

Summary
Disorders arising from impaired assembly of succinate dehydrogenase (SDH) result in a myriad of pathologies, consistent with its unique role in linking the citric acid cycle and electron transport chain. In spite of this critical function, however, only a few factors are known to be required for SDH assembly and function. We show here that two factors, Sdh6 (SDHAF1) and Sdh7 (SDHAF3), mediate maturation of the FeS cluster SDH subunit (Sdh2/SDHB). Yeast and Drosophila lacking SDHAF3 are impaired in SDH activity with reduced levels of Sdh2. Drosophila lacking the Sdh7 ortholog SDHAF3 are hypersensitive to oxidative stress and exhibit muscular and neuronal dysfunction. Yeast studies revealed that Sdh6 and Sdh7 act together to promote Sdh2 maturation by binding to a Sdh1/Sdh2 intermediate, protecting it from the deleterious effects of oxidants. These studies in yeast and Drosophila raise the possibility that SDHAF3 mutations may be associated with idiopathic SDH-associated diseases.
Introduction
Succinate dehydrogenase (SDH) is an integral component of both the mitochondrial respiratory chain and the tricarboxylic acid (TCA) cycle. It catalyzes the two-electron oxidation of succinate to fumarate with the reduction of ubiquinone to ubiquinol (succinate:ubiquinone oxidoreductase). SDH is embedded within the inner membrane (IM) of mitochondria and consists of four nuclear-encoded subunits, designated Sdh1 through Sdh4 in yeast and SDHA through SDHD in mammalian cells. SDH deficiency in humans results in infant encephalomyopathy, myopathy or tumorigenesis in the adult (Finsterer, 2008; Rustin and Rotig, 2002). Loss-of-function mutations in human genes for SDHA, SDHB, SDHC and SDHD are strongly linked with susceptibility to familial paraganglioma, pheochromocytoma, gastrointestinal stromal tumors and renal cell carcinoma (Bardella et al., 2011; Baysal et al., 2000; Feichtinger et al., 2010; Janeway et al., 2011). Tumorigenesis arising from SDH-deficiency is purportedly related to the deleterious effects of supraphysiological levels of succinate that is a known inhibitor of a myriad of αketoglutarate (αKG)-dependent enzymes including prolyl hydroxylases, histone and DNA demethylases (Selak et al., 2005; Xiao et al., 2012).
This tetrameric enzyme contains five redox cofactors including a covalently bound FAD and three iron-sulfur (FeS) clusters in a hydrophilic segment consisting of two subunits Sdh1 and Sdh2 and a heme-containing membrane anchor domain consisting of Sdh3 and Sdh4 subunits (Robinson and Lemire, 1996). The FeS clusters facilitate electron transfer to the ubiquinone-binding site formed between Sdh2 and the membrane subunits (Sun et al., 2005).
Assembly factors are often used to facilitate cofactor insertion in mitochondrial respiratory complexes and mitigate unwanted reactions during biogenesis. Recently, two SDH assembly factors associated with human pathogenesis were identified. Succinate dehydrogenase assembly factor 1 (SDHAF1) was found in a study of infantile mitochondrial diseases in which two families presented with multiple afflicted children with leukoencephalopathy (Ghezzi et al., 2009). Biochemical analyses revealed a SDH deficiency in muscle samples and fibroblasts from these patients along with missense mutations in SDHAF1. Deletion of the yeast ortholog of SDHAF1 (SDH6) resulted in a respiratory deficiency and a specific reduction in SDH activity (Ghezzi et al., 2009). SDH deficiency has subsequently been reported in other patients that carry SDHAF1 mutations (Ohlenbusch et al., 2012). Succinate dehydrogenase assembly factor 2 (SDHAF2 or yeast Sdh5) was shown to be required for the covalent attachment of FAD to the catalytic SDHA (Sdh1) subunit (Hao et al., 2009). Yeast lacking Sdh5 were respiratory deficient due to an absence of SDH activity. Germline loss-of-function mutations in SDHAF2 were identified in SDH-deficient, neuroendocrine paraganglioma tumors (Hao et al., 2009). A number of SDH-deficient pathologies, including Leigh syndrome, gastrointestinal stromal tumors and neuroblastomas, have also been reported that lack mutations in known SDH assembly factors or SDH structural subunits (Feichtinger et al., 2010; Janeway et al., 2011). Thus, additional SDH assembly factors may remain to be discovered, potentially providing insights into the causes of idiopathic SDH-associated diseases.
Sdh6 is a member of the LYR protein family that consists of 10 proteins in the human proteome and four in the yeast proteome (Figure 1A). Within yeast, the founding member is the mitochondrial Isd11 protein that functions in the matrix FeS biogenesis pathway as an effector of the Nfs1 cysteine desulfurase (Adam et al., 2006; Wiedemann et al., 2006). We demonstrated that a second LYR protein Mzm1 is a chaperone for the Rieske FeS subunit of Complex III (Atkinson et al., 2011; Cui et al., 2012). The remaining yeast LYR proteins are Sdh6 and Acn9 (human ortholog ACN9). Although Sdh6 is required for proper SDH activity, its molecular mechanism remains unknown. Moreover, Acn9 (designated Sdh7 in yeast and SDHAF3 in humans and flies) has no known function. Here we show that these two factors are required for SDH biogenesis in eukaryotes. Both Sdh6 and Sdh7 protect Sdh2 maturation from the deleterious effects of endogenous reactive oxygen species. We also report that loss of SDHAF3 in Drosophila cells leads to a marked SDH-deficiency analogous to that in yeast, with defects in muscular and neuronal function in mutant flies. This study identifies functions of two SDH assembly factors, providing a more complete understanding of its critical role in cellular energy production and a potential molecular framework for defining currently idiopathic SDH-associated diseases.
Figure 1. Succinate dehydrogenase deficiency in cells lacking two LYR motif family proteins, Sdh6 and Sdh7.
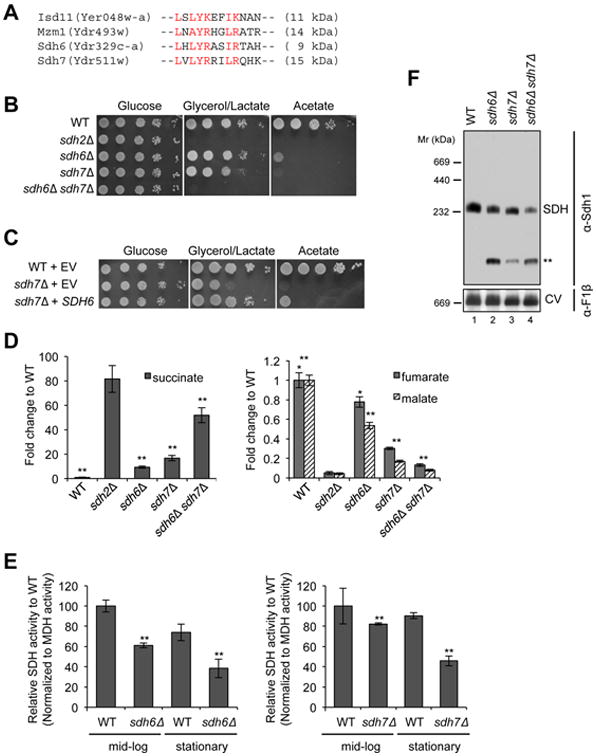
(A) L-X-L/A-Y-R-X-X-L/I-R/K motif conserved in four LYR motif family proteins in yeast. Isd11, a chaperone required for cysteine desulfurase activity in FeS biogenesis pathway (Adam et al., 2006); Mzm1, a protein facilitating the Rieske FeS protein insertion into bc1 (Cui et al., 2012); Sdh6 (SDHAF1); and Sdh7. (B) 10-fold serial dilutions of cells starting from OD600=0.5 were spotted on synthetic complete (SC) media containing different carbon sources as indicated, and incubated at 30°C. (C) 10-fold serial dilutions of cells were spotted on SC media lacking uracil and incubated at 30°C. EV, empty vector (D) Metabolites extracted from cells cultured in synthetic minimal media containing 2% raffinose / 0.2% glucose were analyzed using GC-MS. Cells were harvested at OD600 = 2. Relative levels of metabolites to WT are represented as mean ± SEM (N ≥ 4 biological replicates; *p<0.05; **p<0.005). (E) Relative SDH activity in isolated mitochondria compared to WT. Mitochondria were isolated from cells grown in SC media plus 2% raffinose / 0.2 % glucose for 24 h (midlog) and 48 h (stationary). Data are shown as mean ± SD is shown (N=3; ** p<0.05). (F) Blue-Native (BN) PAGE analysis to visualize protein complexes. Mitochondria isolated from the strains harvested at late-log phase were solubilized with 1% digitonin. After clarification, soluble fractions were separated on BN-PAGE and then transferred to membranes for immunoblotting. Sdh1, a FAD-containing subunit of SDH; F1β, a subunit of ATP synthase (Complex V, CV) in oxidative phosphorylation. The band highlighted by ** is the Sdh1 assembly intermediate. This band is visualized by antisera to Sdh1 but not Sdh2. See also Figures S1 and S2.
Results
Cells lacking Sdh6 and Sdh7 exhibit SDH deficiency
As a first step toward characterizing Sdh6 and Sdh7 function, we examined the growth phenotypes of sdh6Δ or sdh7Δ mutants on non-fermentable carbon sources using S. cerevisiae. Cells lacking Sdh6 or Sdh7 exhibit a partial growth defect on glycerol/lactate medium and a severe growth defect on acetate medium compared to wild-type cells (Figure 1B), which is consistent with previous studies (Ghezzi et al., 2009; McCammon, 1996). We confirmed that the respiratory growth defects of sdh6Δ and sdh7Δ mutants were attributed to deletions of SDH6 and SDH7, as respiratory growth of sdh6Δ and sdh7Δ cells was restored with epitope-tagged Sdh6 and Sdh7, respectively (Figure S1A). Since both Sdh6 and Sdh7 belong to the LYR motif protein family (Figure 1A), we looked for genetic interactions between their encoded proteins. The sdh6Δ sdh7Δ double deletion strain exhibited a marked synthetic growth defect on glycerol/lactate medium (Figure 1B). In addition, the overexpression of SDH6 partially suppressed the respiratory growth defect of sdh7Δ cells (Figure 1C), however, overexpression of SDH7 failed to restore respiratory function of sdh6Δ cells.
Metabolomic profiling was used to identify the biological process impaired in sdh6Δ and sdh7Δ mutants, assaying the levels of approximately 100 polar metabolites. We cultured cells in synthetic minimal medium with 2% raffinose and 0.2% glucose to stationary phase. Metabolites extracted from cells were analyzed using gas chromatography-mass spectrometry (GC-MS) (Figure 1D). Cells lacking either Sdh6 or Sdh7 exhibited elevated succinate levels and attenuated fumarate and malate levels consistent with impaired conversion of succinate to fumarate by SDH in the citric acid cycle. These observations in sdh6Δ cells are consistent with the previous study in fibroblasts harboring a mutated SDHAF1 gene (Ghezzi et al., 2009). Moreover, the sdh6Δ sdh7Δ double mutant showed an enhanced accumulation in succinate, consistent with the synergistic respiratory growth defects in these cells.
To confirm that the increased succinate/fumarate ratio in sdh7Δ mutants was due to impaired SDH function, we quantified SDH activity in mitochondria purified from wild-type (WT) and mutant cells lacking Sdh6 or Sdh7. Mitochondria isolated from sdh6Δ and sdh7Δ cells harvested at mid-log phase exhibited modest diminutions of SDH activity, but the deficit was magnified in stationary phase cells (Figure 1E). The sdh6Δ sdh7Δ double mutant showed markedly decreased SDH activity in mid-log cultures (34% of WT, Figure S1B) relative to the single mutants in accordance with the synergistic respiratory growth defect and TCA cycle intermediates in sdh6Δ sdh7Δ double mutants. Enzymatic activities of pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, aconitase, malate dehydrogenase and bc1 Complex III were unaffected in cells lacking Sdh6 or Sdh7 (Figure S1B,C).
A moderate diminution of the assembled tetrameric SDH complex was seen in mitochondria isolated from late-log cultures of sdh6Δ or sdh7Δ mutants as visualized by BN-PAGE (Figure 1F). The abundance of the SDH complex was further attenuated in sdh6Δ sdh7Δ double mutant cells. Thus, Sdh6 and Sdh7 are required to maintain normal SDH levels and activity in yeast. In addition to reduced levels of tetrameric SDH in the mutant cells, a Sdh1 subcomplex is evident, and the same subcomplex is seen in cells lacking Sdh2 or Sdh4 (** in Figure 1F, Figure S2). Therefore, Sdh6 and Sdh7 appear to function in SDH assembly rather than the regulation of SDH activity.
Maturation of Sdh2 is impaired in the absence of Sdh6 or Sdh7
To characterize the roles of Sdh6 and Sdh7, the steady-state levels of SDH subunits were quantified in mitochondria isolated from mutant cells. The levels of Sdh2 were attenuated in both sdh6Δ and sdh7Δ mutants and further diminished in the sdh6Δ sdh7Δ double mutant (Figure 2A). In contrast, Sdh1 levels were unchanged in the mutant cells and covalent flavinylation of Sdh1 was not significantly altered (Figure 2A). Cells lacking Sdh5 are compromised in Sdh1 flavinylation leading to a reduced stability of Sdh1 (Hao et al., 2009; Kim et al., 2012).
Figure 2. Sdh6 or Sdh7 functions are linked to the Fe/S Sdh2 subunit.
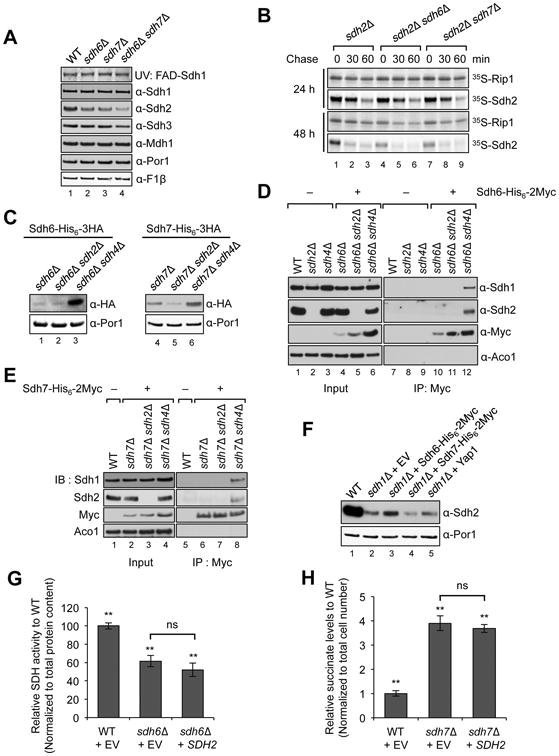
(A) FAD-containing Sdh1 was visualized by UV excitation. Other proteins visualized by immunoblotting include Mdh1, malate dehydrogenase and Por1, porin (loading control). (B) 35S-methionine labeled proteins were incubated with isolated mid-log vs. stationary phase mitochondria for 30 min (pulse), followed by blocking protein import with valinomycin for 30 and 60 min (chase), respectively. Radiolabeled proteins were resolved on SDS-PAGE and detected by autoradiography. (C) Sdh6-His6-3HA or Sdh7-His6-3HA under their own endogenous promoters was expressed from plasmids in cells lacking either Sdh2 or Sdh4 along with endogenous Sdh6 or Sdh7 depleted, respectively. Steady-state levels are shown by immunoblotting. (D) Coimmunoprecipitation of Sdh6-His6-2Myc after crosslinking. Mitochondria were solubilized with 1% digitonin in the presence of 1 mM dithiobis[succinimidylpropionate]. The cross-linking reaction was stopped with Tris buffer (pH 7.4), and the supernatants were absorbed to anti-Myc antibody-conjugated magnetic beads. Bound substances to Myc beads were resolved on SDS-PAGE and detected by immunoblotting. Input, 4% of total lysates; Aco1, FeS aconitase. (E) Standard co-immunoprecipitation of Sdh7-His6-2Myc was performed with isolated mitochondria without crosslinking. Input, 2% of total lysates. (F) Steady-state levels of Sdh2 in sdh1Δ mutants with overexpression of proteins indicated. Yap1, transcription factor up-regulating oxidative stress response genes. (G) SDH activity in sdh6Δ mutants with SDH2 overexpression was detected as described in Figure 1E. Data are represented as mean ± SD (N=3; **p < 0.05; ns, not significant). (H) Succinate levels in sdh7Δ mutants with SDH2 overexpression were measured as described in Figure 1D. Mean ± SEM is shown (N=6; **p < 0.05; ns, not significant). See also Figure S3.
A mitochondrial in vitro protein import assay was used to address a role for Sdh6 and Sdh7 in Sdh2 maturation. The import assay consisted of the in vitro import of 35Smethionine labeled Sdh2 into purified mitochondria isolated from sdh2Δ cells. Sdh2-deficient mitochondria were used to ensure that a pool of unassociated Sdh1 would be available for a stabilizing interaction with imported Sdh2. Furthermore, the use of sdh2Δ cells negates any changes in the mitochondrial membrane potential (which drives protein import) by the loss of Sdh6 or Sdh7. The stability of 35S-Sdh2 as monitored during the chase phase of the reaction was compromised in cells lacking Sdh6 or Sdh7 especially in mitochondria isolated from stationary phase cultures (Figure 2B). This result is consistent with the exacerbated defect in SDH activity seen in mutant cells at late stages of growth. Unlike Sdh2, radioisotope-labeled Rip1, a target of the LYR protein Mzm1, remained unaffected by loss of either Sdh6 or Sdh7.
Sdh1 and Sdh2 accumulate in cells lacking the membrane anchor subunits Sdh3 and/or Sdh4 (Figure S3A) (Kim et al., 2012). We observed that cells depleted of the membrane anchor contain increased steady-state levels of both Sdh6 and Sdh7 (Figure 2C). These results suggest that Sdh6 and Sdh7 may form stalled pre-assembly intermediates with Sdh1 and/or Sdh2. We performed co-immunoprecipitation on epitope-tagged Sdh6 and Sdh7 in mitochondria from WT cells and cells stalled in SDH assembly. A fraction of Sdh1 and Sdh2 were co-purified with either Sdh6 or Sdh7 from cells lacking the membrane anchor domain but not in cells devoid of Sdh2 (Figure 2D-E, Figure S3B). These interactions are seen in the presence and absence of crosslinking prior to resin adsorption. These results suggest that Sdh6 and Sdh7 interact with Sdh2 within a Sdh1/Sdh2 subcomplex, which is known to accumulate in cells lacking the membrane anchor domain (Kim et al., 2012). Moreover, these interactions appear to occur in the mitochondrial matrix, consistent with the known matrix location of Sdh1, Sdh2, and Sdh6 (Ghezzi et al., 2009). Sdh7 is also a matrix protein as revealed by proteinase K treatment of purified mitochondria, which degrades Sdh7 only in the presence of detergents and not upon hypotonic disruption of only the outer membrane. (Figure S3C).
The physical interactions of Sdh6 and Sdh7 with Sdh2 prompted the hypothesis that Sdh6 and Sdh7 may be chaperones for Sdh2. To test this, we first examined the ability of overexpressed Sdh6 or Sdh7 to stabilize the highly labile Sdh2 present in sdh1Δ cells (Kim et al., 2012). Elevated levels of Sdh6, but not Sdh7, led to increased steady-state Sdh2 levels under these conditions (Figure 2F). This result raised a possibility that Sdh6 may function as a chaperone for Sdh2 prior to its interaction with Sdh1. We tested whether SDH2 overexpression in sdh6Δ mutants or sdh7Δ mutants would restore SDH activity and suppress the respiratory growth defects. However, SDH2 overexpression neither restored SDH activity nor reversed succinate accumulation or rescued respiratory growth of sdh6Δ mutants and sdh7Δ mutants (Figure 2G-H, Figure S4A,B).
Once a holo-Sdh2/Sdh1 complex forms, the final step in SDH biogenesis is the addition of the Sdh3/Sdh4 membrane anchor. To address whether Sdh6 and Sdh7 are involved in the recruitment of the membrane anchor, we tested whether co-overexpression of SDH3 and SDH4 would suppress the respiratory defect in the sdh6Δ and sdh7Δ mutant cells. No restoration of growth on acetate medium was observed with elevated cellular levels of Sdh3 and Sdh4 (Figure S4C,D). Thus, Sdh6 and Sdh7 do not likely facilitate the recruitment of the Sdh3/Sdh4 membrane anchor.
We conducted a series of studies to assess whether Sdh6 or Sdh7 have an active role in FeS cluster insertion. These studies failed to reveal a direct role of either factor in FeS cluster insertion. First, affinity purification of Sdh2 in yeast leads to co-purification of Nfu1, Isu1 and Isa2, three key matrix proteins involved in FeS biogenesis (data not shown). However, affinity purification of Sdh6-His6-2Myc or Sdh7-His6-2Myc failed to adsorb Isu1 or Isa2, whereas Sdh2 was associated. Second, whereas overexpression of ISA1 or ISA2 restores respiratory growth in grx5Δ mutant cells (Kim et al., 2010; Rodriguez-Manzaneque et al., 2002), overexpression of ISA1, ISA2, NFU1 or ISU1 failed to suppress the respiratory defect of sdh6Δ or sdh7Δ cells (data not shown). Third, 55Fe incorporation into Sdh2-His6-2Myc was quantified in cells either containing or lacking Sdh6 or Sdh7. Immunocapture of Sdh2 failed to show any clear diminution in 55Fe in the mutant cells (Figure S4E). However, interpretation of the 55Fe study is complicated, since Sdh2 has three distinct FeS centers and Sdh6 or Sdh7 may have a restricted role with one cluster. In addition, ascorbate is used during 55Fe labeling and as a reductant may mimic the exogenous reductants in suppressing the defects in the mutant cells.
Antioxidants ameliorate defects in sdh6Δ mutants and sdh7Δ mutants
Further clues to the function of Sdh6 emerged from a genetic suppressor study in which extragenic suppressors of the acetate growth defect of sdh6Δ mutants were recovered. We generated a high-copy plasmid library with partially digested genomic DNA from sdh6Δ mutants. This library was transformed to sdh6Δ mutants and the transformants that exhibited enhanced growth on acetate medium were collected. Interestingly, multiple independent suppressors were recovered that encoded Yap1, which is a transcriptional activator that induces the expression of a battery of antioxidant genes, including thioredoxin, thioredoxin reductase and glutathione reductase, in response to oxidative stress (Fernandes et al., 1997). The overexpression of YAP1 in subcloned vectors robustly suppressed the acetate growth defect of cells lacking Sdh6 (Figure 3A). Consistent with the antioxidant role of Yap1 expression, supplemental glutathione or N-acetyl-cysteine also restored limited respiratory growth in sdh6Δ mutants (Figure 3B).
Figure 3. Exogenous antioxidants rescue the growth defect of sdh6Δ and sdh7Δ mutants.
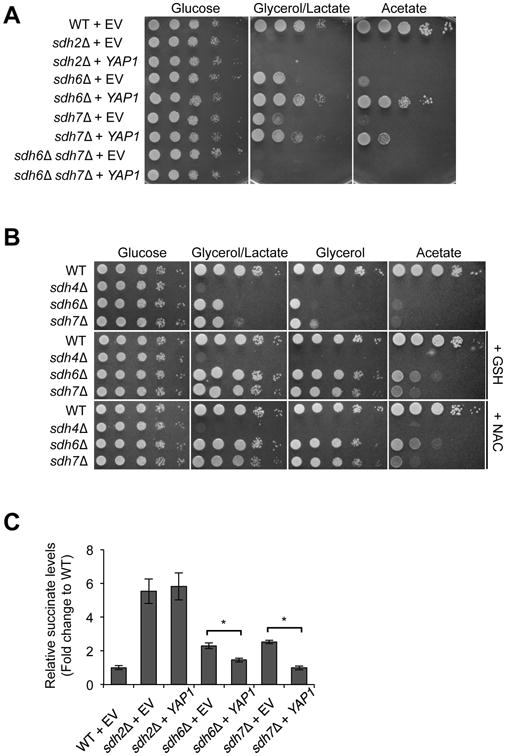
(A) Cells harboring either empty vector (EV) or high-copy YAP1 plasmid were spotted on SC media lacking leucine by 10-fold serial dilutions and incubated at 30°C. (B) 10-fold serial dilutions of cells were spotted on SC medium with the indicated carbon sources ± 5 mM N-acetyl cysteine or 2 mM glutathione and incubated at 30°C. (C) Succinate levels in cells overexpressing YAP1 were measured as described in Figure 1D. Cells were harvested at OD600 = 1. Mean ± SEM is shown (N=6; *p<0.005).
Since we observed a genetic interaction between SDH6 and SDH7 (Figure 1), we tested whether overexpression of YAP1 could also restore respiratory growth in sdh7Δ mutants. Indeed, YAP1 overexpression suppressed the respiratory growth defect of sdh7Δ mutants, although the suppression was less pronounced compared to sdh6Δ mutants (Figure 3A). Moreover, the addition of exogenous reductants restored limited respiratory growth of sdh7Δ mutants (Figure 3B). To confirm that YAP1 overexpression restored SDH activity in sdh6Δ and sdh7Δ single mutant cells, we assessed SDH activity by measuring succinate levels using metabolomics. Indeed, YAP1 overexpression decreased succinate levels in both sdh6Δ and sdh7Δ mutants significantly, while YAP1 overexpression did not affect succinate levels in sdh2Δ mutants as a negative control (Figure 3C). Therefore, we conclude that YAP1 overexpression contributes to the restoration of SDH activity in sdh6Δ and sdh7Δ mutants. Elevated levels of Yap1, however, have no effect on the respiratory growth defects in sdh6Δ sdh7Δ double mutants (Figure 3A).
Sdh2 is stabilized by Sdh6 and Sdh7 under oxidative stress conditions
We hypothesized that Sdh6 and Sdh7 may be important for SDH maturation under oxidative stress conditions. Initially, we tested whether cells lacking Sdh6 or Sdh7 were hypersensitive to the superoxide anion generator paraquat. The respiratory growth defect of sdh6Δ and sdh7Δ single mutants was dramatically exacerbated in the presence of paraquat (Figure 4A). Likewise, steady-state levels of Sdh2 were markedly attenuated in paraquat-treated sdh6Δ and sdh7Δ mutants compared to WT cells (Figure 4B). In contrast, steady-state levels of other mitochondrial FeS containing proteins were not significantly altered by paraquat in the mutant cells (Figure 4B).
Figure 4. sdh6Δ and sdh7Δ mutants are sensitive to oxidative stress.
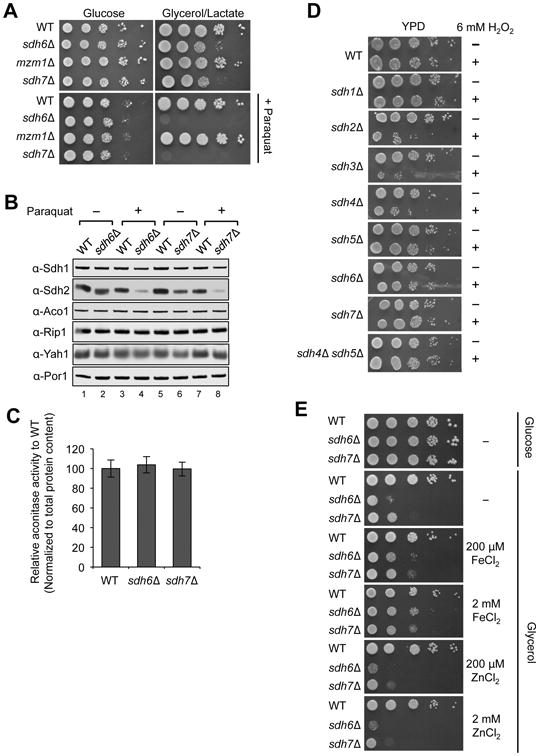
(A) 10-fold serial dilutions of cells were spotted on SC media ± 2 mM paraquat with indicated carbon sources and incubated at 30°C. (B) Steady-state levels of proteins in mitochondria isolated from strains cultured in the presence of 2 mM paraquat. Yah1, ferredoxin of the mitochondrial matrix. (C) Aconitase activity specific to cis-aconitate conversion in isolated mitochondria. Data are shown as mean ± SD (N=3). (D) Pre-cultures grown up to late-log phase in YPD media were diluted 2-fold, followed by addition of 6 mM H2O2 and incubated for 2 h at 30°C. Cells were washed with sterile water and 10-fold serial dilutions were spotteed on YPD plate, followed by incubation at 30°C. (E) Enhanced respiratory growths of sdh6Δ mutants and sdh7Δ mutants with iron supplementation 10-fold serial dilutions of cells were spotted on SC media ± FeCl2 or ZnCl2 as concentrations indicated, and then incubated at 30°C.
The paraquat sensitivity of sdh6Δ and sdh7Δ mutants may arise from either enhanced ROS damage or the accumulation of a pro-oxidant in the mutants. We observed that ROS levels were not changed in untreated sdh6Δ and sdh7Δ mutants using two different assays. First, we quantified aconitase (Aco1) activity. The 4Fe-4S cluster in aconitase is susceptible to ROS damage (Gardner, 2002), thus aconitase activity is an indicator of ROS stress in vivo. No diminution of aconitase activity was seen in sdh6Δ and sdh7Δ mutants compared to WT (Figure 4C). Second, we tested the mutant cells for hydrogen peroxide sensitivity on rich glucose medium. As cells accumulate ROS, they become sensitized to exogenous ROS and subsequently lose viability (Khalimonchuk et al., 2007). After two-hour treatment of cells with 6 mM H2O2, the viability of sdh2Δ, sdh3Δ and sdh4Δ mutant strains was significantly compromised (Figure 4D). However, sdh6Δ and sdh7Δ mutant cells exhibited no growth defects. The lack of impairment in aconitase activity and hydrogen peroxidase sensitivity suggest that ROS levels are not elevated in sdh6Δ and sdh7Δ mutants as compared to WT cells. It is noteworthy that sdh1Δ and sdh5Δ single mutants and the sdh4Δ sdh5Δ double mutant did not show hydrogen peroxide sensitivity compared to sdh2Δ, sdh3Δ and sdh4Δ strains, which suggests that an assembly intermediate containing FAD-Sdh1 may be the source for electron leakage for the generation of ROS in sdh2Δ, sdh3Δ and sdh4Δ single mutants.
The observed hypersensitivity of sdh6Δ and sdh7Δ mutant cells to paraquat may imply that Sdh6 and Sdh7 are shielding the FeS clusters in Sdh2 prior to full assembly. Superoxide inactivation of the 4Fe-4S center in aconitase leads to a dissociation of one iron ion forming an inactive 3Fe-4S center that can be reactivated by supplemental iron salts (Gardner and Fridovich, 1992). We tested whether supplemental iron salts would restore respiratory function to sdh6Δ and sdh7Δ mutant cells. supplemental FeCl2, but not ZnCl2, restored limited glycerol growth to both mutant cells (Figure 4E). These data are consistent with a candidate role of Sdh6 and Sdh7 in FeS cluster protection in SDH maturation.
Drosophila Sdhaf3 mutants are sensitive to oxidative stress
The Drosophila genome encodes a close ortholog of Sdh7 (Figure S5A), but has only a weak candidate homolog of Sdh6. Accordingly, we examined the functions of Sdh7 in Drosophila to determine if its roles in SDH assembly and activity have been conserved through evolution and to define its possible physiological functions. Gene targeting was used to generate a null mutation in the Drosophila sdh7 ortholog (CG14898), which we refer to here as dSdhaf3 (Figure S5B,C). These mutants were outcrossed for six generations to w1118, which was used as a control for most studies. dSdhaf3 mutants progress normally through development and have a normal lifespan when maintained on standard growth media. These animals are, however, sensitive to ethanol (Figure 5A) and oxidative stress, resulting from either paraquat treatment (Figure 5B) or hyperoxia (Figure 5C). The response to hyperoxia is most pronounced, with a 50% reduction in lifespan relative to controls, although SdhB12081 hypomorphic mutants display a more severe effect (Walker et al., 2006). Interestingly, most Sdhaf3 mutants exposed to 100% oxygen for four days held their wings erect, a hallmark of mitochondrial dysfunction and muscle degeneration (DeSimone et al., 1996; Greene et al., 2003). A similar abnormal wing posture was observed in SdhB12081 mutants maintained under normal conditions (10-15%) or exposed to hyperoxia (80-100%).
Figure 5. dSdhaf3 mutants are sensitive to oxidative stress and display reduced levels of SdhB, reduced SDH activity, and motility defects.
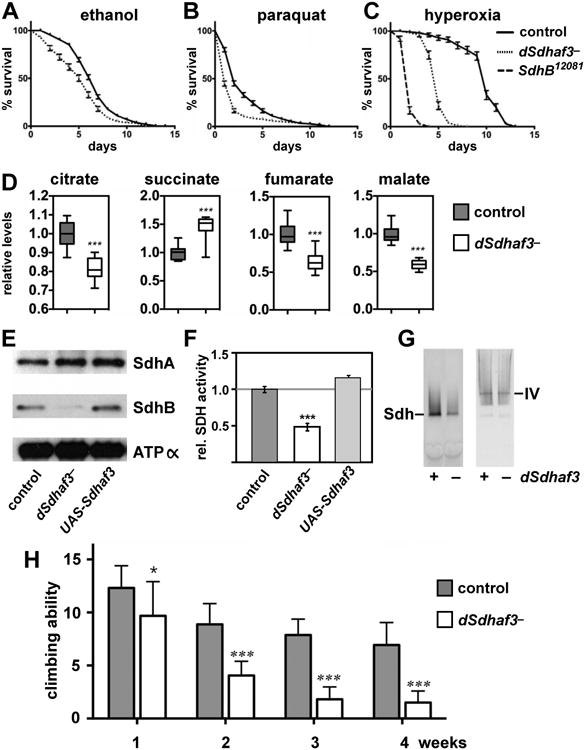
Five-day old w1118 control (solid line) and dSdhaf3 mutant (dotted line) males were transferred to vials with (A) 5% ethanol, 1% agar in PBS, (B) 30 mM paraquat in semi-defined medium, or (C) 100% O2 with standard medium, and living animals were scored daily. Homozygous SdhB12081 mutants (dashed line) were included in the hyperoxia experiment. Each graph was compiled from 3-5 experiments, using a total of 15-21 vials with 20 animals per vial. Error bars represent ±SEM. dSdhaf3 mutants are significantly more sensitive than controls under each condition, p<0.001. (D) GC/MS was used to compare the relative levels of small metabolites in wild-type controls (grey boxes) and dSdhaf3 mutants (white boxes). N=12 samples from two independent experiments with 20 flies/sample (5-day old). ***p<0.001. (E) Proteins were extracted from mitochondria isolated from w1118 controls, dSdhaf3 mutants, or UAS-dSdhaf3/+ transformants, and analyzed by immunoblotting to detect SdhA, SdhB, and ATPα, (subunit of complex V). (F) A continuous colorimetric assay was used to measure SDH enzyme activity in extracts of purified mitochondria from w1118 controls, dSdhaf3 mutants, and UASdSdhaf3/+ transformants. ***p<0.001. (G) Proteins from purified mitochondria were extracted from w1118 controls and dSdhaf3 mutants, fractionated by non-denaturing PAGE, and analyzed for SDH and Complex IV activity. (H) Control w1118 flies and dSdhaf3 mutants were tested for motility in three independent experiments using a total of 18 vials with 20 adults/vial at 1, 2, 3, or 4-weeks of age. Climbing ability is reported as the number of flies that climbed above a line drawn 4 cm above the bottom of the vial five seconds after being tapped to the bottom. *p<0.05, ***p<0.001
dSdhaf3 mutants display reduced SdhB protein levels and SDH activity
If the function of dSdhaf3 has been conserved through evolution, then dSdhaf3 mutants should display a specific defect in SDH function. Consistent with this possibility, metabolomic profiling of dSdhaf3 mutants revealed elevated succinate and reduced levels of fumarate and malate (Figure 5D). Biochemical analysis of mitochondrial extracts from dSdhaf3 mutants demonstrated that dSdhaf3 mutants have normal levels of SdhA, but significantly reduced levels of SdhB (Figure 5E), resulting in an approximate 50% reduction in SDH enzymatic activity (Figure 5F,G), similar to the phenotypes of sdh7Δ yeast. SdhA is also flavinylated normally in dSdhaf3 mutants, as expected (Figure S5E). Combining the hypomorphic SdhB12081 allele with the dSdhaf3 mutation resulted in a dramatic decrease in viability, demonstrating a strong genetic interaction, consistent with the reduced levels of SdhB in dSdhaf3 mutants and confirming the functional interaction between dSdhaf3 and SDH (Figure S6A).
Interestingly, although dSdhaf3 mutants are fully viable and fertile, they display a clear age-dependent reduction in movement (Figure 5H). While mutants at one week of adult life show no difference in motility relative to controls, mutants display an approximate 50% reduction in movement by two weeks of age and a more severe motility defect at later stages (Figure 5H). Mutants are also significantly more sensitive to paralysis by two weeks of age, relative to controls (Figure S5F). Thus, like its counterpart in yeast, dSdhaf3 is required to maintain normal SDH levels and activity, and is required for proper wing muscle function and motility in Drosophila.
Antioxidant and genetic rescue of dSdhaf3 mutants
Consistent with the ability of antioxidants to suppress the growth defects in sdh7Δ yeast mutants, either a dietary (N-acetyl cysteine) or genetic (Sod2 expression) reduction in oxidative stress rescued the hyperoxia sensitivity of dSdhaf3 mutants (Figure 6A,B). Unlike the yeast studies, however, overexpression of the putative Sdh6 homolog (encoded by CG34229), using Act5C-GAL4 to drive a UAS-CG34229 transgene in dSdhaf3 mutants, had no effect on their sensitivity to hyperoxia (Figure S6B). This result, however, is difficult to interpret because there is only limited sequence homology between CG34229 and Sdh6. We conclude that this functional interaction between Sdh6 and Sdh7 may not be conserved through evolution or, alternatively, that CG34229 is not a functional homolog of Sdh6.
Figure 6. dSdhaf3 function is required in the muscles and nervous system.
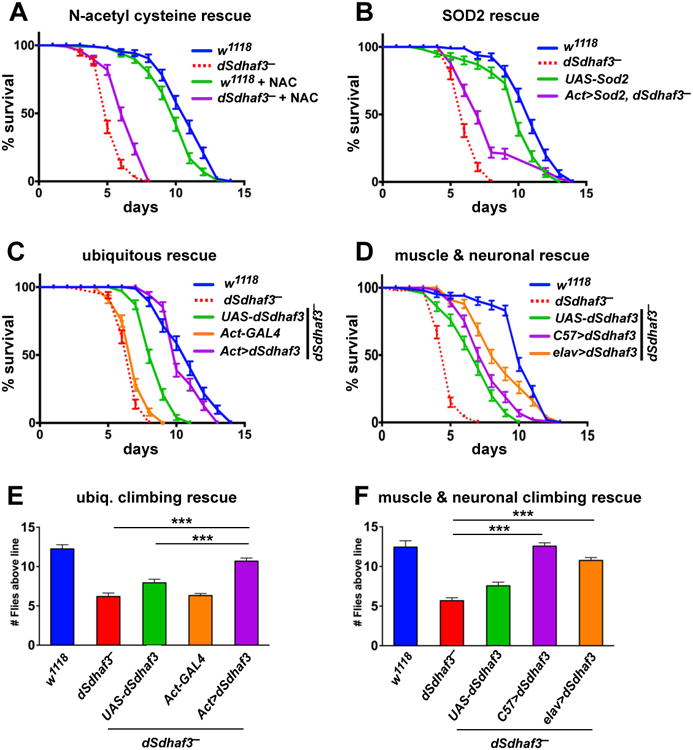
Five-day old w1118 control (blue solid line) and dSdhaf3 mutant (red dotted line) males were transferred to vials with 100% O2 with standard medium, and living animals were scored daily. (A) 0.1% N-acetyl cysteine was added to the culture medium for a quarter of the vials. (B) Expression of Sod2 using the ubiquitous Act5C-GAL4 driver (Act>Sod2; purple line) partially rescues the hyperoxia sensitivity of dSdhaf3 mutants. Both NAC treatment and Sod2 expression significantly rescue the hyperoxia sensitivity of dSdhaf3 mutants, p<0.001. (C) Expression of wild-type dSdhaf3 using the ubiquitous Act5C-GAL4 driver (Act>dSdhaf3; purple line) rescues the hyperoxia sensitivity of dSdhaf3 mutants (p<0.001). (D) The muscle-specific C57-GAL4 driver provides minor, but significant (p<0.01), rescue of the hyperoxia sensitivity of dSdhaf3 mutants (purple line) relative to the control that carries the UAS-dSdhaf3 transgene alone (green line), while the CNS-specific elav-GAL4 driver provides more efficient rescue (orange line)(p<0.001). Expression of wild-type dSdhaf3 by using either the ubiquitous Act5C-GAL4 driver (E, purple), the muscle-specific C57-GAL4 driver (F, purple), or the CNS-specific elavGAL4 driver (F, orange) rescues the climbing defect in dSdhaf3 mutants. The Act>dSdhaf3 rescue in C and E was performed in females, other rescue studies were performed in males (A,B,D,F). The apparent partial rescue of dSdhaf3 mutants by a single copy of the UAS-dSdhaf3 transgene (C,D, green line) appears to be due to genetic background since UAS-dSdhaf3 transformants have normal levels of SdhB and SDH activity (E,F). Each graph was compiled from two experiments with a total of 10 vials with 20 animals per vial. ***p<0.001
The wing posture and motility defects in dSdhaf3 mutants suggested that these animals suffer from muscular and neuronal dysfunction. Consistent with this model, widespread expression of WT dSdhaf3 (Act>dSdhaf3) in dSdhaf3 mutants fully rescues their sensitivity to hyperoxia (Figure 6C), while muscle-specific (C57>dSdhaf3) or neuronal-specific (elav>dSdhaf3) expression provides partial rescue (Figure 6D). Similar effects are seen on the climbing defects in dSdhaf3 mutants (Figure 6E,F). Genetic rescue in the fat body (Cg-GAL4) or intestine (Mex-GAL4), however, provided no significant rescue (data not shown). Widespread overexpression of dSdhaf3 in wild-type flies has no significant effect on their resistance to hyperoxia (data not shown). Taken together, we conclude that dSdhaf3 function is conserved through evolution and that proper SDH levels and activity are required for resistance to oxidative stress as well as muscular and neuronal function, consistent with their dependence on mitochondrial oxidative phosphorylation.
SDHB is specifically impaired in human cells deficient in wild-type SDHAF1
The human Sdh6 ortholog, SDHAF1, was shown previously to be important for SDH activity and abundance in fibroblasts (Ghezzi et al., 2009); however, its mechanism of action remained undefined. We addressed whether SDHAF1 protects SDHB (the human Sdh2 ortholog) from ROS damage similar to yeast Sdh6. First, we determined steady-state levels of SDH structural subunits in SDHAF1-depleted HEK293 cells by siRNA knockdown. SDHB and SDHC levels were significantly reduced upon SDHAF1 depletion (Figure 7A). SDHA levels, however, remained unaffected. Next, we tested the effects of paraquat on SDHAF1-depleted HEK293 cells. In accordance with the yeast and fly data, SDHB levels were further attenuated in SDHAF1-deficient cells (Figure 7B). The sensitivity to paraquat also suggests that SDHAF1 resembles Sdh6 in protecting holo-SDHB from ROS damage.
Figure 7. SDHB is destabilized in human cells with reduced levels of SDHAF1.
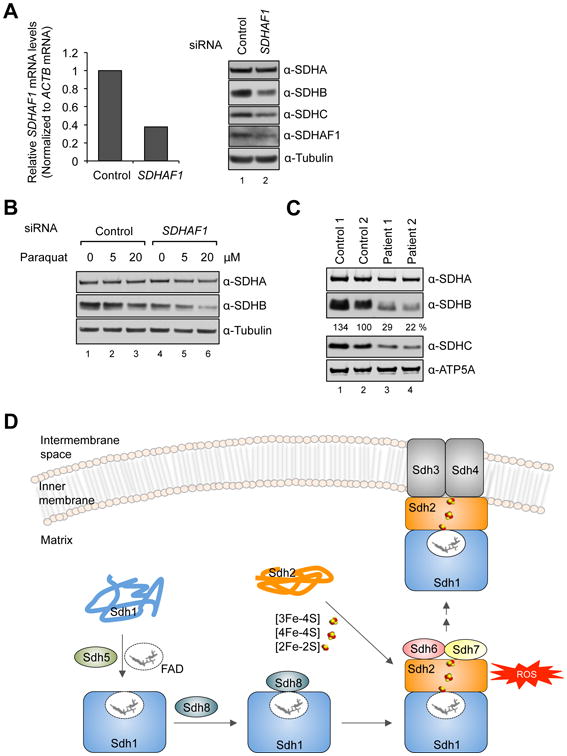
(A) Relative SDHAF1 mRNA levels in HEK293 cells 72 h after SDHAF1 knockdown using siRNA (left panel) and steady-state levels of proteins from total cell lysates (right panel). (B) HEK293 cells were treated with either control siRNA or SDHAF1 siRNA. Paraquat was added to cultures 24 h after siRNA transfection. Total cell lysates were obtained 48 h after paraquat treatment. (C) Steady-state levels of proteins in mitochondria isolated from control fibroblasts and patient fibroblasts harboring mutations on SDHAF1 (Ohlenbusch et al., 2012). The indicated percentages are relative levels of SDHB normalized to ATP5A levels by densitometry. (D) Model of the role of Sdh6(SDHAF1) and Sdh7(SDHAF3) in maturation of Sdh2(SDHB). Sdh6 and Sdh7 associate with Sdh2 within a Sdh1/Sdh2 intermediate. Sdh1 maturation requires covalent flavinylation by Sdh5 followed by formation of the Sdh1/Sdh2 subcomplex that is chaperoned by Sdh8 (see accompanying paper VanVraken et al. 2014).
We also examined the steady-state SDHB levels in patient fibroblasts with a known SDHAF1 mutation (Ohlenbusch et al., 2012). Both SDHB and SDHC levels were diminished in mitochondria isolated from the patient fibroblasts, whereas SDHA levels remained unaffected compared to controls (Figure 7C). SDHB protein levels were <50% of wild-type controls, and SDH enzyme activity was 52% and 40% in patients 1 and 2 relative to control values. Thus, the limited SDHB levels in the patient cells likely contribute to reduced SDH function.
Discussion
The present work demonstrates that maturation of the FeS subunit of Sdh2 (SDHB) requires the participation of two assembly factors, Sdh6 (SDHAF1) and Sdh7 (SDHAF3). These factors are shown to guide Sdh2 maturation within the mitochondrial matrix in the midst of endogenous oxidants. Yeast, flies and mammalian cells lacking one of these factors are impaired in SDH activity and assembly with Sdh2 exhibiting a heightened susceptibility to oxidants. Normal oxidative metabolism in the mitochondria leads to the formation of superoxide anions from the one electron reduction of O2. Superoxide anions can readily dissociate FeS clusters, rendering the assembly of FeS cluster centers in mitochondrial enzymes susceptible to oxidative damage. The present studies in yeast, flies and mammalian cells suggest that Sdh6 and Sdh7 shield one or more of the three FeS clusters in Sdh2 from oxidants during assembly.
Yeast studies reveal that Sdh6 and Sdh7 act in concert in the maturation of Sdh2 with a limited redundancy in function. Yeast lacking either factor show a marked SDH deficiency in late-log cultures that rely on oxidative metabolism. Under these conditions, the mutant cells contain a reduced level of the assembled tetrameric enzyme. These cells exhibit a hypersensitivity to the superoxide generator paraquat. The respiratory defect of these mutants is readily suppressed by overexpression of the Yap1 transcriptional activator of oxidative stress genes or exogenous reductants. These studies highlight the role of Sdh6 and Sdh7 in shielding Sdh2 maturation from deleterious effects of oxidants.
Flies lacking the Sdh7 ortholog SDHAF3, likewise, are hypersensitive to paraquat and hyperoxia. The mutant flies show diminished levels of active SDH and, as a result, accumulate succinate. The dSdhaf3 mutants are viable and fertile; yet display impaired movement that intensifies with age. The erect wing phenotype exhibited under hyperoxic conditions and the motility defects evident in aged mutant flies are consistent with muscular and neuronal dysfunction. Moreover, neuronal or muscle-specific expression of wild-type dSdhaf3 is sufficient to partially rescue the hyperoxia sensitivity of the mutants, demonstrating the importance of SDH function in these tissues that rely heavily on OXPHOS. The hyperoxia sensitivity of dSdhaf3 mutants is also partially suppressed by dietary N-acetyl cysteine or overexpression of the matrix manganosuperoxide dismutase Sod2. These effects of antioxidants mimic the rescue of yeast sdh7Δ mutant oxidative growth and demonstrate the apparent close conservation of Sdh7/SDHAF3 function through evolution.
Conservation in Sdh6/SDHAF1 function between yeast and humans also exists. Attenuation of SDHAF1 in HEK293 culture cells leads to a hypersensitivity in the stability of the FeS SDHB subunit to paraquat. SDHB instability is also seen in two SDHAF1 patient fibroblast lines. One implication of the observed antioxidant rescue of the defect of sdh6Δ yeast cells and dSdhaf3 mutant flies is the potential use of antioxidant therapeutics for patients afflicted with SDHAF1 (and perhaps SDHAF3) mutations. Patients with SDHAF1 mutations have presented with SDH-deficient leukoencephalopathy (Ghezzi et al., 2009; Ohlenbusch et al., 2012), and have a survival window that may be amenable to antioxidant therapy. Two case studies were reported that attempted to alleviate clinical symptoms in SDH-deficient patients harboring SDHAF1 mutations by supplementing riboflavin and CoQ10. The clinical outcomes, however, were not significantly improved (Jain-Ghai et al., 2013). More recently, a candidate therapeutic, EPI-743, is being tested for treatment of Leigh syndrome patients (Martinelli et al., 2012). EPI-743 is a vitamin E quinone, which is orally bioavailable and crosses the blood-brain barrier (Shrader et al., 2011). Future studies will focus on the efficacy of antioxidants with SDHAF1 patient fibroblasts. In addition, it is important to note that every gene encoding an SDH subunit or known assembly factor is causally associated with human disease. We thus anticipate that SDHAF3 mutations will be associated with one or more previously idiopathic SDH-associated diseases and propose that SDHAF1 and SDHAF3 are candidate susceptibility factors for undefined SDH-deficient tumors.
The present work provides insights into the physiological function of Sdh6 and Sdh7 in Sdh2 maturation. Sdh6 and Sdh7 are shown to bind to the Sdh1/Sdh2 assembly intermediate that accumulates in mutants lacking the SDH membrane anchor. In addition, both Sdh6 and Sdh7 accumulate in the membrane anchor mutant cells. Sdh6 was found to impart stabilization to Sdh2 in cells lacking the FAD subunit Sdh1, suggesting that at least Sdh6 has a specific interface for Sdh2. These assembly factors do not appear to be apo-Sdh2 chaperones, since elevated levels of Sdh2 do not suppress the respiratory defects of sdh6Δ or sdh7Δ mutants. Sdh6 and Sdh7 appear to be key chaperones in holo-Sdh2 maturation during oxidative growth. However, a recent report implicated SDHAF1 in an active role in FeS cluster insertion (Maio et al., 2014).
The observation that the two LYR proteins Sdh6 and Sdh7 function with the FeS cluster Sdh2 subunit has significant implications for the uncharacterized LYR proteins present in the human proteome. Our studies raise the possibility that future functional studies of LYRM1, LYRM2, LYRM5 and LYRM9 will reveal novel roles in maturation of mammalian-specific FeS cluster enzymes.
Experimental Procedures
Yeast strains and plasmids
All S. cerevisiae strains and plasmids used are listed in Table S1 and Table S2, respectively. Culture media and conditions are described in the supplemental Experimental Procedures in detail.
Mitochondrial enzymatic activity assay
Succinate dehydrogenase (SDH) and aconitase activity assays were performed as described previously (Atkinson et al., 2011). For SDH activity, quinone-mediated reduction of dichlorophenolindophenol (DCPIP) upon succinate oxidation was measured with isolated mitochondria spectrophotometrically at 600 nm. Aconitase activity was measured with 100 μM cis-aconitate in 50 mM Tris (pH 7.4) at 240 nm in soluble fractions of mitochondria disrupted by repetitive freeze-thaw. For malate dehydrogenase (MDH) activity, soluble mitochondrial fractions were obtained using sonication. Oxidation of 0.2 mM NADH was monitored in the presence of 2 mM oxaloacetate in 100 mM Tris (pH 7.4) at 340 nm (Hayes et al., 1991).
Mitochondrial protein import assay
Mitochondrial protein import assay was performed as described previously (Wagener et al., 2011). Briefly. SDH2 and RIP1 open reading frames were subcloned in pGEM-4Z for in vitro transcription/translation, respectively. Radiolabeled precursor proteins were obtained using reticulocyte lysate (Promega) in the presence of 35S-Met. Precursors were imported into 75 μg of isolated mitochondria in 50 mM HEPES-KOH (pH 7.2) buffer containing 0.6 M sorbitol, 0.5 mg/ml BSA, 2 mM potassium phosphate, 75 mM KCl, 10 mM magnesium acetate, 2 mM EDTA, 2.5 mM MnCl2, 2 mM ATP, 2 mM NADH, 10 mM creatine phosphate, 0.1 mg/ml creatine kinase, 2.5 mM malate, 2.5 mM succinate for 30 min at 25°C for pulse. Import was stopped by adding 5 μM valinomycin and then chased for the periods of time indicated. Non-imported precursors were degraded by proteinase K on ice. Samples were separated on SDS-PAGE and detected by autoradiography.
Co-immunoprecipitation
Mitochondria were solubilized in 10 mM sodium phosphate (pH 7.4), 500 mM NaCl, 1 mM EDTA, 1% digitonin and 1× protease inhibitor cocktail (Roche) for 30 min on ice. Crosslinking was performed with solubilization by adding 1 mM of Dithiobis[succinimidylpropionate] (Pierce) for 30 min at RT. Supernatants after centrifugation at 14,000 × g were incubated with magnetic anti-Myc beads (Cell Signaling Tech.) for 4 h at 4°C. Beads were washed with 10 mM sodium phosphate (pH 7.4), 500 mM NaCl, 1 mM EDTA, 0.1% digitonin and 1 mM PMSF. After washing three times, bound substances were recovered by boiling with 2× SDS-PAGE sample buffer, which was subjected to immunoblotting.
Drosophila strains
Flies were maintained on standard Bloomington Stock Center medium with malt at 25°C. The following stocks were obtained from the Bloomington Stock Center: SdhAHP21216/CyO (Bloomington # 22087), SdhB12081/CyO (Walker et al., 2006) da-Gal4 (Wodarz et al., 1995), Act5C-Gal4/CyO (Bloomington # 25374). The UAS-dSdhaf3 transformant were generated as described in the supplemental Experimental Procedures.
Statistics
Yeast data were analyzed using Microsoft Excel 2011. Data are presented as mean ± SD or mean ± SEM as indicated. Statistical significance was evaluated using Student's test. p < 0.05 was considered significant. Statistical analysis and graphical presentation for Drosophila studies were performed using PRISM software. Student's t test was used for pairwise comparisons and one-way analysis of variance (ANOVA) was used for multiple comparisons. Fly metabolomic data are graphically represented as box plots, with the box representing the lower and upper quartiles, the horizontal line representing the median, and the bars representing the minimum and maximum data points. All other data are shown as the mean ± SEM.
Supplementary Material
Highlights.
SDH maturation is dependent on two Sdh2-specific assembly factors
Cells lacking SDHAF1 and SDHAF3 are sensitive to endogenous oxidants
Drosophila lacking SDHAF3 exhibit muscular and neuronal dysfunction
Acknowledgments
We thank the Bloomington Stock Center for providing fly stocks, and FlyBase for information used for this study. We acknowledge support of funds in conjunction with grant P30 CA042014 awarded to Huntsman Cancer Institute. DKB was supported by the NIH Genetics Predoctoral Training Grant T32 GM007464. This research was supported by NIH RO1 ES03817 (DRW) and 1R01 GM094232 (CST).
Footnotes
Author Contributions: UN designed the experiments and performed the cellular and biochemical analyses with yeast and mammalian cells and wrote the paper. WY designed the experiments and performed the genetic experiments with Drosophila, JC performed the metabolomics analyses, DKB initiated the Drosophila study. KB provided the SDHAF1 patient fibroblast cells, JR provided the tissue culture facility, CST conceived the Drosophila studies, interpreted results and contributed to the writing and financial support. DRW conceived the yeast and mammalian study and design, contributed to writing the paper, interpreted results, provided financial support and finalized the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam AC, Bornhovd C, Prokisch H, Neupert W, Hell K. The Nfs1 interacting protein Isd11 has an essential role in FeS cluster biogenesis in mitochondria. EMBO J. 2006;25:174–183. doi: 10.1038/sj.emboj.7600905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson A, Smith P, Fox JL, Cui TZ, Khalimonchuk O, Winge DR. The LYR Protein Mzm1 Functions in the Insertion of the Rieske FeS Protein in Yeast Mitochondria. Mol Cell Biol. 2011;31:3988–3996. doi: 10.1128/MCB.05673-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011;1807:1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers, et al. Mutations in SDHD, a mitochondrial complex II gene in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Cui TZ, Smith PM, Fox JL, Khalimonchuk O, Winge DR. Late-Stage Maturation of the Rieske FeS Protein: Mzm1 Stabilizes Rip1 but Does Not Facilitate Its Translocation by the AAA ATPase Bcs1. Mol Cell Biol. 2012;32:4400–4409. doi: 10.1128/MCB.00441-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSimone S, Coelho C, Roy S, VijayRaghavan K, White K. ERECT WING, the Drosophila member of a family of DNA binding proteins is required in imaginal myoblasts for flight muscle development. Development. 1996;122:31–39. doi: 10.1242/dev.122.1.31. [DOI] [PubMed] [Google Scholar]
- Feichtinger RG, Zimmermann F, Mayr JA, Neureiter D, Hauser-Kronberger C, Schilling FH, Jones N, Sperl W, Kofler B. Low aerobic mitochondrial energy metabolism in poorly- or undifferentiated neuroblastoma. BMC Cancer. 2010;10:149. doi: 10.1186/1471-2407-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes L, Rodrigues-Pousada C, Struhl K. Yap, a novel family of eight bZIP proteins in Saccharomyces cerevisiae with distinct biological functions. Mol Cell Biol. 1997;17:6982–6993. doi: 10.1128/mcb.17.12.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol. 2008;39:223–235. doi: 10.1016/j.pediatrneurol.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Gardner PR. Aconitase: sensitive target and measure of superoxide. Meth Enzymol. 2002;349:9–23. doi: 10.1016/s0076-6879(02)49317-2. [DOI] [PubMed] [Google Scholar]
- Gardner PR, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem. 1992;267:8757–8763. [PubMed] [Google Scholar]
- Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmuller H, D'Adamo P, Gasparini P, Strom TM, Prokisch H, et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009;43:259–265. doi: 10.1038/ng.378. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MK, Luethy MH, Elthon TE. Mitochondrial malate dehydrogenase from corn: purification of multiple forms. Plant Physiol. 1991;97:1381–1387. doi: 10.1104/pp.97.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain-Ghai S, Cameron JM, Al Maawali A, Blaser S, MacKay N, Robinson B, Raiman J. Complex II deficiency—a case report and review of the literature. Am J Med Genet. 2013;161A:285–294. doi: 10.1002/ajmg.a.35714. [DOI] [PubMed] [Google Scholar]
- Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalimonchuk O, Bird A, Winge DR. Evidence for a pro-oxidant intermediate in the assembly of cytochrome oxidase. J Biol Chem. 2007;282:17442–17449. doi: 10.1074/jbc.M702379200. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Jeong MY, Na U, Winge DR. Flavinylation and assembly of succinate dehydrogenase are dependent on the C-terminal tail of the flavoprotein subunit. J Biol Chem. 2012;287:40670–40679. doi: 10.1074/jbc.M112.405704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KD, Chung WH, Kim HJ, Lee KC, Roe JH. Monothiol glutaredoxin Grx5 interacts with Fe-S scaffold proteins Isa1 and Isa2 and supports Fe-S assembly and DNA integrity in mitochondria of fission yeast. Biochem Biophys Res Commun. 2010;392:467–472. doi: 10.1016/j.bbrc.2010.01.051. [DOI] [PubMed] [Google Scholar]
- Maio N, Singh A, Uhrigshardt H, Saxena N, Tong WH, Rouault TA. Cochaperone Binding to LYR Motifs Confers Specificity of Iron Sulfur Cluster Delivery. Cell Metab. 2014;19:445–457. doi: 10.1016/j.cmet.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi-Vici C, Pontrelli G, Corsetti T, Livadiotti S, Kheifets V, et al. EPI-743 reverses the progression of the pediatric mitochondrial disease--genetically defined Leigh Syndrome. Mol Genet Metab. 2012;107:383–388. doi: 10.1016/j.ymgme.2012.09.007. [DOI] [PubMed] [Google Scholar]
- McCammon MT. Mutants of Saccharomyces cerevisiae with defects in acetate metabolism. Genet. 1996;144:57–69. doi: 10.1093/genetics/144.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlenbusch A, Edvardson S, Skorpen J, Bjornstad A, Saada A, Elpeleg O, Gartner J, Brockmann K. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J Rare Dis. 2012;7:69. doi: 10.1186/1750-1172-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Lemire BD. Covalent attachment of FAD to the yeast succinate dehydrogenase flavoprotein requires import into mitochondria, presequence removal, and folding. J Biol Chem. 1996;271:4055–4060. doi: 10.1074/jbc.271.8.4055. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Manzaneque MT, Tamarit J, Belli G, Ros J, Herrero E. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol Biol Cell. 2002;13:1109–1121. doi: 10.1091/mbc.01-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustin P, Rotig A. Inborn errors of complex II--unusual human mitochondrial diseases. Biochim Biophys Acta. 2002;1553:117–122. doi: 10.1016/s0005-2728(01)00228-6. [DOI] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Shrader WD, Amagata A, Barnes A, Enns GM, Hinman A, Jankowski O, Kheifets V, Komatsuzaki R, Lee E, Mollard P, et al. alpha-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg Med Chem Lett. 2011;21:3693–3698. doi: 10.1016/j.bmcl.2011.04.085. [DOI] [PubMed] [Google Scholar]
- Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121:1043–1057. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Wagener N, Ackermann M, Funes S, Neupert W. A Pathway of Protein Translocation in Mitochondria Mediated by the AAA-ATPase Bcs1. Mol Cell. 2011;44:191–202. doi: 10.1016/j.molcel.2011.07.036. [DOI] [PubMed] [Google Scholar]
- Walker DW, Hajek P, Muffat J, Knoepfle D, Cornelison S, Attardi G, Benzer S. Hypersensitivity to oxygen and shortened lifespan in a Drosophila mitochondrial complex II mutant. Proc Natl Acad Sci USA. 2006;103:16382–16387. doi: 10.1073/pnas.0607918103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N, Urzica E, Guiard B, Muller H, Lohaus C, Meyer HE, Ryan MT, Meisinger C, Muhlenhoff U, Lill R, Pfanner N. Essential role of Isd11 in mitochondrial iron-sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 2006;25:184–195. doi: 10.1038/sj.emboj.7600906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodarz A, Hinz U, Engelbert M, Knust E. Expression of crumbs confers apical character on plasma membrane domains of ectodermal epithelia of Drosophila. Cell. 1995;82:67–76. doi: 10.1016/0092-8674(95)90053-5. [DOI] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, et al. Inhibition of αKG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Devel. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.