fluorogenic assay. (A1) Correlation of fluorescence intensity and anisotropy and (B1) correlation of lifetime and anisotropy. For A1 and B1, the color (from cold color to warm color, i.e., from blue to red) of the data points represents the time sequence from the start to the end of the fluorogenic enzymatic turnover event. (A2) The correlation plots of fluorescence intensity and anisotropy with identified rotational correlation times. (B2) Correlation of lifetime and anisotropy with identified rotational correlation times.
In Figure 5 B2, the regions 1, 2, 3′, and 3″ are centered at (r, τf) = (0.01, 3.1 ns), (0.15, 3.7 ns), (0.24, 3.1 ns), and (0.26, 4.0 ns), respectively. On the basis of eq 6, the corresponding rotational correlation times, τr, of the regions 1, 2, 3′, and 3” are calculated to be 0.10, 3.30, 9.54, and 17.93 ns, respectively. The rotation correlation time of 0.10 ns from region 1 is consistent with that of the freely rotating resorufin in aqueous solution.61,67 Region 3″ has the longest rotation correlation time, which is close to the rotation correlation time of HRP in solution, 18.3 ns, since the τr is proportional to the molecular weight and increases about 1 ns for each 2400 Da increase in molecular weight.68 In region 3″, the resorufin is most likely bound with HRP tightly in the form of an enzyme–product complex, and there is essentially no measurable relative rotation between resorufin and HRP. In regions 2 and 3′, the calculated rotational correlation times are shorter than the rotational correlation time of HRP in the solution. The shorter rotation correlation time most likely comes from the less tight spatial confinement of the unbounded or loosely bound nascent resorufin at the HRP enzymatic active site. Therefore, the apparently different τr can be associated with the enzyme conformational dynamics associated with the enzyme–product interactions. Here, the relatively shorter rotation correlation time in region 2 can be ascribed to a relatively loose state of HRP, which makes more free space for the rotation of nascent resorufin. The relatively longer rotation correlation time in region 3′ could be ascribed to a relatively tight state of the HRP, which makes the rotation time of the resorufin molecule much closer to that of the enzyme HRP. In other words, when the confinement space of the active site becomes larger, the enzyme–product interaction is looser, and the product molecule has higher flexibility of movements, which constitutes a shorter τr, and vice versa. The assignment of loose state and tight state of HRP from the rotation correlation time of nascent resorufin molecules is also consistent with the quenching effect of the heme group to the florescence lifetime of resorufin.54,69,70 As in the loose states, the average distance from the nascent resorufin to the heme group is larger than that in tight states, which results in the decrease of the quenching effect from the heme and thus makes the lifetime longer than that in tight states. Furthermore, the local dielectric constant variation also influences the fluorescence lifetime of the enzymatic product that is embedded in the active site of the HRP enzyme. Because the average refractive index of proteins and amino acids is larger than that of water,71,72 the tight state of the conformation gives a larger local dielectric constant. The increase in local dielectric constant at the enzymatic active site induces a shorter lifetime since the spontaneous emission rate scales with the square of the refractive index based on a simple density-of-states argument.73−75 Therefore, we note that HRP has breathing-type conformation motions between the tight states and loose states of the enzyme active site under enzyme–product interactions before the nascent product molecule is released from the enzyme into the solution.
To further identify the conformational dynamics of the product releasing from the enzyme active site, we have analyzed the distributions of the fluorescence lifetime and anisotropy at the rising edges and falling edges of about 211 fluorescence bursting spikes from the trajectories shown in Figure 2. Here, the signal rising edge corresponds to the fluorescent product generated at a single-molecule reactant-to-product turnover, and the signal falling edge corresponds to the product releasing from the active site into the solution and diffusing away from the confocal volume. Figure 6 shows the distributions of the lifetimes and anisotropies of the products from the rising edges and the falling edges of each fluorescence spike. Comparing the lifetime distributions from the rising and falling edges of the fluorescence spikes (Figure 6 A1 and Figure 6 B1), we notice that the mean lifetime of the falling part is about 0.38 ns shorter than that of the rising part, while the anisotropy distributions are almost the same. On the basis of the influence of the quenching effect and local dielectric constant on the fluorescence lifetime of the nascent product as discussed above, the most possible explanation for the shorter lifetime of the single resorufin molecules at the falling edges is that the product molecules are buried in tight states compared to the rising edge.
Figure 6.

Single-molecule fluorescence lifetime and anisotropy distributions of the rising edges and falling edges of single HRP-catalyzed oxidation of amplex red reaction fluorescence turnover events. (A1) The lifetime distribution of rising edges with mean at 3.75 ns and standard deviation 1.26 ns. (A2) The anisotropy distribution of rising edges with mean at 0.17 and standard deviation 0.14 ns. (B1) The lifetime distribution of falling edges with mean at 3.37 ns and standard deviation 1.3. (B2) The anisotropy distribution of falling edges with mean at 0.15 and standard deviation 0.16.
Rotation correlation time of the enzymatic reaction product, resorufin, at the falling edges of the turnover fluorogenic signals is a sensitive parameter to reveal the enzyme–product interactions right at the product releasing events. Figure 7 shows the distribution of the rotation correlation time of the resorufin at the falling edges and the distribution peaks at 2 ns that are close to the rotation correlation time of the loose enzyme–product states in region 2 of Figure 5B2, and these product events are most likely related to the enzyme active site opening up to release the fluorescent product into the solution. Nevertheless, about 20% of the product releasing events are associated with the rotation correlation time larger than 9 ns, which suggests that the product releasing events occurred at a strong enzyme–product interaction so that the fluorescent product rotational motions are at the protein motion time scale. Furthermore, the fluorescence lifetime of the product releasing events is also associated with shorter fluorescence lifetime, as we have discussed above, which also suggests a tight enzyme–substrate interaction at the events of product releasing. The correlated shorter lifetime and longer rotation correlation time of the products at the falling edges support our attribution that there is a significant pathway in which the product molecules are spilled out from the enzymatic active site, driven by a squeezing effect by the breath-type motions between loose and tight active-site conformation states; i.e., there is a significant portion of product molecules that are released from a tight active-site conformational state rather than from an open active-site conformational state. This most remarkable phenomenon provides a new insight into the enzymatic reaction dynamics and mechanism—specifically on the product releasing mechanism and dynamics.
Figure 7.
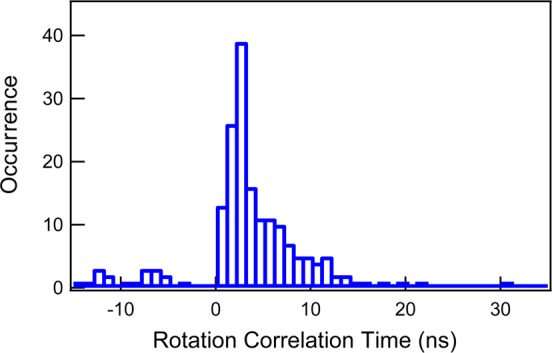
Distribution of rotation correlation time of the product at the signal falling edges that correspond to the moments of releasing the product molecules from the enzyme into the solution.
It is well-known that enzymatic reactions involve multiple kinetic steps, and each step involves complex molecular interactions, inhomogeneous conformational changes, and fluctuations of confined local environment.6−9 Both the overall dynamics and mechanism of the enzymatic reaction have been well investigated in ensemble-averaged experiments about the HRP enzyme, although there are different fluorogenic enzymatic reaction mechanisms proposed in the recent literature (see Supporting Information).23−26,30,85−89 Here, the catalytic networks of the HRP-catalyzed amplex red have been further characterized by our single-molecule fluorogenic assay. Rotational correlation times of the product during the releasing process have shown the enzyme undergoing breathing-type conformational motions fluctuating between loose states and the tight states. On the basis of the crystal structure of HRP, these breathing-type configurational motions may come from the active-site segment motion as shown in Figure 8A. As the enzyme changes to the loose states, the water-soluble substrate is facilitated to transport into the active site. Conformation fluctuations between the loose states and tight states occur during the products releasing. Nevertheless, we propose a new mechanism of the HRP enzymatic reaction (Figure 8B): There are multiple conformations involved in the enzymatic reaction enzyme–substrate and enzyme–product interactions as well as the product releasing from the active site, and the conformational fluctuations occur in consecutive and parallel pathways in the mechanism with both the active-site open releasing product pathway and the active-site close spilling product pathway.7 Overall, the active-site close spilling product pathway reported in this work is the most significant new knowledge for the HRP enzymatic reaction dynamics and mechanism. We anticipated that the “spilling” product releasing pathway widely existed in other enzymatic reaction product releasings, especially for the enzyme active-site associated with large conformational motions in enzymatic reaction turnovers.
Figure 8.

Proposed dynamics of single HRP-catalyzed oxidation of amplex red reaction. (A) The breathing motions of the enzymatic reaction active site. (B) The schematic of the two pathways of product release. Our experimental observation suggests that about 20% product releasing is involved in the spilling pathways.
The enzymatic reaction rate could be influenced by different factors, such as temperature, pH, enzyme concentration, substrate concentration, local environment, and the presence of any inhibitors or activators.1,3 Beyond the ensemble-averaged reaction kinetic scope, the enzymatic reaction turnover rate fluctuation for a single enzyme is often observed, which is characterized as the dynamic disorder.2,6,34,76−81 In our experiment, only the slow product releasing events, giving more than 600 detected photons, were used for analyzing the lifetimes, anisotropy, and rotational correlation times of the rising edges and the falling edges. The detected turnover rate in our single-molecule fluorogenic enzymatic assay is typically about 0.2 events/s. Compared to the turnover rate of the conventional enzymatic reaction assay at mM or μM substrate concentrations,9,30,82 our measured turnover rates are slow, mostly due to the low concentration of 200 nM substrate used in our experiments and also due to that only the high fluorescence photon counts events were detected and analyzed. We used the low concentration for two reasons: (1) This low concentration of substrate effectively controls the background increasing from laser-induced and autocatalytically enhanced photo-oxidation of amplex red to resorufin, and (2) in this work, we focus on studying the enzymatic reaction product releasing event analysis but not the enzymatic activity or enzymatic reaction rates; therefore, isolated single events are ideal for our detailed analysis of lifetime, anisotropy, and rotational correlation time for both the rising edge and falling edge of the fluorogenic turnover event. Nevertheless, the substrate concentration is the dominate factor in the diffusion-controlled reaction processes, and we further note that we cannot completely rule out the effect of the local steric hindrance around the tethered enzyme,83 which may play a role in the influence of the turnover rate. The local steric hindrance can affect the motions of the enzymes, both the substrate accessing to the active site and product releasing from the active site may be slowed, although the statistics of analyzing hundreds of single fluorogenic turnover events should essentially remove the significance of the steric hindrance effect for our examination on the fluorescence lifetime, anisotropy, and rotational correlation time distributions.
One of the highest aims of studying enzymology is to obtain a fundamental understanding of why the enzymes are capable of catalyzing a biochemical reaction by millions or even billions of times.84,85 Probing and characterizing the enzymatic reaction active site has been one of the central efforts in modern enzymology. In this work, we focus on resolving the product releasing pathway analysis as it is one of the critical processes in an enzymatic reaction, as described by the Michaelis–Menten mechanism; however, the significance of being able to probe the nascent fluorescent product formation at an enzymatic reaction site is much more profound and beyond just illuminating the product releasing pathways because the nascent fluorescent product molecule at the active site serve as an ideal and sensitive probe at the fluctuations and distributions of the chemical bonding configuration, the local static electric field, local force field, solvation local environment, and the fluctuation dynamics of the enzymatic reaction transition state.8,10,12,86−99 Clearly, there are significant challenges for single-molecule spectroscopy to probe the details of these important physical properties at the active site and at the exact time of enzymatic reaction of forming the product, and the recent developments of high time-resolved and spatial resolved single-molecule imaging and spectroscopy with the multiple physical parameter selectivity and sensitivity are definitely promising for carrying out these high efforts.34,36,37,100−120
Conclusions
We have used an enzymatic reaction, horseradish peroxidase-catalyzed oxidation of amplex red, as a model system to carry out single-molecule enzymatic conformational dynamics studies. Using our combined and correlated single-molecule fluorescence intensity, anisotropy, and lifetime measurements, we are able to identify the specific pathways with distinctive specifications on the conformational dynamics. Broad distributions of florescence intensity, anisotropy, and lifetime beyond their standard deviation values identify the presence of intermediate conformational states associated with the product releasing step. Rotation correlation time and correlation between the florescence lifetime and anisotropy confirm the existence of a tightly confined product–enzyme complex prior to the releasing step. The resorufin anion state is responsible for the increased lifetime value of resorufin confined in the enzyme’s active site. The lifetime fluctuations in the active site result from conformational fluctuations of the resorufin–enzyme complex between loose and tight states. Although only the enzyme–product complexes are investigated in this experiment, the conclusion could also be used to understand the conformational dynamics of enzymes during catalysis. All the conformation fluctuations between the loose states and tight states happen before the product releasing. The lifetime and the rotation correlation times of the products at the falling edges have shown that there is a significant pathway during which the product molecules are spilled out from the enzymatic active site, driven by a squeezing effect; i.e., a significant portion of product molecules are released from a tight active-site conformational state beside an open active-site conformational state. This most interesting phenomenon provides a new insight into the enzymatic reaction dynamics and mechanism, and the information is uniquely obtainable from our combined time-resolved single-molecule spectroscopic measurements and analyses. This multiple conformations occur in consecutive and parallel in the mechanism, which would help to understand the rugged multidimensional standard free energy.
Acknowledgments
This work is supported by NIH/NIGMS and Ohio Eminent Scholar endowment. We thank Yufan He for his stimulating discussions on sample preparation and data analysis.
Supporting Information Available
Additional information about the catalytic mechanism of horseradish peroxidase with hydrogen peroxide and the crystal structure of the active site; movie about single-molecule fluorogenic turnovers from the tethered HRP enzyme; complementary fluorogenic assay results of tethered single-molecule HRP enzymes. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Segel I.Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems; John Wiley & Sons: New York, 1993. [Google Scholar]
- English B. P.; Min W.; van Oijen A. M.; Lee K. T.; Luo G.; Sun H.; Cherayil B. J.; Kou S.; Xie X. S. Ever-fluctuating single enzyme molecules: Michaelis-Menten equation revisited. Nat. Chem. Biol. 2005, 2287–94. [DOI] [PubMed] [Google Scholar]
- Cornish-Bowden A.Fundamentals of enzyme kinetics; John Wiley & Sons: New York, 2013. [Google Scholar]
- Boehr D. D.; Dyson H. J.; Wright P. E. An NMR perspective on enzyme dynamics. Chem. Rev. 2006, 10683055–3079. [DOI] [PubMed] [Google Scholar]
- Yon J.; Perahia D.; Ghelis C. Conformational dynamics and enzyme activity. Biochimie 1998, 80133–42. [DOI] [PubMed] [Google Scholar]
- Lu H. P. Probing single-molecule protein conformational dynamics. Acc. Chem. Res. 2005, 387557–565. [DOI] [PubMed] [Google Scholar]
- Benkovic S. J.; Hammes G. G.; Hammes-Schiffer S. Free-Energy Landscape of Enzyme Catalysis. Biochemistry 2008, 47113317–3321. [DOI] [PubMed] [Google Scholar]
- Garcia-Viloca M.; Gao J.; Karplus M.; Truhlar D. G. How enzymes work: analysis by modern rate theory and computer simulations. Science 2004, 3035655186–195. [DOI] [PubMed] [Google Scholar]
- Edman L.; Földes-Papp Z.; Wennmalm S.; Rigler R. The fluctuating enzyme: a single molecule approach. Chem. Phys. 1999, 247111–22. [Google Scholar]
- Gao J.; Ma S.; Major D. T.; Nam K.; Pu J.; Truhlar D. G. Mechanisms and free energies of enzymatic reactions. Chem. Rev. 2006, 10683188–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Thai V.; Lei M.; Ott M.; Wolf-Watz M.; Fenn T. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 4507171838–844. [DOI] [PubMed] [Google Scholar]
- Bruice T. C. Computational approaches: reaction trajectories, structures, and atomic motions. Enzyme reactions and proficiency. Chem. Rev. 2006, 10683119–3139. [DOI] [PubMed] [Google Scholar]
- Blank K.; De Cremer G.; Hofkens J. Fluorescence-based analysis of enzymes at the single-molecule level. Biotechnol. J. 2009, 44465–479. [DOI] [PubMed] [Google Scholar]
- Rachofsky E. L.; Osman R.; Ross J. A. Probing structure and dynamics of DNA with 2-aminopurine: effects of local environment on fluorescence. Biochemistry 2001, 404946–956. [DOI] [PubMed] [Google Scholar]
- Viseu T.; Hungerford G.; Coelho A.; Ferreira M. Dye-host interactions for local effects recognition in homogeneous and nanostructured media. J. Phys. Chem. B 2003, 1074813300–13312. [Google Scholar]
- Schröder G. F.; Alexiev U.; Grubmüller H. Simulation of fluorescence anisotropy experiments: probing protein dynamics. Biophys. J. 2005, 8963757–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista M. R.; Martínez L. Dynamics of Nuclear Receptor Helix-12 Switch of Transcription Activation by Modeling Time-Resolved Fluorescence Anisotropy Decays. Biophys. J. 2013, 10571670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallée R. A.; Van Der Auweraer M.; De Schryver F. C.; Beljonne D.; Orrit M. A microscopic model for the fluctuations of local field and spontaneous emission of single molecules in disordered media. ChemPhysChem 2005, 6181–91. [DOI] [PubMed] [Google Scholar]
- Alcala J. R.; Gratton E.; Prendergast F. Fluorescence lifetime distributions in proteins. Biophys. J. 1987, 514597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivian J. T.; Callis P. R. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001, 8052093–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callis P. R.; Liu T. Quantitative prediction of fluorescence quantum yields for tryptophan in proteins. J. Phys. Chem. B 2004, 108144248–4259. [Google Scholar]
- Liu R.; Hu D.; Tan X.; Lu H. P. Revealing two-state protein-protein interactions of calmodulin by single-molecule spectroscopy. J. Am. Chem. Soc. 2006, 1283110034–10042. [DOI] [PubMed] [Google Scholar]
- Veitch N. C. Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry 2004, 653249–259. [DOI] [PubMed] [Google Scholar]
- Berglund G. I.; Carlsson G. H.; Smith A. T.; Szöke H.; Henriksen A.; Hajdu J. The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002, 4176887463–468. [DOI] [PubMed] [Google Scholar]
- Carlsson G. H.; Nicholls P.; Svistunenko D.; Berglund G. I.; Hajdu J. Complexes of horseradish peroxidase with formate, acetate, and carbon monoxide. Biochemistry 2005, 442635–642. [DOI] [PubMed] [Google Scholar]
- Veitch N. C.; Smith A. T. Horseradish peroxidase. Adv. Inorg. Chem. 2000, 51, 107–162. [Google Scholar]
- Hassler K.; Rigler P.; Blom H.; Rigler R.; Widengren J.; Lasser T. Dynamic disorder in horseradish peroxidase observed with total internal reflection fluorescence correlation spectroscopy. Opt. Express 2007, 1595366–5375. [DOI] [PubMed] [Google Scholar]
- Edman L.; Rigler R. Memory landscapes of single-enzyme molecules. Proc. Natl. Acad. Sci. U.S.A. 2000, 97158266–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas-Aragonès M.; Engelkamp H.; Claessen V. I.; Sommerdijk N. A.; Rowan A. E.; Christianen P. C.; Maan J. C.; Verduin B. J.; Cornelissen J. J.; Nolte R. J. A virus-based single-enzyme nanoreactor. Nat. Nanotechnol. 2007, 210635–639. [DOI] [PubMed] [Google Scholar]
- Gorris H. H.; Walt D. R. Mechanistic aspects of horseradish peroxidase elucidated through single-molecule studies. J. Am. Chem. Soc. 2009, 131176277–6282. [DOI] [PubMed] [Google Scholar]
- Luong A. K.; Gradinaru C. C.; Chandler D. W.; Hayden C. C. Simultaneous time-and wavelength-resolved fluorescence microscopy of single molecules. J. Phys. Chem. B 2005, 1093315691–15698. [DOI] [PubMed] [Google Scholar]
- Michalet X.; Kapanidis A. N.; Laurence T.; Pinaud F.; Doose S.; Pflughoefft M.; Weiss S. The power and prospects of fluorescence microscopies and spectroscopies. Annu. Rev. Biophys. Biomol. Struct. 2003, 321161–182. [DOI] [PubMed] [Google Scholar]
- Novotny L. Single molecule fluorescence in inhomogeneous environments. Appl. Phys. Lett. 1996, 69253806–3808. [Google Scholar]
- Lu H. P.; Xie X. S. Single-molecule spectral fluctuations at room temperature. Nature 1997, 3856612143–146. [Google Scholar]
- Wazawa T.; Ishii Y.; Funatsu T.; Yanagida T. Spectral fluctuation of a single fluorophore conjugated to a protein molecule. Biophys. J. 2000, 7831561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell P.; Berger S.; Kensch O.; Felekyan S.; Antonik M.; Wöhrl B.; Restle T.; Goody R.; Seidel C. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase: primer/template complexes. Proc. Natl. Acad. Sci. U.S.A. 2003, 10041655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling C.; Berger S.; Brand L.; Fries J.; Schaffer J.; Volkmer A.; Seidel C. Data registration and selective single-molecule analysis using multi-parameter fluorescence detection. J. Biotechnol. 2001, 863163–180. [DOI] [PubMed] [Google Scholar]
- Zheng D.; Kaldaras L.; Lu H. P. Total internal reflection fluorescence microscopy imaging-guided confocal single-molecule fluorescence spectroscopy. Rev. Sci. Instrum. 2012, 831013110–013110–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D.; Lu H. P. Single-molecule nanosecond anisotropy dynamics of tethered protein motions. J. Phys. Chem. B 2003, 1072618–626. [Google Scholar]
- Lu H. P. Revealing time bunching effect in single-molecule enzyme conformational dynamics. Phys. Chem. Chem. Phys. 2011, 13156734–6749. [DOI] [PubMed] [Google Scholar]
- Cross A. J.; Fleming G. R. Analysis of time-resolved fluorescence anisotropy decays. Biophys. J. 1984, 46145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahlmann K.; Hell S. Electric field depolarization in high aperture focusing with emphasis on annular apertures. J. Microsc. 2000, 200159–67. [DOI] [PubMed] [Google Scholar]
- Hall P.; Selinger B. Better estimates of exponential decay parameters. J. Phys. Chem. 1981, 85202941–2946. [Google Scholar]
- Zander C.; Sauer M.; Drexhage K.; Ko D.-S.; Schulz A.; Wolfrum J.; Brand L.; Eggeling C.; Seidel C. Detection and characterization of single molecules in aqueous solution. Appl. Phys. B: Laser Opt. 1996, 635517–523. [Google Scholar]
- Bajzer Ž.; Therneau T. M.; Sharp J. C.; Prendergast F. G. Maximum likelihood method for the analysis of time-resolved fluorescence decay curves. Eur. Biophys. J. 1991, 205247–262. [DOI] [PubMed] [Google Scholar]
- Barber P.; Ameer-Beg S.; Pathmananthan S.; Rowley M.; Coolen A. A Bayesian method for single molecule, fluorescence burst analysis. Biomed. Opt. Express 2010, 141148–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G.-H.; Legresley S. E.; Snyder N.; Aubry P. D.; Antonik M. Single-Molecule Analysis And Lifetime Estimates Of Heterogeneous Low-Count-Rate Time-Correlated Fluorescence Data. Appl. Spectrosc. 2011, 659981–990. [DOI] [PubMed] [Google Scholar]
- Horng M.-L.; Gardecki J.; Maroncelli M. Rotational dynamics of coumarin 153: Time-dependent friction, dielectric friction, and other nonhydrodynamic effects. J. Phys. Chem. A 1997, 10161030–1047. [Google Scholar]
- Gustavsson T.; Cassara L.; Marguet S.; Gurzadyan G.; Van der Meulen P.; Pommeret S.; Mialocq J.-C. Rotational diffusion of the 7-diethylamino-4-methylcoumarin C1 dye molecule in polar protic and aprotic solvents. Photochem. Photobiol. Sci. 2003, 23329–341. [DOI] [PubMed] [Google Scholar]
- Nie S.; Chiu D. T.; Zare R. N. Probing individual molecules with confocal fluorescence microscopy. Science 1994, 1018–1018. [DOI] [PubMed] [Google Scholar]
- Jones G.; Jackson W. R.; Halpern A. M. Medium effects on fluorescence quantum yields and lifetimes for coumarin laser dyes. Chem. Phys. Lett. 1980, 722391–395. [Google Scholar]
- Vallée R.; Van der Auweraer M.; Paul W.; Binder K. Fluorescence lifetime of a single molecule as an observable of meta-basin dynamics in fluids near the glass transition. Phys. Rev. Lett. 2006, 9721217801. [DOI] [PubMed] [Google Scholar]
- Braeken E.; De Cremer G.; Marsal P.; Pèpe G.; Müllen K.; Vallée R. A. Single molecule probing of the local segmental relaxation dynamics in polymer above the glass transition temperature. J. Am. Chem. Soc. 2009, 1313412201–12210. [DOI] [PubMed] [Google Scholar]
- Ryder A. G.; Power S.; Glynn T. J. Fluorescence lifetime based pH sensing using Resorufin. Proc. SPIE 2003, 4876, 827–835. [Google Scholar]
- Flamigni L.; Venuti E.; Camaioni N.; Barigelletti F. A spectroscopic investigation of the temperature and solvent sensitivities of resorufin. J. Chem. Soc., Faraday Trans. 2 1989, 85121935–1943. [Google Scholar]
- Ghosh D. K.; Ray K.; Rogers A. J.; Nahm N. J.; Salerno J. C. FMN fluorescence in inducible NOS constructs reveals a series of conformational states involved in the reductase catalytic cycle. FEBS J. 2012, 27971306–1317. [DOI] [PubMed] [Google Scholar]
- Salerno J. C.; Ray K.; Poulos T.; Li H.; Ghosh D. K. Calmodulin activates neuronal nitric oxide synthase by enabling transitions between conformational states. FEBS Lett. 2012, 587144–47. [DOI] [PubMed] [Google Scholar]
- Eftink M. R.; Ghiron C. A. Fluorescence quenching studies with proteins. Anal. Biochem. 1981, 1142199–227. [DOI] [PubMed] [Google Scholar]
- Vallée R.; Tomczak N.; Kuipers L.; Vancso G.; Van Hulst N. Single molecule lifetime fluctuations reveal segmental dynamics in polymers. Phys. Rev. Lett. 2003, 913038301. [DOI] [PubMed] [Google Scholar]
- Templeton E. F. G.; Quitevis E. L.; Kenney-Wallace G. A. Picosecond reorientational dynamics of resorufin: correlations of dynamics and liquid structure. J. Phys. Chem. 1985, 89153238–3243. [Google Scholar]
- Kurnikova M.; Balabai N.; Waldeck D.; Coalson R. Rotational relaxation in polar solvents. Molecular dynamics study of solute-solvent interaction. J. Am. Chem. Soc. 1998, 120246121–6130. [Google Scholar]
- Ladokhin A. S. Fluorescence spectroscopy in peptide and protein analysis. Encyclopedia of analytical chemistry; John Wiley & Sons Ltd.: Chichester, 2000; pp 5762–5779. [Google Scholar]
- Fan C.; Plaxco K. W.; Heeger A. J. High-efficiency fluorescence quenching of conjugated polymers by proteins. J. Am. Chem. Soc. 2002, 124205642–5643. [DOI] [PubMed] [Google Scholar]
- Thompson R. B.; Lakowicz J. R. Fiber optic pH sensor based on phase fluorescence lifetimes. Anal. Chem. 1993, 657853–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widengren J.; Kudryavtsev V.; Antonik M.; Berger S.; Gerken M.; Seidel C. A. Single-molecule detection and identification of multiple species by multiparameter fluorescence detection. Anal. Chem. 2006, 7862039–2050. [DOI] [PubMed] [Google Scholar]
- Sisamakis E.; Valeri A.; Kalinin S.; Rothwell P. J.; Seidel C. A. Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 2010, 475, 455–514. [DOI] [PubMed] [Google Scholar]
- Hay C. E.; Marken F.; Blanchard G. Effects of Electrolyte Concentration on the Rotational Dynamics of Resorufin. J. Phys. Chem. A 2010, 1144912875–12880. [DOI] [PubMed] [Google Scholar]
- Brunet J. E.; Vargas V.; Gratton E.; Jameson D. M. Hydrodynamics of horseradish peroxidase revealed by global analysis of multiple fluorescence probes. Biophys. J. 1994, 662446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T. K.; Mazumdar S. Conformational substates of apoprotein of horseradish peroxidase in aqueous solution: a fluorescence dynamics study. J. Phys. Chem. 1995, 993513283–13290. [Google Scholar]
- Thompson R. B.; Lakowicz J. R. Fiber optic pH sensor based on phase fluorescence lifetimes. Anal. Chem. 1993, 657853–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMeekin T. L.; Groves M. L.; Hipp N. J.. Refractive indices of amino acids, proteins, and related substances. In Amino Acids and Serum Proteins; American Chemical Society: Washington DC, 1964; pp 54–66. [Google Scholar]
- Zhao H.; Brown P. H.; Schuck P. On the distribution of protein refractive index increments. Biophys. J. 2011, 10092309–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magde D.; Wong R.; Seybold P. G. Fluorescence Quantum Yields and Their Relation to Lifetimes of Rhodamine 6G and Fluorescein in Nine Solvents: Improved Absolute Standards for Quantum Yields. Photochem. Photobiol. 2002, 754327–334. [DOI] [PubMed] [Google Scholar]
- Magde D.; Rojas G. E.; Seybold P. G. Solvent dependence of the fluorescence lifetimes of xanthene dyes. Photochem. Photobiol. 1999, 705737–744. [Google Scholar]
- Strickler S.; Berg R. A. Relationship between absorption intensity and fluorescence lifetime of molecules. J. Chem. Phys. 1962, 37, 814–822. [Google Scholar]
- Lu H. P.; Xun L.; Xie X. S. Single-molecule enzymatic dynamics. Science 1998, 28253951877–1882. [DOI] [PubMed] [Google Scholar]
- Kou S.; Cherayil B. J.; Min W.; English B. P.; Xie X. S. Single-molecule michaelis-menten equations. J. Phys. Chem. B 2005, 1094119068–19081. [DOI] [PubMed] [Google Scholar]
- Claessen V. I.; Engelkamp H.; Christianen P. C.; Maan J. C.; Nolte R. J.; Blank K.; Rowan A. E. Single-biomolecule kinetics: the art of studying a single enzyme. Annu. Rev. Anal. Chem. 2010, 3, 319–340. [DOI] [PubMed] [Google Scholar]
- Zwanzig R. Rate processes with dynamical disorder. Acc. Chem. Res. 1990, 235148–152. [Google Scholar]
- Karplus M. Aspects of protein reaction dynamics: Deviations from simple behavior. J. Phys. Chem. B 2000, 104111–27. [Google Scholar]
- Lu H. P. Single-molecule spectroscopy studies of conformational change dynamics in enzymatic reactions. Curr. Pharm. Biotechnol. 2004, 53261–269. [DOI] [PubMed] [Google Scholar]
- Agresti J. J.; Antipov E.; Abate A. R.; Ahn K.; Rowat A. C.; Baret J.-C.; Marquez M.; Klibanov A. M.; Griffiths A. D.; Weitz D. A. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl. Acad. Sci. U.S.A. 2010, 10794004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrup S. H.; Erickson H. P. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc. Natl. Acad. Sci. U.S.A. 1992, 8983338–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer T.Kinetics of single-substrate enzyme-catalyzed reactions. In Understanding enzymes; Prentice Hall/Ellis Horwood: Hertfordshire, 1995; pp 107–127. [Google Scholar]
- Purich D. L.Enzyme Kinetics and Mechanism, Part F: Detection and Characterization of Enzyme Reaction Intermediates: Methods in Enzymology; Academic Press: New York, 2002; Vol. 354. [Google Scholar]
- Onuchic J. N.; Wolynes P. G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004, 14170–75. [DOI] [PubMed] [Google Scholar]
- Bryngelson J. D.; Onuchic J. N.; Socci N. D.; Wolynes P. G. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins: Struct., Funct., Bioinf. 1995, 213167–195. [DOI] [PubMed] [Google Scholar]
- Washel A.; Levitt M. Theoretical studies of enzymatic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [DOI] [PubMed] [Google Scholar]
- Washel A.Computer modeling of chemical reactions in enzymes and solution; John Wiley and Sons: New York, 1989. [Google Scholar]
- Jia S. H.; Li Y.; Parodo J.; Kapus A.; Fan L.; Rotstein O. D.; Marshall J. C. Pre–B cell colony–enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J. Clin. Invest. 2004, 11391318–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q.; Wang J. Kinetics and statistical distributions of single-molecule conformational dynamics. J. Phys. Chem. B 2009, 11351517–1521. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Liu H.; Yang W. Free energy calculation on enzyme reactions with an efficient iterative procedure to determine minimum energy paths on a combined ab initio QM/MM potential energy surface. J. Chem. Phys. 2000, 11283483–3492. [Google Scholar]
- Gilmartin M. A.; Hart J. P. Sensing with chemically and biologically modified carbon electrodes. A review. Analyst 1995, 12041029–1045. [DOI] [PubMed] [Google Scholar]
- Whitford P. C.; Onuchic J. N.; Wolynes P. G. Energy landscape along an enzymatic reaction trajectory: hinges or cracks?. HFSP J. 2008, 2261–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Thai V.; Lei M.; Ott M.; Wolf-Watz M.; Fenn T.; Pozharski E.; Wilson M. A.; Petsko G. A.; Karplus M. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 4507171838–844. [DOI] [PubMed] [Google Scholar]
- Burner U.; Obinger C. Transient-state and steady-state kinetics of the oxidation of aliphatic and aromatic thiols by horseradish peroxidase. FEBS Lett. 1997, 4112269–274. [DOI] [PubMed] [Google Scholar]
- Gao J.; Truhlar D. G. Quantum mechanical methods for enzyme kinetics. Annu. Rev. Phys. Chem. 2002, 531467–505. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Chu X.; Longhi S.; Roche P.; Han W.; Wang E.; Wang J. Multiscaled exploration of coupled folding and binding of an intrinsically disordered molecular recognition element in measles virus nucleoprotein. Proc. Natl. Acad. Sci. U.S.A. 2013, 11040E3743–E3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Oliveira R. J.; Chu X.; Whitford P. C.; Chahine J.; Han W.; Wang E.; Onuchic J. N.; Leite V. B. Topography of funneled landscapes determines the thermodynamics and kinetics of protein folding. Proc. Natl. Acad. Sci. U.S.A. 2012, 1093915763–15768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühnemuth R.; Seidel C. A. Principles of single molecule multiparameter fluorescence spectroscopy. Single Mol. 2001, 24251–254. [Google Scholar]
- Eggeling C.; Widengren J.; Brand L.; Schaffer J.; Felekyan S.; Seidel C. A. Analysis of photobleaching in single-molecule multicolor excitation and Förster resonance energy transfer measurements. J. Phys. Chem. A 2006, 11092979–2995. [DOI] [PubMed] [Google Scholar]
- Moerner W.; Fromm D. P. Methods of single-molecule fluorescence spectroscopy and microscopy. Rev. Sci. Instrum. 2003, 7483597–3619. [Google Scholar]
- Kinkhabwala A.; Yu Z.; Fan S.; Avlasevich Y.; Müllen K.; Moerner W. Large single-molecule fluorescence enhancements produced by a bowtie nanoantenna. Nat. Photonics 2009, 311654–657. [Google Scholar]
- Tamarat P.; Maali A.; Lounis B.; Orrit M. Ten years of single-molecule spectroscopy. J. Phys. Chem. A 2000, 10411–16. [DOI] [PubMed] [Google Scholar]
- Kulzer F.; Orrit M. Single-molecule optics. Annu. Rev. Phys. Chem. 2004, 55, 585–611. [DOI] [PubMed] [Google Scholar]
- Ha T.; Enderle T.; Chemla D.; Selvin P.; Weiss S. Single molecule dynamics studied by polarization modulation. Phys. Rev. Lett. 1996, 77193979–3982. [DOI] [PubMed] [Google Scholar]
- Rasnik I.; McKinney S. A.; Ha T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat. Methods 2006, 311891–893. [DOI] [PubMed] [Google Scholar]
- Selvin P. R.; Ha T.. Single-molecule techniques; CSHL Press: Long Island, New York,2008.
- Wu S.; Ho W. Two-photon-induced hot-electron transfer to a single molecule in a scanning tunneling microscope. Phys. Rev. B 2010, 828085444. [Google Scholar]
- Ho W. Single-molecule chemistry. J. Chem. Phys. 2002, 1172411033–11061. [Google Scholar]
- Yang H.; Xie X. S. Probing single-molecule dynamics photon by photon. J. Chem. Phys. 2002, 1172410965–10979. [Google Scholar]
- Cang H.; Wong C. M.; Xu C. S.; Rizvi A. H.; Yang H. Confocal three dimensional tracking of a single nanoparticle with concurrent spectroscopic readouts. Appl. Phys. Lett. 2006, 8822223901. [Google Scholar]
- Perkins T. T. Optical traps for single molecule biophysics: a primer. Laser Photon. Rev. 2009, 31–2203–220. [Google Scholar]
- Lu H. P. Sizing up single-molecule enzymatic conformational dynamics. Chem. Soc. Rev. 2014, 4341118–1143. [DOI] [PubMed] [Google Scholar]
- Chong S.; Min W.; Xie X. S. Ground-state depletion microscopy: detection sensitivity of single-molecule optical absorption at room temperature. J. Phys. Chem. Lett. 2010, 1233316–3322. [Google Scholar]
- Kruithof M.; Chien F.-T.; Routh A.; Logie C.; Rhodes D.; van Noort J. Single-molecule force spectroscopy reveals a highly compliant helical folding for the 30-nm chromatin fiber. Nat. Struct. Mol. Biol. 2009, 165534–540. [DOI] [PubMed] [Google Scholar]
- Min W.; Freudiger C. W.; Lu S.; Xie X. S. Coherent nonlinear optical imaging: beyond fluorescence microscopy. Annu. Rev. Phys. Chem. 2011, 62, 507–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celebrano M.; Kukura P.; Renn A.; Sandoghdar V. Single-molecule imaging by optical absorption. Nat. Photon 2011, 5295–98. [Google Scholar]
- Donhauser Z.; Mantooth B.; Kelly K.; Bumm L.; Monnell J.; Stapleton J.; Price D.; Rawlett A.; Allara D.; Tour J. Conductance switching in single molecules through conformational changes. Science 2001, 29255252303–2307. [DOI] [PubMed] [Google Scholar]
- Brinks D.; Hildner R.; van Dijk E. M.; Stefani F. D.; Nieder J. B.; Hernando J.; van Hulst N. F. Ultrafast dynamics of single molecules. Chem. Soc. Rev. 2014, 4382476–2491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.