

Abstract
Rheumatoid arthritis (RA) is a chronic autoinflammatory disease that affects 1-2% of the world population and is characterized by widespread joint inflammation. IL-1 is an important mediator of cartilage destruction in rheumatic diseases1, but our understanding of the upstream mechanisms leading to IL-1β production in rheumatoid arthritis is limited by the absence of suitable RA mouse models in which inflammasomes contribute to pathology. Myeloid-cell-specific deletion of the RA-susceptibility gene A20/TNFAIP3 in mice (A20myel-KO mice) triggers a spontaneous erosive polyarthritis that resembles RA in patients2. Notably, RA in A20myel-KO mice was not rescued by tumor necrosis factor receptor 1 (TNF-R1) deletion, but we showed it to crucially rely on interleukin-1 receptor (IL-1R) signaling. Macrophages lacking A20 had increased basal and LPS-induced expression levels of the inflammasome adaptor Nlrp3 and proIL-1β. As a result, A20-deficiency in macrophages significantly enhanced Nlrp3 inflammasome-mediated caspase-1 activation, pyroptosis and IL-1β secretion by soluble and crystalline Nlrp3 stimuli. In contrast, activation of the Nlrc4 and AIM2 inflammasomes was not altered. Importantly, increased Nlrp3 inflammasome activation contributed to RA pathology in vivo, because deletion of Nlrp3 and caspase-1 markedly protected against RA-associated inflammation and cartilage destruction in A20myel-KO mice. These results reveal A20 as a novel negative regulator of Nlrp3 inflammasome activation, and describe A20myel-KO mice as the first experimental model to study the role of inflammasomes in RA pathology.
Keywords: inflammasome, Nlrp3, A20, caspase-1, IL1R1
A20 was deleted in myeloid cells by crossing A20flox/flox mice into Lysosyme M (LysM)-Cre recombinase-expressing mice. Unlike wild-type macrophages, A20myel-KO macrophages failed to induce A20 mRNA and protein expression in response to the TLR4 ligand LPS (Fig. 1a, b), demonstrating the effectiveness of LysM-driven deletion of A20 in myeloid cells. Arthritis development in A20myel-KO mice was shown to be TNFR1-independent, whereas deletion of MyD88 markedly protected against RA pathology2. As this signaling adaptor operates downstream of both TLRs and IL-1R, we crossed IL1R1−/− mice into A20myel-KO mice to assess the contribution of IL-1 signaling to arthritis pathogenesis. As expected, wild-type mice (A20flox/floxIL1R1+/+) did not develop arthritis, whereas A20myel-KO mice spontaneously developed an arthritic phenotype (Fig. 1c). The incidence of A20myel-KO mice developing arthritis was 100% (Fig. 1d). In sharp contrast, A20myel-KOIL1R1−/− mice were virtually devoid of clinical signs of arthritis (Fig. 1c, d). In agreement, clinical scoring of disease severity confirmed A20myel-KOIL1R1+/+ mice to develop severe arthritic disease (high clinical score) whereas A20myel-KOIL1R1−/−mice were markedly protected (clinical score 0). A20myel-KO littermates that were heterozygous for IL1R1 expression showed an intermediate arthritic phenotype between those of A20myel-KOIL1R1+/+ and A20myel-KOIL1R1−/− mice (Fig. 1d, e). These clinical assessments were supported by a histological examination of representative ankle joints. Hematoxylin and eosin-stained tissue sections of diseased A20myel-KOIL1R1+/− mice showed significant synovial and periarticular inflammation and high levels of infiltrated mononuclear cells, which was associated with extensive cartilage and bone destruction (Fig. 1f). In marked contrast, ankle joints of A20myel-KOIL1R1−/− littermates were strongly protected from arthritic histopathology and contained significantly reduced numbers of infiltrating inflammatory cells (Fig. 1f). Semi-quantitative scoring of these histological parameters confirmed that the severity of arthritis was substantially lower in A20myel-KOIL1R1−/− mice relative to A20myel-KOIL1R1+/− littermates (Fig. 1g and Extended Data Table 1). These results demonstrate that IL-1 production is detrimental for arthritis pathogenesis in mice with a myeloid cell-restricted deletion in A20.
Figure 1. Il1r1-deficiency rescues the arthritis phenotype of A20myel-KO mice.

a, b, A20 and β-actin mRNA (a) and protein (b) levels of LPS-stimulated BMDMs. c, Hind paws of 20 weeks old mice. d, e, A20fl/flIL1R1+/+ (n=19), A20myel-KOIL1R1+/+ (n=8), A20myel-KOIL1R1+/− (n=15) and A20myel-KOIL1R1−/− (n=9) mice aged 21-30 weeks were clinically scored for arthritis incidence (d) and severity (e). f, Ankle joints sections stained with haematoxylin and eosin; magnification: 40× (top) and 100x (bottom). g, Histological scores of ankle sections of A20myel-KOIL1R1+/− (n=10) and A20myel-KOIL1R1−/− (n=8). P-values in e and g were determined by Student’s t-test.
Macrophages are a prime source of proIL-1β, and generally depend on caspase-1 for maturation and secretion of the biologically active cytokine. Caspase-1 is produced as a cytosolic zymogen, the activation of which is controlled by different inflammasomes3. To study the role of A20 in inflammasome signaling, we assessed caspase-1 processing in bone marrow-derived macrophages (BMDMs) of wild-type and A20myel-KO mice. Notably, caspase-1 activation was substantially increased in LPS-primed A20myel-KO macrophages that were treated with soluble (ATP and nigericin) or crystalline (silica) stimuli of the Nlrp3 inflammasome compared to wild-type BMDMs (Fig. 2a). Concurrently, the levels of secreted IL-1β in the culture medium of ATP- and nigericin-treated A20myel-KO macrophages were about twice those of wild-type cells, and silica triggered nearly four times higher levels of secreted IL-1β (Fig. 2b). Enhanced caspase-1 autoprocessing (Fig. 2c, d) and IL-1β secretion (Fig. 2e, f) by the Nlrp3 inflammasome was evident within 10 minutes after ATP or nigericin addition, and continued to increase in a time-dependent fashion. Similarly, the induction of caspase-1-dependent pyroptosis was enhanced in A20myel-KO macrophages (Fig. 2g, h). Despite their hypersensitivity for Nlrp3 inflammasome activation, A20myel-KO macrophages failed to process caspase-1, and secrete IL-1β and IL-18 when treated with LPS, ATP or nigericin alone (Extended Data figure 1). The increased responsiveness of A20myel-KO macrophages towards inflammasome activation was restricted to the Nlrp3 inflammasome because caspase-1 processing and pyroptotic cell death by the Nlrc4 inflammasome were similarly induced in wild-type and A20myel-KO macrophages that had been infected with Salmonella Typhimurium (S. Typhimurium) (Fig. 2i, j). Similarly, stimulation of the AIM2 inflammasome with cytosolic dsDNA did not result in differential levels of caspase-1 processing and pyroptosis induction in wild-type and A20myel-KO macrophages (Fig. 2k, l). It is worth noting, however, that despite normal caspase-1 activation and pyroptosis levels in response to S. Typhimurium infection and dsDNA transfection, secretion of IL-1β in response to these treatments was consistently higher in A20myel-KO macrophages compared to wild-type controls (Fig. 2m, n), which could be explained by increased induction of proIL-1β mRNA in A20myel-KO macrophages (data not shown). Together, these results demonstrate that A20 negatively regulates activation of caspase-1 by the Nlrp3 inflammasome, but not by the Nlrc4 and AIM2 inflammasomes.
Figure 2. Hyperactivation of the Nlrp3, but not the Nlrc4 and AIM2 inflammasomes, in A20-deficient macrophages.

a-h, BMDMs were stimulated as described in Methods. Lysates were immunoblotted for caspase-1 (a, c, d) and supernatants analyzed for IL-1β (b, e, f) and LDH (g, h). i-n, BMDMs were treated as described in Methods. Lysates were immunoblotted for caspase-1 (i, k) and supernatants analyzed for LDH (j, l) and IL-1β (m, n). Black arrow, procaspase-1; white arrow, p20. Data represent mean ± s.d. of 1 out of 3 biological replicates, with 3 technical replicates each (*P < 0.05; ***P < 0.001; Student’s t-test).
Activation of the Nlrp3 inflammasome in wild-type macrophages is tightly regulated at different levels. A priming signal (referred to as step 1 and usually provided by TLRs) upregulates Nlrp3 expression levels along with the inflammasome substrate proIL-1β via the pro-inflammatory transcription factor NF-κB4. A20 negatively regulates LPS-induced NF-κB activation (refs.5-8 and Extended Data figure 2a), which also was reflected in increased secretion of the NF-κB-dependent cytokines IL-6 and TNF in A20myel-KO macrophages (Extended Data figure 2b, c). We further showed A20-deficiency to markedly enhance LPS-induced mRNA expression levels of Nlrp3 (Fig. 3a) and proIL-1β (Fig. 3b). In contrast, LPS-induced transcript levels of caspase-1 and the inflammasome adaptor ASC were respectively mildly upregulated and normal in A20myel-KO macrophages (Extended Data figure 3a, b). Analysis of protein expression levels confirmed Nlrp3 and proIL-1β to be significantly higher in LPS-primed A20myel-KO macrophages compared to wild-type cells, whereas caspase-1 and ASC were not differentially modulated in the two genotypes (Fig. 3c).
Figure 3. A20 inhibits Nlrp3 inflammasome priming.

a, b, Nlrp3 (a) and proIL-1β (b) mRNA levels of LPS-treated BMDMs. c, Expression of the indicated proteins in BMDMs 6 h after LPS treatment. d, e, Rapid Nlrp3 inflammasome activation as described in Methods. Expression of the indicated proteins (d), and secreted cytokines (e) were determined. f-i, A20myel-KO BMDMs were treated as indicated. Nlrp3 mRNA levels (f), caspase-1 expression (g), secreted IL-1β (h) and LDH activity (i) were determined. Black arrow, procaspase-1; white arrow, p20. Data represent mean ± s.d. of 1 out of 3 biological replicates, with 3 technical replicates each (*P < 0.05; ***P < 0.001; Student’s t-test).
TLR stimulation during brief time intervals (10 minutes or less) was recently shown to license activation of the Nlrp3 inflammasome independently of new protein synthesis9-12. Rapid Nlrp3 inflammasome activation resulted in procaspase-1 processing and secretion of pre-synthesized proIL-18 in the absence of the NF-κB dependent cytokines IL-1β, TNF and IL-69-11. To address whether A20 modulated rapid Nlrp3 inflammasome activation, cells were exposed to ATP or nigericin after being primed with LPS for 10 minutes. As reported9-12, wild-type BMDMs activated caspase-1 (Fig. 3d) and secreted significant amounts of IL-18, but not IL-1β, TNF or IL-6 (Fig. 3e). Moreover, we noted Nlrp3 protein levels were lowered after stimulation in both wild-type and A20myel-KO macrophages (Fig. 3d), supporting the notion that acute Nlrp3 inflammasome activation occurred independently of LPS-induced new protein synthesis. Both caspase-1 processing and IL-18 secretion were markedly increased in A20myel-KO macrophages in the absence of substantial IL-1β, TNF and IL-6 secretion (Fig. 3d, e). This was likely due to increased basal expression of Nlrp3 and proIL-18 in these cells (Fig. 3a, d). In agreement, basal mRNA levels of Nlrp3, proIL-1β and proIL-18 were increased in untreated A20myel-KO macrophages (Fig. 3a, b and Extended Data figure 3c). Together, these results suggest that A20 negatively regulates Nlrp3 inflammasome signaling by suppressing NF-κB-dependent production of Nlrp3 and the inflammasome substrates proIL-1β and proIL-18. In agreement, the pharmacological IKK inhibitor BMS-345541 significantly reduced Nlrp3 levels in LPS-primed A20myel-KO macrophages (Fig. 3f). Moreover, both BMS-345541 and the selective IKK2 inhibitor TCPA-1 significantly reduced ATP- and nigericin-induced caspase-1 autoprocessing, IL-1β secretion and pyroptosis induction in LPS-primed A20myel-KO macrophages (Fig. 3g-i).
Having established A20 as a negative regulator of Nlrp3 inflammasome activation, we hypothesized that excessive Nlrp3 activation might drive RA pathology in A20-deficient mice upstream of IL1R1. To test this hypothesis, Nlrp3−/− mice were crossed into A20myel-KO mice and the levels of 4 RA-relevant cytokines (IL-1α, IL-1β, IL-6 and TNF) were monitored. Although IL-1α levels were not significantly different in A20-sufficient and A20myel-KO mice (Extended Data figure 4a), the latter mice had significantly higher levels of IL-1β in circulation (Fig. 4a). In addition, the levels of IL-6 and TNF were also significantly higher in A20myel-KO mice compared to A20flox/flox littermates (Extended Data figure 4b, c). Notably, deletion of Nlrp3 in A20myel-KO mice markedly reduced IL-1β secretion to baseline levels of A20flox/flox mice, thereby demonstrating that the Nlrp3 inflammasome contributes critically to excessive IL-1β production in A20myel-KO mice in vivo (Fig. 4a). Intriguingly, A20myel-KONlrp3−/− mice also were protected from excessive IL-6 production, suggesting that high IL-6 levels are consequent to excessive inflammasome-mediated IL-1β production (Extended Data figure 4b). In contrast, TNF production was not significantly affected in A20myel-KONlrp3−/− and A20myel-KOCasp1−/− mice (Extended Data figure 4c). In agreement, TNF-R1 signaling was previously shown to be dispensable for RA pathology in A20myel-KO mice, whereas IL-6 neutralization provided protection2.
Figure 4. Nlrp3 and caspase-1 deletion rescues arthritis in A20myel-KO mice.

a, IL1β serum levels of A20fl/flNlrp3+/+ (n=10), A20myel-KONlrp3+/+ (n=10) and A20myel-KONlrp3−/− (n=10) mice aged 20-35 weeks. b, Hind paws of 30 weeks old mice. c, d, A20myel-KONlrp3+/+ (n = 20) and A20myel-KONlrp3−/−(n=17) mice were scored for arthritis incidence (d) and severity (e). e, Representative ankle joints sections; magnification: 40× (top) and 100x (bottom). f, Histological scores of ankle sections of A20myel-KONlrp3+/+ (n=5) and A20myel-KONlrp3−/− (n=5) mice. g, micro-CT images of hind paws of A20myel-KONlrp3+/+ and A20myel-KONlrp3−/− mice. Solid arrow, bone erosion; empty arrow, intact cartilage and bone. h, IL-1β serum levels of A20fl/flCasp1/11+/+ (n=11), A20myel-KOCasp1/11+/+ (n=26) and A20myel-KOCasp1/11−/− (n=16) mice aged 20-35 weeks. i, Hind paws of 25 weeks old mice. j, k, 15-30 weeks old A20myel-KOCasp1/11+/+ (n=12), A20myel-KOCasp1/11+/−(n=15) and A20myel-KOCasp1/11−/− (n=24) mice were clinically scored for arthritis incidence (j) and severity (k). l, Representative ankle joints sections; magnification: 40× (top) and 100x (bottom). P-values were determined by Student’s t-test (a, d, f, k) and Mann-Whitney test (h).
Based on these findings, we assessed the contribution of Nlrp3 to RA pathogenesis in A20myel-KO mice. Swelling and redness of the hind paws of A20myel-KONlrp3+/+ mice became evident around 11 weeks of age (Fig. 4b), and had afflicted all animals of this genotype when they were 20 weeks old (Fig. 4c). Disease severity continued to progress, and became increasingly pronounced in A20myel-KONlrp3+/+ mice of 21-40 weeks old (Fig. 4d). In contrast, A20myel-KONlrp3−/−mice of similar age were markedly protected from RA, and their hind paws had a normal appearance and lacked clinical signs of RA pathology (Fig. 4b-d). Histological analysis of the ankle joints of these mice showed significantly reduced synovial and periarticular inflammation, and substantially less infiltrated mononuclear cells compared to tissue sections of A20myel-KONlrp3+/+ mice of comparable age (Fig. 4e, f and Extended Data Table 2). In agreement, three-dimensional micro-CT imaging showed that the extent of bone erosion in hind paws of representative A20myel-KONlrp3+/+ and A20myel-KONlrp3−/− mice was markedly different. Unlike in Nlrp3-deficient A20myel-KO mice, hind paws of Nlrp3-sufficient mice exhibited severe loss of bone density in the metatarsal region (Fig. 4g), demonstrating a key role for Nlrp3 in RA pathology. We also analyzed the impact of caspase-1/11-deficiency on IL-1β secretion and RA pathology in A20myel-KO mice. IL-1β levels in circulation were significantly reduced in A20myel-KOCasp1/11−/−mice compared to A20myel-KOCasp1/11+/+ and A20flox/flox mice (Fig. 4h). As in A20myel-KONlrp3−/−mice, serum levels of IL-6 were markedly reduced in A20myel-KOCasp1/11−/− mice, whereas TNF production was not significantly different compared to A20myel-KOCasp1/11+/+ mice (Extended Data figure 4). Moreover, hind paws of all analyzed A20myel-KOCasp1/11+/+ mice were clearly inflamed and swollen (Fig. 4i-k). In contrast, 50% of A20myel-KOCasp1/11−/− mice were devoid of clinical signs of arthritis, and disease symptoms in the remaining A20myel-KOCasp1/11−/− mice were very mild (Fig. 4i-k). In agreement, analysis of heamatoxylin & eosin-stained joint sections of arthritic A20myel-KOCasp1/11+/− mice showed massive mononuclear cell infiltration associated with marked articular, periarticular and synovial inflammation and reactive fibrosis. In contrast, joints of A20myel-KOCasp1/11−/− mice were markedly protected from RA histopathology (Fig. 4l).
Collectively, these results demonstrate that A20 puts a brake on Nlrp3 inflammasome activation by reducing basal and LPS-induced Nlrp3 expression levels (Extended Data Fig. 5). Moreover, we showed that excessive Nlrp3 inflammasome activation drives arthritis pathogenesis in A20myel-KO mice. Arthritis in humans is a complex disease that may be caused by different combinations of genetic and environmental triggers. As such, available RA mouse models may each be relevant to distinct subsets of RA patients, or suitable to study certain aspects of RA pathogenesis. Pathology in the widely used collagen- and albumin-induced mouse arthritis models occurs independently of inflammasome signaling13,14. In contrast, we demonstrated here that arthritis pathology in A20myel-KO mice critically relies on the Nlrp3 inflammasome/IL-1 signaling axis. Because both A20/Tnfaip315-18 and Nlrp319-21 are RA-susceptibility genes, this suggests that A20myel-KO mice might be a suitable pre-clinical model for validating the effectiveness of experimental RA therapies targeting inflammasomes and/or IL-1 signaling.
Methods
Mice
A20myel-KO mice with a lysosome M-Cre-targeted deletion of A20 in myeloid cells were described2. Nlrp3−/−22 and Casp1−/−23 mice have been described, and were kindly provided by Dr. V. Dixit (Genentech) and Dr. R. Flavell (Yale University). IL1R1−/−24 mice were purchased from Jackson Laboratories and bred in-house. In vivo experiments were controlled with age- and sex-matched littermates. The sample size was chosen so as to validate statistical analyses. Mice were housed in individually ventilated cages and kept under pathogen-free conditions at animal facilities of Ghent University. All animal experiments were conducted with permission of the Ethical committees on laboratory animal welfare of Ghent University.
Macrophage differentiation and stimulation
Bone marrow-derived macrophages (BMDMs) were generated by culturing bone marrow cells in IMDM containing 10% heat-inactivated FBS, 30% L cell-conditioned medium, 100 U ml−1 penicillin, and 100 mg ml−1 streptomycin at 37°C in a humidified atmosphere containing 5% CO2. 6 days later, cells were collected and seeded in 12-well plates in IMDM containing 10% heat-inactivated FBS and 1% non-essential amino acids in the presence of antibiotics. The next day BMDMs were either left untreated or treated with 5 μg ml−1 ultrapure LPS from Salmonella minnesota (Invivogen) for 3 h followed by 5 mM ATP (Roche) or 20 μM nigericin (Sigma-Aldrich) for 30 min or 0.5 mg ml−1 Silica (Min-U-Sil 5, US Silica) for 6 h. BMDMs were also separately infected with Salmonella enterica serovar Typhimurium at the indicated multiplicity of infection (MOI) for 3 h, the last hour of which in the presence of gentamycin (50 μg ml−1). To activate the AIM2 inflammasome, BMDMs were left untreated or treated with 5 μg ml−1 LPS for 3 h. After removal of LPS, BMDMs were exposed to Lipofectamine 2000 (mock, Life Technologies) or Lipofectamine with 1.25 ug ml−1 pCDNA3 plasmid DNA for 19 h. To induce rapid Nlrp3 inflammasome activation, BMDMs were stimulated with LPS for 10 min, and then treated with ATP or nigericin for 45 min. For inhibition of NF-κB, A20myel-KO BMDMs were preincubated for 30 min with 1.25 or 2.5 μM of the selective IKK inhibitors BMS-34554125 or TPCA-126 (Sigma-Aldrich) before stimulation with LPS.
Western blotting
Cell lysates and culture supernatants were incubated with cell lysis buffer (20 mM Tris HCl pH 7.4, 200 mM NaCl, 1% NP-40) and denatured in laemmli buffer. Subsequently the protein samples were boiled at 95°C for 10 min and separated by SDS-PAGE. Separated proteins were transferred to PVDF membranes. Blocking, incubation with antibody and washing of the membrane were done in PBS supplemented with 0.05% Tween-20 (v/v) and 3% (w/v) non-fat dry milk. Immunoblots were incubated overnight with primary antibodies against caspase-1 (AG-20B-0042-C100, Adipogen), Nlrp3 (AG-20B-0014-C100, Adipogen), ASC (AG-25B-0006, Adipogen), IL-1β (GTX74034, Genetex), IL-18 (5180R-100, Biovision), IkBα (9242S, Cell Signaling), Phospho-IkBα (2859S, Ser32) (Cell Signaling), β-actin (NB600-501H, Novus Biologicals) and A20 (sc-166692, Santa Cruz Biotechnology). Horseradish peroxidase-conjugated goat anti-mouse (115-035-146, Jackson Immunoresearch Laboratories) or anti-rabbit secondary antibody (111-035-144, Jackson Immunoresearch Laboratories) was used to detect proteins by enhanced chemiluminescence (Thermo Scientific).
Cytokine analysis and LDH measurement
Cytokine levels in culture medium and serum were determined by magnetic bead-based multiplex assay using Luminex technology (Bio-Rad) and IL1β ELISA (R&D Systems), according to the manufacturers’ instructions. Cell death levels were determined by LDH assay in culture medium according to the manufacturer’s instructions (Promega).
RT-PCR
Total RNA was isolated from BMDMs using RNeasy kit (Qiagen). cDNA was synthesized using the QuantiTect Reverse Transcription Kit (Qiagen). A20 mRNA expression in wild-type and A20myel-KO BMDMs, treated with 5 μg ml−1 LPS for 6 h, was determined by RT-PCR using the following primers: 5′-CTCGGAACTTTAAATTCCGC-3′ and 5′-GGGTAAGTTAGCTTCATCC-3′. To analyse mRNA levels of Nlrp3, proIL-1β, ASC and caspase-1, wild-type and A20myel-KO BMDMs were left untreated or treated with 5 μg ml−1 LPS for 3 h and Quantitative RT-PCR (qRT-PCR) was performed using LightCycler 480 SYBR Green I Master Mix Kit (Roche Applied Science) in a LightCycler 480 real-time PCR machine (Roche Applied Science). The cycling conditions were 95°C for 5 min followed by 45 cycles of 95°C for 10 s, 60°C for 15 s, and 72°C for 20 s. Gene expression levels were normalized to GAPDH. qRT-PCR Sense/antisense primers used: Mouse GAPDH, 5′-GGTGAAGGTCGGTGTGAACG-3′ and 5′-CTCGCTCCTGGAAGATGGTG-3′; Mouse Nlrp3, 5′-ATTACCCGCCCGAGAAAGG-3′ and 5′-TCGCAGCAAAGATCCACACAG-3′; Mouse proIL-1β, 5′-TGGGCCTCAAAGGAAAGA-3′ and 5′-GGTGCTGATGTACCAGTT-3′; Mouse ASC, 5′-CTTGTCAGGGGATGAACTCAAAA-3′ and 5′-GCCATACGACTCCAGATAGTAGC-3′; Mouse Casp1, 5′-ACAAGGCACGGGACCTATG-3′ and 5′-TCCCAGTCAGTCCTGGAAATG-3′. Quantitative analysis of mouse proIL-18 mRNA was performed using Taqman gene expression assay (Mm00434225_m1, Applied Biosystems).
Clinical scoring
Mice were randomly scored in a blinded fashion for development of peripheral arthritis once a week and scores were subsequently linked to genotypes. A score ranging from 0 to 3 was assigned to each paw, with 0 being normal, 0.5 being swelling of one or more toes, 1 being mild swelling of the wrist and/or ankle or carpus and/or tarsus, 2 being moderate swelling of the wrist and/or ankle or carpus and/or tarsus or mild swelling of both, and 3 being severe swelling of the entire paw.
Micro-CT Imaging
Micro-CT micrographs of paws fixed in formalin were made using an ex vivo micro-CT scanner (LocusSP Specimen CT, GE Healthcare) at 28-μm isotropic voxel size, with 720 projections, an integration time of 1,700 msec, photon energy of 80 keV, and a current of 70 μA.
Histology and histological scoring
Murine paws were dissected, fixed in 4% formaldehyde, decalcified in 5% formic acid until bones were pliable. Paraffin sections were stained with H&E for evaluation of inflammation and bone erosions. Histological sections were evaluated by two blinded assessors, and scores were determined by combining assessment of 2 parameters, calcaneal erosion and inflammation at the synovio-entheseal complex, each ranging from 0 (normal) to 3. (erosion: 0 none, 1 minimal, 2 intermediate, 3 into bone marrow; Inflammation: 0 none, 1: minimal 1-2 cell layers, 2: 2-5 cell layers, 3: >5 cell layers).
Statistics
GraphPad Prism 5.0 software was used for data analysis. Data are shown as mean with standard deviation (s.d.). Data were compared by an unpaired 2-tailed Student’s t-test when values followed a Gaussian distribution, or with the non-parametric Mann-Whitney U test. P < 0.05 was considered to indicate statistical significance.
Supplementary Material
a-c, Wild-type and A20myel-KO BMDMs were stimulated with 5 μg ml−1 LPS for 3 h, treated with 5 mM ATP or 20 μM nigericin for 60 min, or stimulated with 5 μg ml−1 LPS for 3 h and then treated with 5 mM ATP or 20 μM nigericin for 60 min. Cell extracts were immunoblotted for caspase-1 (a) and culture supernatants were analyzed for secretion of IL-1β (b) and IL-18 (c). Black arrows on Western blots denote procaspase-1 (p45) and white arrows denote the processed p20 subunit (p20). Elisa data are shown as mean ± s.d. of 1 out of 3 biological replicates, with 3 technical replicates each (***P < 0.001; Student’s t-test).
{kind=link}
a, Wild-type and A20myel-KO BMDMs were incubated with 5 μg ml−1 LPS for the indicated durations before cell extracts were prepared and immunoblotted with the indicated antibodies. b,c, Wild-type and A20myel-KO BMDMs were stimulated with 5 μg ml−1 LPS for 3 h, treated with 5 mM ATP or 20 μM nigericin for 60 min, or stimulated with 5 μg ml−1 LPS for 3 h and then treated with 5 mM ATP or 20 μM nigericin for 60 min. Culture supernatants were analyzed for IL-6 secretion (b) and TNF secretion (c). ELISA data are shown as mean ± s.d. of 1 out of 3 biological replicates, with 3 technical replicates each (***P < 0.001; Student’s t-test).
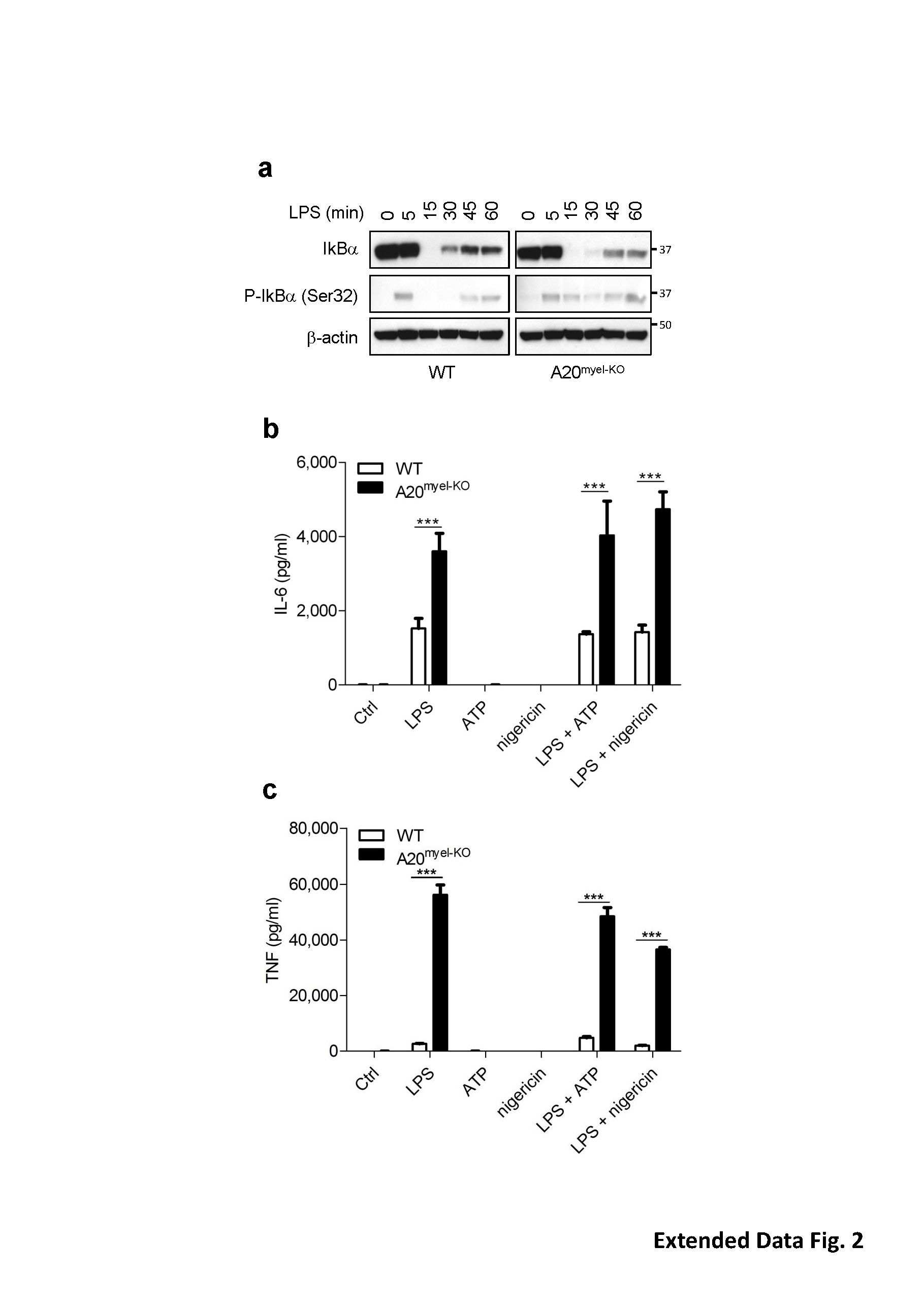{kind=link}
a-c, Wild-type and A20myel-KO BMDMs were stimulated with 5 μg ml−1 LPS for 3 h before mRNA levels of caspase-1 (a), ASC (b) and proIL-18 (c) were analyzed by qRT-PCR. Data are shown as mean ± s.d. of 1 out of 3 biological replicates, with 3 technical replicates each (*P < 0.05; ***P < 0.001; Student’s t-test).
{kind=link}
a-c, Levels of IL-1α (a), IL-6 (b) and TNF (c) in serum of A20fl/fl (n=10), A20myel-KO (n=10), A20myel-KOCASP1/11−/− (n=12) and A20myel-KONlrp3−/− (n=9) mice between 20 and 35 weeks of age. P-values were determined by Mann-Whitney (b) and Student’s t-test (c).
{kind=link}
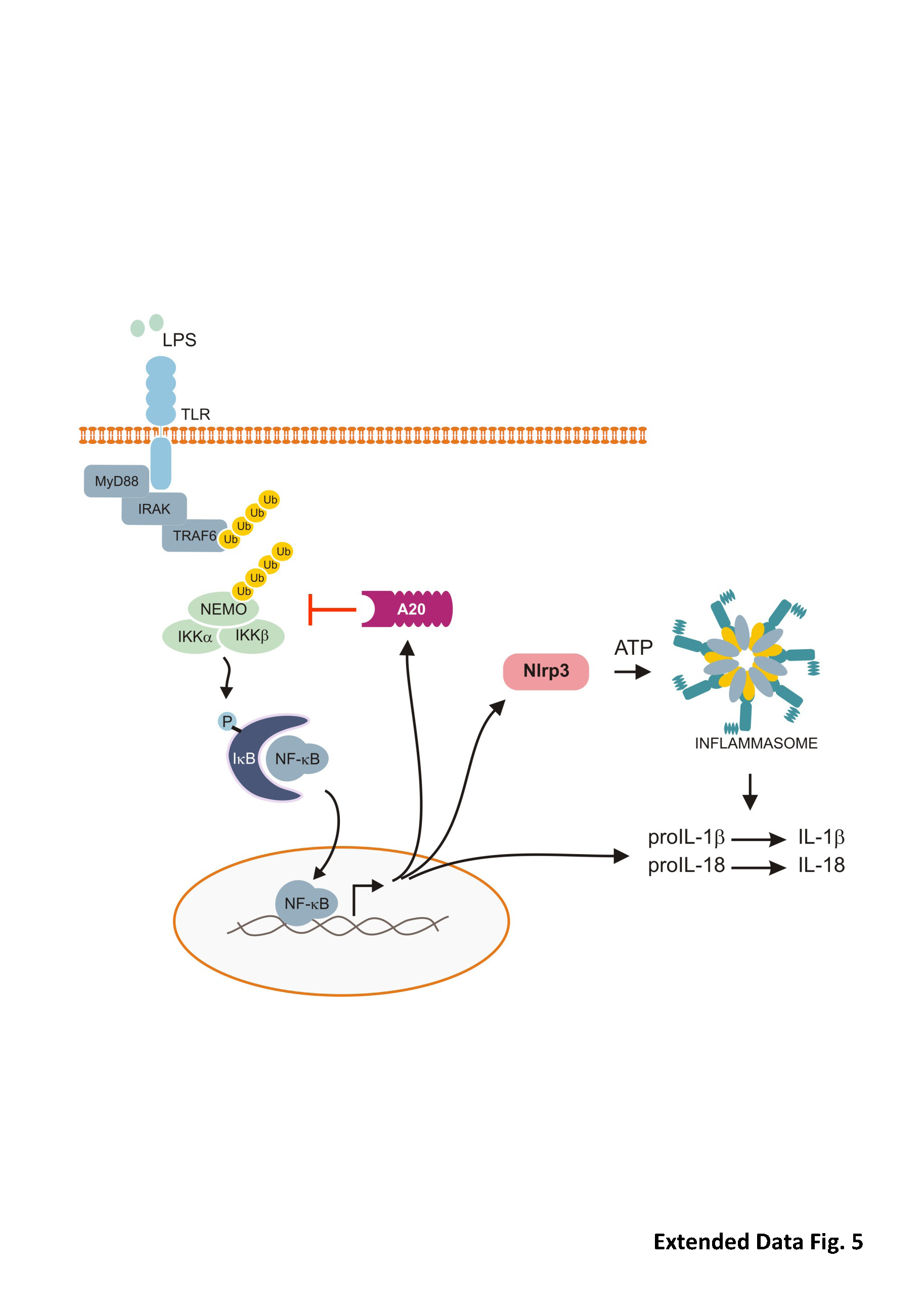
The NLR member Nlrp3, the inflammatory cytokine proIL-1β and the ubiquitin-editing enzyme A20 are expressed at low levels in resting macrophages. Binding of the TLR4 ligand LPS to its receptor triggers phosphorylation and rapid degradation of IκBα, allowing translocation of NF-κB to the nucleus, and NF-κB-mediated upregulation of proIL-1β, proIL-18, Nlrp3 and A20. A20 prevents excessive Nlrp3 inflammasome activation by dampening basal and LPS-induced NF-κB-mediated upregulation of Nlrp3. As such, A20 reduces the pool of Nlrp3 that is available for inflammasome assembly. In addition, it limits the levels of the inflammasome substrates proIL1β and proIL-18.
{kind=link}
Raw data of histological scores of ankle sections of A20myel-KOIL1R1+/− (n=10) and A20myel-KOIL1R1−/− mice (n=8). Arthritic histopathology of ankles was scored on 2 parameters, calcaneal erosion and inflammation at the synovio–entheseal complex, as described in the Methods section.
{kind=link}
Raw data of histological scores of ankle sections of A20myel-KONlrp3+/+ (n=5) and A20myel-KONlrp3−/− mice (n=5). Arthritic histopathology of ankles was scored on 2 parameters, calcaneal erosion and inflammation at the synovio–entheseal complex, as described in the Methods section.
{kind=link}
Acknowledgements
We thank Richard Flavell (Yale University) and Vishva Dixit (Genentech) for generous supply of mutant mice. LVW is a postdoctoral fellow with the Fund for Scientific Research-Flanders. This work was supported by the Ghent University Concerted Research Actions (grant BOF14/GOA/013) and grants from the European Research Council (Grant 281600) and the Fund for Scientific Research (FWO)-Flanders (grants G030212N, 1.2.201.10.N.00 and 1.5.122.11.N.00) to M.L., and by FWO research grants (Odysseus-G091908, G061910N and G016812N) and the Ghent University ‘Group-ID MRP’ to G.V.L., R.B. and D.E. T-D.K is supported by grants from the National Institute of Health (Grants AR056296, CA163507 and AI101935) and the American Lebanese Syrian Associated Charities (ALSAC).
Abbreviations
- RA
rheumatoid arthritis
- TNFAIP3
Tumor necrosis factor, alpha-induced protein 3
- caspase
cysteinyl aspartate-specific protease
- NLR
NOD-like receptor
- IL1R1
Interleukin 1 receptor, type I
- wt
wild-type
Footnotes
The authors declare no competing financial interests.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Burger D, Dayer JM, Palmer G, Gabay C. Is IL-1 a good therapeutic target in the treatment of arthritis? Best Pract Res Clin Rheumatol. 2006;20:879–896. doi: 10.1016/j.berh.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Matmati M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet. 2011;43:908–912. doi: 10.1038/ng.874. [DOI] [PubMed] [Google Scholar]
- 3.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 4.Bauernfeind FG, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 6.Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009;30:383–391. doi: 10.1016/j.it.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 9.Fernandes-Alnemri T, et al. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol. 2013;191:3995–3999. doi: 10.4049/jimmunol.1301681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juliana C, et al. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin KM, et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. 2014;111:775–780. doi: 10.1073/pnas.1320294111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schroder K, et al. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology. 2012;217:1325–1329. doi: 10.1016/j.imbio.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 13.Ippagunta SK, et al. Inflammasome-independent role of apoptosis-associated speck-like protein containing a CARD (ASC) in T cell priming is critical for collagen-induced arthritis. J Biol Chem. 2010;285:12454–12462. doi: 10.1074/jbc.M109.093252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolly L, et al. Inflammatory role of ASC in antigen-induced arthritis is independent of caspase-1, NALP-3, and IPAF. J Immunol. 2009;183:4003–4012. doi: 10.4049/jimmunol.0802173. [DOI] [PubMed] [Google Scholar]
- 15.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dieguez-Gonzalez R, et al. Analysis of TNFAIP3, a feedback inhibitor of nuclear factor-kappaB and the neighbor intergenic 6q23 region in rheumatoid arthritis susceptibility. Arthritis Res Ther. 2009;11:R42. doi: 10.1186/ar2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plenge RM, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomson W, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ben Hamad M, et al. Association study of CARD8 (p.C10X) and NLRP3 (p.Q705K) variants with rheumatoid arthritis in French and Tunisian populations. Int J Immunogenet. 2012;39:131–136. doi: 10.1111/j.1744-313X.2011.01070.x. [DOI] [PubMed] [Google Scholar]
- 20.Kastbom A, et al. Genetic variation in proteins of the cryopyrin inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project) Rheumatology. 2008;47:415–417. doi: 10.1093/rheumatology/kem372. [DOI] [PubMed] [Google Scholar]
- 21.Mathews RJ, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2013-203276. [DOI] [PubMed] [Google Scholar]
- 22.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 23.Schott WH, et al. Caspase-1 is not required for type 1 diabetes in the NOD mouse. Diabetes. 2004;53:99–104. doi: 10.2337/diabetes.53.1.99. [DOI] [PubMed] [Google Scholar]
- 24.Glaccum MB, et al. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol. 1997;159:3364–3371. [PubMed] [Google Scholar]
- 25.Burke JR, et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 26.Podolin PL, et al. Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IkappaB Kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell Proliferation. J Pharmacol Exp Ther. 2005;312:373–381. doi: 10.1124/jpet.104.074484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a-c, Wild-type and A20myel-KO BMDMs were stimulated with 5 μg ml−1 LPS for 3 h, treated with 5 mM ATP or 20 μM nigericin for 60 min, or stimulated with 5 μg ml−1 LPS for 3 h and then treated with 5 mM ATP or 20 μM nigericin for 60 min. Cell extracts were immunoblotted for caspase-1 (a) and culture supernatants were analyzed for secretion of IL-1β (b) and IL-18 (c). Black arrows on Western blots denote procaspase-1 (p45) and white arrows denote the processed p20 subunit (p20). Elisa data are shown as mean ± s.d. of 1 out of 3 biological replicates, with 3 technical replicates each (***P < 0.001; Student’s t-test).
a, Wild-type and A20myel-KO BMDMs were incubated with 5 μg ml−1 LPS for the indicated durations before cell extracts were prepared and immunoblotted with the indicated antibodies. b,c, Wild-type and A20myel-KO BMDMs were stimulated with 5 μg ml−1 LPS for 3 h, treated with 5 mM ATP or 20 μM nigericin for 60 min, or stimulated with 5 μg ml−1 LPS for 3 h and then treated with 5 mM ATP or 20 μM nigericin for 60 min. Culture supernatants were analyzed for IL-6 secretion (b) and TNF secretion (c). ELISA data are shown as mean ± s.d. of 1 out of 3 biological replicates, with 3 technical replicates each (***P < 0.001; Student’s t-test).
a-c, Wild-type and A20myel-KO BMDMs were stimulated with 5 μg ml−1 LPS for 3 h before mRNA levels of caspase-1 (a), ASC (b) and proIL-18 (c) were analyzed by qRT-PCR. Data are shown as mean ± s.d. of 1 out of 3 biological replicates, with 3 technical replicates each (*P < 0.05; ***P < 0.001; Student’s t-test).
a-c, Levels of IL-1α (a), IL-6 (b) and TNF (c) in serum of A20fl/fl (n=10), A20myel-KO (n=10), A20myel-KOCASP1/11−/− (n=12) and A20myel-KONlrp3−/− (n=9) mice between 20 and 35 weeks of age. P-values were determined by Mann-Whitney (b) and Student’s t-test (c).
The NLR member Nlrp3, the inflammatory cytokine proIL-1β and the ubiquitin-editing enzyme A20 are expressed at low levels in resting macrophages. Binding of the TLR4 ligand LPS to its receptor triggers phosphorylation and rapid degradation of IκBα, allowing translocation of NF-κB to the nucleus, and NF-κB-mediated upregulation of proIL-1β, proIL-18, Nlrp3 and A20. A20 prevents excessive Nlrp3 inflammasome activation by dampening basal and LPS-induced NF-κB-mediated upregulation of Nlrp3. As such, A20 reduces the pool of Nlrp3 that is available for inflammasome assembly. In addition, it limits the levels of the inflammasome substrates proIL1β and proIL-18.
Raw data of histological scores of ankle sections of A20myel-KOIL1R1+/− (n=10) and A20myel-KOIL1R1−/− mice (n=8). Arthritic histopathology of ankles was scored on 2 parameters, calcaneal erosion and inflammation at the synovio–entheseal complex, as described in the Methods section.
Raw data of histological scores of ankle sections of A20myel-KONlrp3+/+ (n=5) and A20myel-KONlrp3−/− mice (n=5). Arthritic histopathology of ankles was scored on 2 parameters, calcaneal erosion and inflammation at the synovio–entheseal complex, as described in the Methods section.