

Abstract
The viral reservoir represents a critical challenge facing HIV-1 eradication strategies1–5. However, it remains unclear when and where the viral reservoir is seeded during acute infection and the extent to which it is susceptible to early antiretroviral therapy (ART). Here we show that the viral reservoir is seeded very early following mucosal SIV infection of rhesus monkeys and prior to systemic viremia. We initiated suppressive ART in groups of monkeys on days 3, 7, 10, and 14 following intrarectal SIVmac251 infection. Treatment on day 3 blocked the emergence of viral RNA and proviral DNA in peripheral blood and also substantially reduced levels of proviral DNA in lymph nodes and gastrointestinal mucosa as compared with treatment at later timepoints. In addition, treatment on day 3 abrogated the induction of SIV-specific humoral and cellular immune responses. Nevertheless, following discontinuation of ART after 24 weeks of fully suppressive therapy, virus rebounded in all animals, although animals treated on day 3 exhibited a delayed viral rebound as compared with animals treated on days 7, 10 and 14. The time to viral rebound correlated with total viremia during acute infection and with proviral DNA at the time of ART discontinuation. These data demonstrate that the viral reservoir is seeded very early following intrarectal SIV infection of rhesus monkeys, during the “eclipse” phase, and prior to viremia. This strikingly early seeding of the refractory viral reservoir raises important new challenges for HIV-1 eradication strategies.
The viral reservoir in memory CD4+ T cells in HIV-1-infected individuals cannot be eliminated by current antiretroviral drugs or HIV-1-specific immune responses1–5. This archive of replication-competent virus is the source of viral rebound in nearly all HIV-1-infected individuals who discontinue ART3,5 and represents a critical hurdle for HIV-1 eradication strategies6,7. The temporal dynamics of seeding the viral reservoir has not previously been defined but has been presumed to occur during peak viremia in acute HIV-1 infection. To evaluate the impact of early ART on the viral reservoir, we initiated suppressive ART at various timepoints following mucosal SIV infection of rhesus monkeys.
We inoculated 20 Indian origin adult rhesus monkeys (Macaca mulatta) that did not express the protective MHC class I alleles Mamu-A*01, Mamu-B*08, and Mamu-B*17 with 500 TCID50 SIVmac2518–10 by the intrarectal route. We initiated ART on days 3, 7, 10, and 14 following infection with a pre-formulated cocktail of tenofovir, emtricitabine, and dolutegravir (see Methods), and a control group received no ART (n=4/group). ART was administered daily by subcutaneous injection for 24 weeks. Treatment on day 3 following infection resulted in no detectable viremia (<50 RNA copies/ml)11 at any timepoint in 4 of 4 monkeys (Fig. 1a). In contrast, treatment on days 7, 10, and 14 abruptly interrupted the exponential growth of the virus and reduced plasma viral RNA to undetectable levels within 3–4 weeks. The mean levels of plasma viral RNA at the time of ART initiation in these groups of monkeys were 5.88 log copies/ml (day 7), 7.11 log copies/ml (day 10), and 7.50 log copies/ml (day 14), which were comparable with the levels of plasma viral RNA in untreated controls at these timepoints (Fig. 1b). Viral dynamics modeling12 revealed an initial exponential growth rate of 1.5 ± 0.5 per day, corresponding to a basic reproductive ratio of R0 = 9.5 ± 5.1 (see Methods; Extended Data Fig. 1; Extended Data Table 1). An exponential decay rate of plasma viremia following ART initiation of 0.60 ± 0.17 per day was observed in all the treated groups, corresponding to a 1.3 ± 0.4 day half-life of infected cells (Extended Data Fig. 1).
Figure 1. Viral decay kinetics after treatment with ART.

Log plasma viral RNA (copies/ml) in rhesus monkeys infected with SIVmac251 and following initiation of ART on days 3, 7, 10, and 14 of infection (a) or with no ART (b). Assay sensitivity is 50 RNA copies/ml. Red arrows indicate initiation of ART. Black dots below x-axis indicate sampling timepoints.
Following initial control of viremia, all animals treated with ART exhibited undetectable plasma viral loads (<50 RNA copies/ml) for the full 24 week course of suppressive therapy with no detectable viral blips (Fig. 1a), demonstrating the potency and consistency of this ART regimen. Moreover, ultrasensitive plasma viral load assays at week 20 also proved negative (<6 RNA copies/ml)13 in all animals (Extended Data Fig. 2). In addition, viral sequences from stimulated PBMC from ART suppressed, SIV-infected monkeys using the same ART regimen revealed no viral sequence evolution over 6 months in a separate study (J.B.W., unpublished data). Furthermore, treatment intensification studies in SIV-infected rhesus monkeys in which the protease inhibitor darunavir was added to the current ART regimen did not lead to improved virologic control (R.G., unpublished data). Taken together, these data suggest that the ART regimen that was utilized in the present study was fully suppressive.
We next assessed the development of SIV-specific humoral and cellular immune responses in these animals. Animals treated on day 3 following infection developed no detectable SIV Env-specific antibody responses by ELISA (Fig. 2a) and no detectable SIV Env-, Pol-, or Gag-specific T lymphocyte responses by IFN-γ ELISPOT assays (Fig. 2b) at weeks 4, 10, and 20 or 24 of infection. In contrast, animals treated on days 7, 10, and 14 developed detectable but lower SIV-specific humoral and cellular immune responses as compared with untreated controls, presumably as a result of reduced antigenic stimulus following ART initiation. Multiparameter intracellular cytokine staining (ICS) assays confirmed that SIV Gag-specific CD8+ and CD4+ T lymphocyte responses were undetectable in animals treated on day 3 and were lower in animals treated on days 7, 10, and 14 as compared with untreated controls (Extended Data Figs. 3–4). Gag-specific CD8+ and CD4+ T lymphocytes in animals treated on days 7, 10, and 14 also exhibited reduced immune activation and proliferation as measured by Ki67 expression as compared with untreated controls (Extended Data Fig. 4). These data demonstrate that initiation of ART on day 3 blocked the emergence of plasma viremia and abrogated the induction of SIV-specific humoral and cellular immune responses.
Figure 2. SIV-specific humoral and cellular immune responses during ART.

Env-specific ELISA antibody titers at weeks 0, 4, 10, and 24 (a) and Env-, Pol-, and Gag-specific IFN-γ ELISPOT responses at weeks 0, 4, 10, and 20 in SIV-infected monkeys that initiated ART on days 3, 7, 10, and 14 of infection or with no ART (b). Mean responses are shown (N=4 animals per group). Error bars reflect standard errors.
We next determined the impact of early ART on levels of proviral DNA14 in peripheral blood mononuclear cells (PBMC), lymph node mononuclear cells (LNMC), and gastrointestinal mucosa mononuclear cells (GMMC) over the course of 24 weeks of treatment with suppressive ART. In animals that initiated ART on day 3, there was a striking anatomic discordance with no proviral DNA detected in PBMC at any timepoint (<3 DNA copies/106 cells) (Fig. 3a). In contrast, clear but low levels of proviral DNA were detected in inguinal LNMC and in colorectal GMMC in these animals, although proviral DNA declined to undetectable or nearly undetectable levels in 3 of 4 of these animals by week 24. In monkeys treated with ART on days 7, 10, and 14, proviral DNA was readily detected in PBMC as well as in LNMC and GMMC (Fig. 3b–d). Moreover, in animals treated with ART on days 10 and 14, proviral DNA in LNMC appeared to stabilize by week 12 with minimal subsequent decline, consistent with a stable viral reservoir (Fig. 3c–d). In untreated animals, proviral DNA was markedly higher than in ART treated animals with minimal decline (Fig. 3e). Analysis of sorted cell subpopulations demonstrated that proviral DNA was found primarily in central memory and transitional memory CD4+ T lymphocytes in lymph nodes on day 3 and in both PBMC and lymph nodes on day 7 following SIV infection (Extended Data Fig. 5).
Figure 3. Proviral DNA during ART.

Log proviral DNA (copies/106 cells) in peripheral blood mononuclear cells (PBMC), lymph node mononuclear cells (LNMC), and gastrointestinal mucosa mononuclear cells (GMMC) in monkeys that initiated ART on days 3 (a), 7 (b), 10 (c), and 14 (d) of infection or with no ART (e). Comparisons of mean levels of log proviral DNA/106 cells at the time of ART discontinuation (week 24) in ART treated and untreated (NT) monkeys in PBMC (f), LNMC (g), and GMMC (h) are also shown (N=4 animals/group). Assay sensitivity is 3 DNA copies/106 cells. P-values reflect one-sided t-tests. Error bars reflect standard errors.
These data indicate that initiation of ART on day 3 reduced levels of proviral DNA at week 24 by at least 2.2 logs in PBMC (P<0.0001; Fig. 3f), 1.0 log in LNMC (P=0.004; Fig. 3g), and 0.9 log in GMMC (P=NS; Fig. 3h) as compared with initiation of ART on day 14. The high variability in the GMMC samples likely reflected sampling variation in this anatomic compartment. Compared to untreated animals, initiation of ART on day 3 reduced levels of proviral DNA at week 24 by at least 3.3 logs in PBMC (P<0.0001; Fig. 3f) and 3.1 logs in LNMC (P<0.0001; Fig. 3g).
At week 24, ART was discontinued, and animals were monitored twice weekly for evidence of viral rebound. Viral rebound, defined as plasma viral RNA >50 copies/ml, occurred in all animals (Fig. 4a). In particular, viral rebound occurred in 4 of 4 animals that initiated ART on day 3, albeit with 3-fold delayed kinetics as compared with animals that initiated ART at later timepoints (median 21 days to viral rebound in the day 3 treated animals compared with 7 days in the day 14 treated animals; P<0.001; Fig. 4b). The median log setpoint viral load following rebound, defined as viral loads on days 56–112 following ART discontinuation, was also 1.04 log RNA copies/ml lower for all the ART treated animals as compared with untreated controls (4.59 log RNA copies/ml for all treatment groups combined vs. 5.63 log RNA copies/ml for untreated animals; P=0.01; Fig. 4c), suggesting a benefit to early ART, although no significant differences were observed among setpoint viral loads in the day 3, 7, 10, and 14 treatment groups. Setpoint viral loads in the untreated animals were comparable with historical controls9,10. Taken together, these data show that the persistent viral reservoir was seeded by day 3 of infection and led to viral rebound in all animals following ART discontinuation.
Figure 4. Viral rebound kinetics after ART discontinuation.

Log plasma viral RNA (copies/ml) in animals following discontinuation of ART at week 24 in monkeys that initiated ART on days 3, 7, 10, and 14 of infection (a). Assay sensitivity is 50 RNA copies/ml. Median times to viral rebound, defined as the first timepoint at which plasma viral RNA was >50 copies/ml, is also shown by the red bars (b). Median setpoint viral loads following viral rebound, defined as day 56–112 following ART discontinuation, are also shown by the red bars in ART treated monkeys compared with untreated monkeys (c). P-values reflect one-sided t-tests.
To gain mechanistic insight into the kinetics of the viral rebound, we utilized viral dynamics modeling15,16 (see Methods; Extended Data Fig. 6; Extended Data Table 2). Initiation of ART on day 3 as compared with days 7, 10, and 14 resulted in lower modeled residual viral loads at the time of ART discontinuation (P=0.01) and a trend towards a greater viral growth rate R0 during viral rebound (P=0.06), but no difference in post-rebound setpoint viral loads (Extended Data Fig. 7). Average R0 during viral rebound was 4.2 ± 1.8 in the day 3 treated animals as compared with 2.3 ± 0.6 in the day 14 treated animals (P=0.05; Extended Data Fig. 7), presumably reflecting the partially effective SIV-specific immune responses in the latter group (Fig. 2). Total plasma viremia during acute infection was interpolated and calculated as the area under the curve for pre-ART viral loads (AUC VL) (Extended Data Fig. 8). Consistent with recent findings in acute HIV-1 infection in humans16, the AUC VL during acute infection correlated with levels of proviral DNA in PBMC (P<0.0001; Fig. 5a), LNMC (P=0.005; Fig. 5b), and GMMC (P=0.04; data not shown) at the time of ART discontinuation. Moreover, both the AUC VL (P<0.0001; Fig. 5c) and proviral DNA in PBMC at the time of ART discontinuation (P=0.003; Fig. 5d) correlated inversely with the interpolated time to viral rebound. These data suggest that total plasma viremia during acute infection and proviral DNA immediately prior to ART discontinuation may predict the time to viral recrudescence.
Figure 5. Viral dynamics and correlations.

Correlations of area under the curve for pre-ART viral loads (AUC VL) with proviral DNA in PBMC (a) and LNMC (b) at the time of ART discontinuation are shown. Correlations of AUC VL (c) and proviral DNA in PBMC prior to ART discontinuation (d) with the interpolated time to viral rebound are also shown. R2 and P-values were calculated from correlation analyses, and trendlines were calculated using total least squares regression. Animals with undetectable proviral DNA were plotted at the detection limit of 3 DNA copies (0.48 log)/106 cells.
In this study, we show that the viral reservoir can be seeded substantially earlier than previously recognized. Following intrarectal SIV infection of rhesus monkeys, the viral reservoir was seeded during the first few days of infection, during the “eclipse phase”, and prior to detectable viremia, probably in the mucosal and lymphoid tissues that represent the first sites of viral replication17. Consistent with this finding, we observed proviral DNA in lymph nodes and in gastrointestinal mucosa but not in PBMC in animals treated on day 3 following infection (Fig. 3a; Extended Data Fig. 5). The observation that the viral reservoir can be seeded prior to detectable viremia suggests that substantial pathogenesis occurs in tissues in the first few days following mucosal virus exposure and prior to virus replication in peripheral blood, which has important implications for HIV-1 therapeutics and eradication strategies.
Our data are concordant with recent clinical studies that have demonstrated that early ART can reduce the size of the viral reservoir and delay or reduce viral rebound following ART discontinuation in humans18–22. Our findings similarly show that early ART decreased proviral DNA in blood and tissues (Fig. 3f–h) and both delayed and reduced viral rebound (Fig. 4b–c) following ART discontinuation in SIV-infected rhesus monkeys. Moreover, our observations extend prior studies that have shown effective post-exposure prophylaxis with short courses of ART when initiated 24 hours following SIV infection in monkeys23,24. However, in the present study, initiation of suppressive ART even as early as day 3 of infection failed to eliminate the viral reservoir and did not prevent viral rebound despite 24 weeks of effective therapy. In addition, our data (Fig. 4a) are consistent with clinical studies that have shown that the vast majority HIV-1-infected individuals who initiate ART during acute infection show viral rebound following discontinuation of ART25–27.
Our findings contrast with the sustained remission and potential cure of a viremic baby that was treated with ART at 31 hours of life28. It is possible that this baby was inoculated parenterally with maternal cells instead of mucosally with virus, resulting in rapid viremia without a previremic eclipse phase of viral replication in mucosal and lymphoid tissues. The positive outcome in this baby might therefore have reflected the route of transmission, the lack of an eclipse phase in tissues, the very rapid initiation of ART, and/or the paucity of memory CD4+ T lymphocytes in the neonatal immune system29.
Clinical studies are required to confirm our observations, since important differences exist between SIV infection of rhesus monkeys and HIV-1 infection of humans. For example, the SIV dose used in the present study in monkeys was selected to limit the number of transmitted/founder viruses but also to infect all animals8 and thus was substantially higher than typical HIV-1 doses in humans. Nevertheless, the higher challenge dose has been shown to shorten the eclipse period and to lead to earlier plasma viremia8. Additional virologic and immunologic differences may also exist between SIV-infected rhesus monkeys and HIV-1-infected humans.
The strikingly early seeding of the viral reservoir within the first few days of infection is sobering and presents new challenges to HIV-1 eradication efforts. If HIV-1 similarly seeds a persistent viral reservoir in mucosal and lymphoid tissues during the eclipse phase of infection and prior to viremia following sexual exposures in humans, then it will be very difficult to initiate ART prior to reservoir seeding, since viremia is typically utilized for the clinical diagnosis of acute HIV-1 infection. Taken together, our data suggest that extremely early initiation of ART, extended ART duration for years, and most likely additional interventions that activate the viral reservoir will be required for HIV-1 eradication. Moreover, an improved understanding of the virologic parameters that predict viral rebound following ART discontinuation will help guide future HIV-1 eradication efforts.
Methods
Animals
20 outbred, Indian-origin, young adult, male and female rhesus monkeys (Macaca mulatta) were genotyped and selected as negative for the protective MHC class I alleles Mamu-A*01, Mamu-B*08, and Mamu-B*17. Animals expressing susceptible and resistant TRIM5α alleles were distributed amongst the groups. Animals were otherwise randomly allocated to groups. All monkeys were housed at Bioqual, Rockville, MD. Animals were infected with 500 TCID50 of our SIVmac251 challenge stock8–10 by the intrarectal route. Monkeys were bled up to two times per week for viral load determinations. Assays were performed blinded. All animal studies were approved by the appropriate Institutional Animal Care and Use Committee (IACUC).
ART regimen
The pre-formulated antiretroviral therapy (ART) cocktail contained two reverse transcriptase inhibitors, 20 mg/mL tenofovir (TFV) and 50 mg/mL emtricitabine (FTC) plus 2.5 mg/mL of the integrase inhibitor dolutegravir (DTG) in a solvent containing 25% (v/v) polyethylene glycol 400 (PEG-400), 15% (w/v) captisol and 0.075 N sodium hydroxide (NaOH) in water. This ART cocktail was administered once daily at 1 mL/kg body weight via the subcutaneous route. The cocktail was prepared by mixing DTG stock solution (10 mg/mL of DTG in PEG-400), TFV and FTC stock solution (80 mg/mL of TFV and 200 mg/mL of FTC in 0.3 N NaOH), and 30% (w/w) captisol solution at a 1:1:2 (v:v:v) ratio. The final solution was clear and at pH ~6. It was sterile filtered, aliquoted into sterile glass vials in a biosafety cabinet, and frozen at −20°C until used.
Cellular immune assays
SIV-specific cellular immune responses were assessed by IFN-γ ELISPOT assays and multiparameter ICS assays essentially as previously described9. 12-color ICS assays were performed with the Aqua green-fluorescent reactive dye (Invitrogen, L23101) and predetermined titers of mAbs (Becton-Dickinson) against CD3 (SP34; Alexa Fluor 700), CD4 (OKT4; BV711, Biolegend), CD8 (SK1; allophycocyanin-cyanine 7 [APC-Cy7]), CD28 (L293; BV610), CD95 (DX2; allophycocyanin [APC]), CD69 (TP1.55.3; phycoerythrin-Texas red [energy-coupled dye; ECD]; Beckman Coulter), gamma interferon (IFN-γ) (B27; phycoerythrin-cyanine 7 [PE-Cy7]), Ki67 (B56; fluorescein isothiocyanate [FITC]), CCR5 (3A9; phycoerythrin [PE]), CCR7(3D12; Pacific Blue), and PD-1(EH21.1; peridinin chlorophyll-A-cyanine 5.5 [PerCP-Cy5.5]). IFN-γ backgrounds were consistently <0.01% in PBMC.
Humoral immune assays
SIV Env-specific antibody responses were assessed by a direct ELISA essentially as previously described9.
Viral RNA assays
Viral RNA was isolated from cell-free plasma using a viral RNA extraction kit (Qiagen) and was quantitated essentially as previously described11. RNA was isolated by phenol-chloroform purification followed by ethanol precipitation. All purified RNA preparations were quantified by optical density. Quantitative RT-PCR was conducted in a 2-step process. First, RNA was reverse transcribed in parallel with an SIV-gag RNA standard using the gene-specific primer sGag-R 5′CACTAGGTGTCTCTGCACTATCTGTTTTG-3′. All samples were then treated with RNAse H (Stratagene) for 20 minutes at 37°C. Primer sequences were adapted from those described11 including the forward primer s-Gag-F: 5′-P: 5′-CTTCCTCAGTGTGTTTCACTTTCTCTTCTGCG-3′, linked to Fam and BHQ (Invitrogen, Carlsbad, CA). All reactions were carried out on a 7300 ABI Real-Time PCR system (Applied Biosystems) in triplicate according to the manufacturer’s protocols. Ultrasensitive viral load assays were performed essentially as described13.
Proviral DNA assays
Lymph node and gastrointestinal mucosal biopsies were processed as single cell suspensions, and levels of tissue-specific proviral DNA were quantitated as previously described14. Total cellular DNA was isolated from 5x106 cells using a QIAamp DNA Blood Mini kit (Qiagen). The absolute quantification of viral DNA in each sample was determined by qPCR using primers specific to a conserved region SIVmac239. All samples were directly compared to a linear virus standard and the simultaneous amplification of a fragment of human GAPDH gene. The sensitivity of linear standards was compared against the 3D8 cell line as a reference standard as described14. PCR assays were performed with 100–200 ng sample DNA.
Viral dynamics modeling and statistical analyses
Plasma viral load levels pre-treatment, during treatment, and following treatment interruption were fit to a viral dynamics model to determine key parameters12. In order to derive individual level estimates with limited data points for each animal, we used a simplified version of the full system of differential equations describing the viral dynamics model, which had fewer parameters and could be solved analytically. All fits were evaluated using the nlinfit function in the MATLAB Statistical Toolbox. For acute infection in all animals treated on days 7, 10 or 14, and for rebound in all animals, data points below the limit of detection were treated as missing data (except for the last undetectable value after infection or interruption, which we took to equal the detection limit of 50 copies/ml). Treating undetectable viral load values as left-censored data using a maximum likelihood approach did not noticeably change the results.
Pre-treatment and during treatment
We fit the viral dynamics observed following infection and treatment up to week 24 using a three-parameter piecewise function:
| (1) |
where tART, the time that treatment was started, was defined based on the cohort as 3, 7, 10, or 14 days. Here v0 is the effective infectious viral load from which the infection starts (v(0)), a is the growth rate of viral load during acute infection, and d is the decay rate of viral load once treatment is started (if treatment is effective, the lifespan of infected cells is approximately 1/d). In essence, the model allows viral load to increase exponentially until treatment begins, and then decrease exponentially until it is below the detection limit. This approximates the observed dynamics well because a stable setpoint was not reached prior to treatment, and insufficient data points above the detection limit existed during viral decay to observe biphasic behavior.
The basic reproductive ratio of the infection, defined as the average number of new infected cells produced by a single infected cell during early infection, can be calculated from these parameters as R0 = a/d +1. However, others have reported12 that due to the intracellular delay between infection and the production of virus, this formula underestimates R0. To correct for this effect, we use a modified expression, R0 = (a/d +1)(ap +1), where p is the average length of the delay (which we take to be 1 day).
The area-under-the-curve of viral loads (AUC VL) was used as a measure of the total number of cells infected15,16. The AUC VL was used as a measure of the total amount of infections occurring, which is a reasonable approximation for acute SIV infection since CD4+ T cell levels do not change consistently or significantly. We assumed that ART was fully effective so that there were no new infections following ART initiation, leading to the following formula:
| (2) |
No fits were conducted for animals treated after 3 days, since all observed viral load values were below the limit of detection. To estimate AUC VL for these, we used the average values of v0 and a from the remaining 12 animals in the above formula with tART = 3.
Following ART discontinuation
We fit the viral dynamics observed following ART discontinuation (starting at week 24) using a separate three-parameter function:
| (3) |
Here t is the time since interruption. The parameter vr is the infectious residual viral load from which the infection starts (v(0)), r is the growth rate of viral load during rebound, and vmax is the viral setpoint reached after rebound. This logistic equation is an approximation to the full viral dynamics model that smooths out both high frequency fluctuations in viral load and lags in uninfected CD4+ T cell dynamics (such as overshooting the setpoint)15. Using these fits, we can interpolate the time at which viral load crossed the threshold of 50 copies/ml to define the rebound time. Similar to pre-treatment, we can calculate the post-rebound basic reproductive ratio as R0 = (r/d +1)(a p +1), where d is the viral decay measured during treatment and p is the average length of the intracellular delay between infection and virion production which is assumed to be exponentially distributed (and which we take to be 1 day).
Statistics
All comparisons between cohorts were done using unpaired, equal variance, one-tailed t-tests. Correlation analysis was carried out in using the MATLAB corrcoef function (Pearson correlation). The standardized major axis method was employed for regression analysis (also known as “geometric mean regression” or “reduced major axis regression”), using the gmregress function for the MATLAB Statistical Toolbox (available at http://www.mathworks.com/matlabcentral/fileexchange/27918-gmregress).
Extended Data
Extended Data Figure 1.
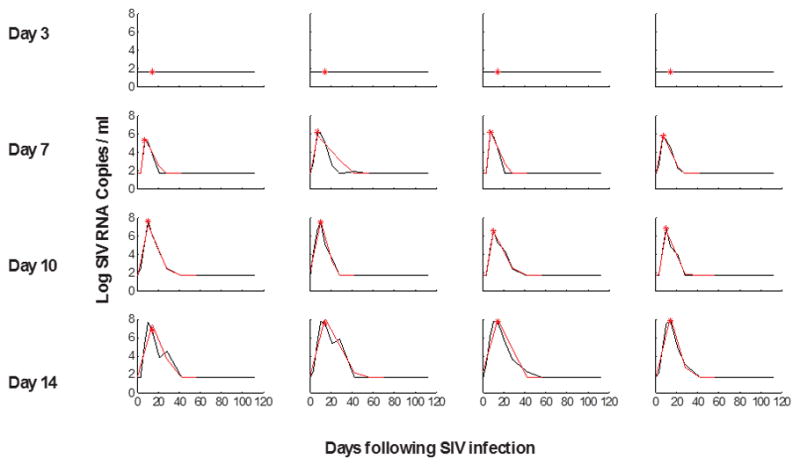
Viral dynamics modeling of initial viral growth and decay following ART initiation.
Red lines indicate fitted values from the model in monkeys that initiated ART on days 3, 7, 10, and 14 of infection and were utilized for AUC VL calculations. The red asterisk indicates the time of treatment initiation.
Extended Data Figure 2.
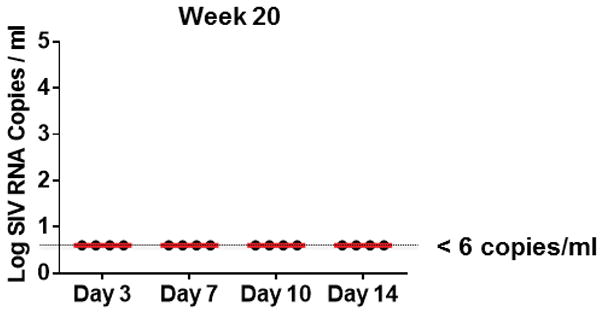
Ultrasensitive plasma viral loads in monkeys during ART.
Log plasma viral RNA (copies/ml) at week 20 in rhesus monkeys infected with SIVmac251 and following initiation of ART on days 3, 7, 10, and 14 of infection. Assay sensitivity is 6 RNA copies/ml.
Extended Data Figure 3.
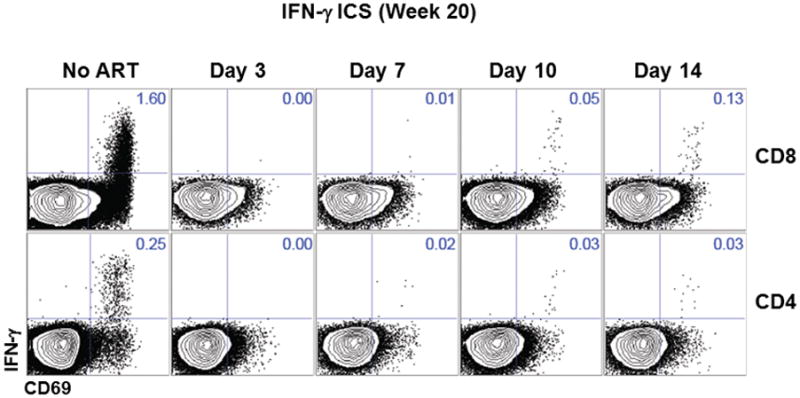
Intracellular cytokine staining raw data of Gag-specific CD8+ and CD4+ T cells.
Representative data for the magnitude of Gag-specific IFN-γ+ CD8+ and CD4+ T cell responses at week 20 in monkeys that initiated ART on days 3, 7, 10, and 14 of infection or with no ART.
Extended Data Figure 4.
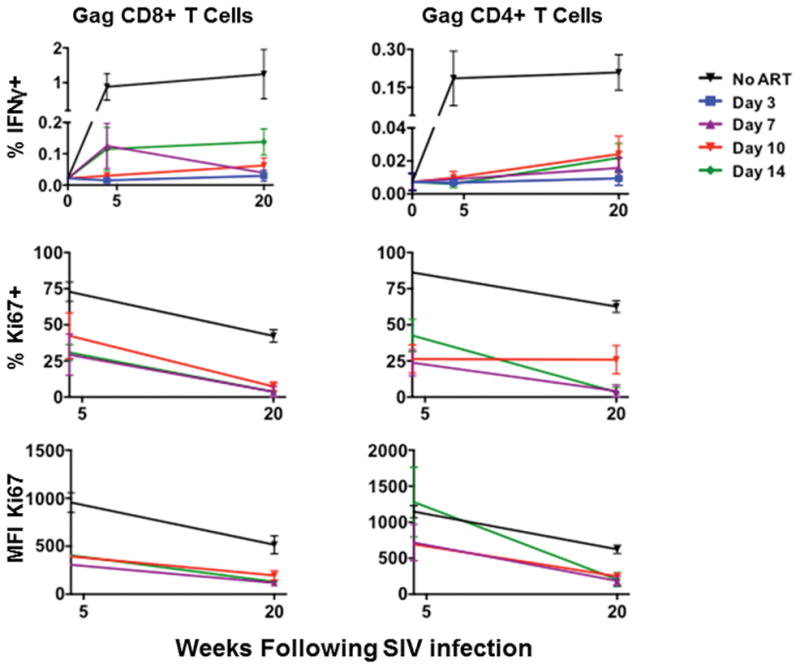
Intracellular cytokine staining of Gag-specific CD8+ and CD4+ T cells.
Summary data for the magnitude of Gag-specific IFN-γ+ CD8+ and CD4+ T cell responses and Ki67 expression at week 4 and week 20 in monkeys that initiated ART on days 3, 7, 10, and 14 of infection or with no ART (N=4 animals per group).
Extended Data Figure 5.
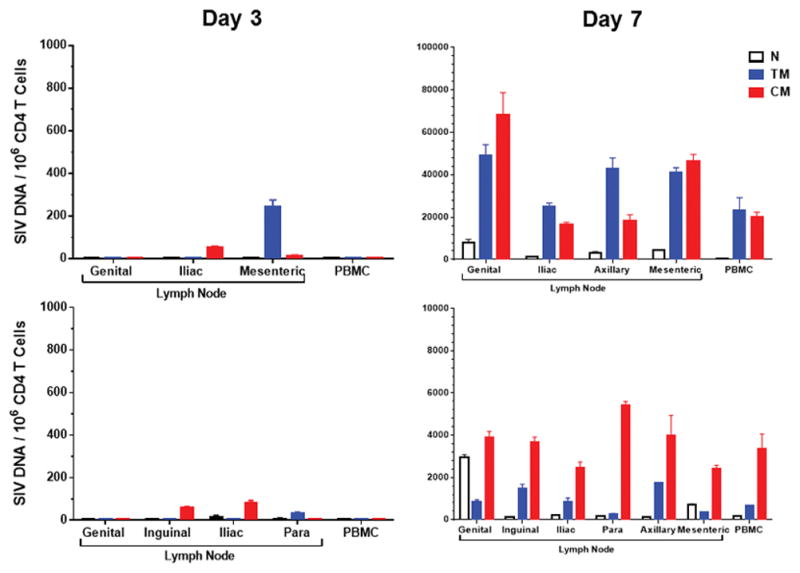
Proviral DNA in CD4+ T cell subpopulations during ART.
Log proviral DNA (copies/106 CD4 T cells) in sorted naïve (N), transitional effector memory (TM), and central memory (CM) CD4 T cell subpopulations from PBMC and from genital, inguinal, iliac, para-aortic, axillary, and/or mesenteric lymph nodes obtained from two animals necropsied on day 3 and two animals necropsied on day 7 following mucosal SIVmac251 infection.
Extended Data Figure 6.
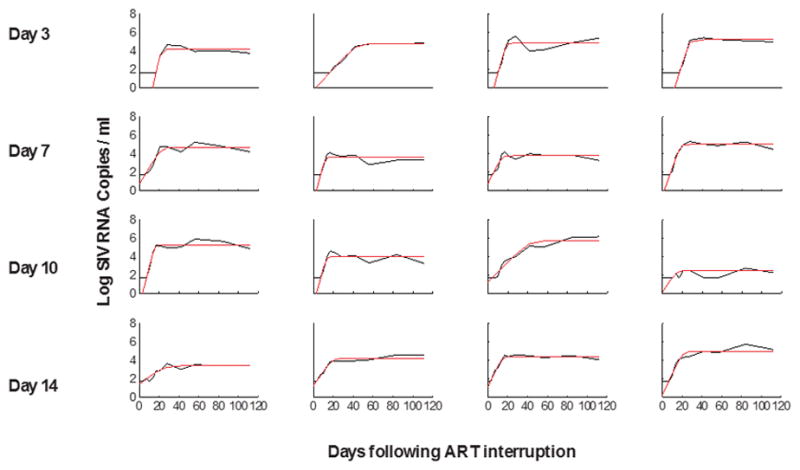
Viral dynamics modeling of viral rebound following ART discontinuation.
Red lines indicate fitted values from the model in monkeys that initiated ART on days 3, 7, 10, and 14 of infection.
Extended Data Figure 7.
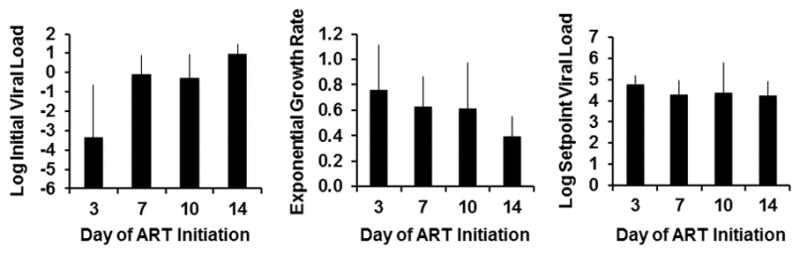
Viral kinetics and setpoint viral loads following ART discontinuation.
Log initial viral loads, exponential viral growth rate, and log setpoint viral loads following viral rebound derived from the model fits in groups of monkeys that initiated ART on days 3, 7, 10, and 14 of infection (N=4 animals per group).
Extended Data Figure 8.
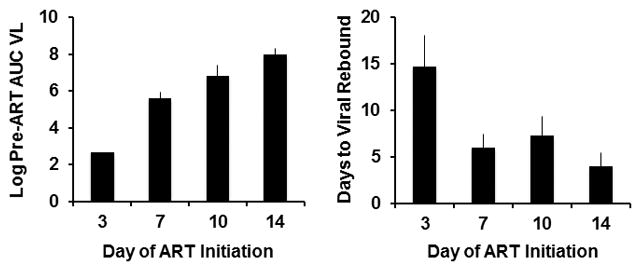
Early ART impacts AUC VL and time to viral rebound.
Log AUC VL and interpolated time of viral rebound derived from the model fits are shown in monkeys that initiated ART on days 3, 7, 10, and 14 of infection (N=4 animals per group).
Extended Data Table 1.
Best-fit parameters for viral dynamics pre-ART and during ART.
Fitted parameters include v0, the effective infectious viral load from which the infection starts, a, the growth rate of viral load during acute infection, and d, the decay rate of viral load once treatment is started. See the Methods section, including Equations 1 and 2, for detailed descriptions.
| Fitted parameters | Derived parameters | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Days until ART started | log10(v0) | a | d | Half-life (In(2)/d) | R0 | log10(AUC) |
| 3 | - | - | - | - | - | 2.6 |
| 3 | - | - | - | - | - | 2.6 |
| 3 | - | - | - | - | - | 2.6 |
| 3 | - | - | - | - | - | 2.6 |
| 7 | −1.16 | 2.19 | 0.50 | 1.38 | 17.10 | 5.1 |
| 7 | 1.44 | 1.45 | 0.29 | 2.37 | 14.57 | 5.7 |
| 7 | −1.77 | 2.66 | 0.58 | 1.19 | 20.30 | 5.9 |
| 7 | 1.34 | 1.49 | 0.56 | 1.24 | 9.13 | 5.7 |
| 10 | 1.27 | 1.38 | 0.63 | 1.11 | 7.65 | 7.1 |
| 10 | 2.16 | 1.25 | 0.91 | 0.76 | 5.33 | 7.5 |
| 10 | −0.25 | 1.55 | 0.51 | 1.36 | 10.32 | 6.3 |
| 10 | −0.24 | 1.57 | 0.60 | 1.15 | 9.23 | 6.4 |
| 14 | 1.76 | 0.94 | 0.62 | 1.12 | 4.86 | 7.5 |
| 14 | 1.79 | 1.05 | 0.49 | 1.42 | 6.45 | 8.2 |
| 14 | 2.30 | 0.94 | 0.53 | 1.30 | 5.36 | 8.0 |
| 14 | 1.54 | 1.11 | 0.91 | 0.76 | 4.69 | 8.3 |
|
| ||||||
| Mean | 0.85 | 1.46 | 0.60 | 1.26 | 9.58 | NA |
| STD | 1.35 | 0.51 | 0.17 | 0.41 | 5.16 | NA |
Extended Data Table 2.
Best-fit parameters for viral dynamics following ART discontinuation.
Fitted parameters include vr, the infectious residual viral load from which the infection starts, r, the growth rate of viral load during rebound, and vmax, the viral setpoint reached after rebound. See the Methods section, including Equation 3, for detailed descriptions.
| Fitted parameters | Derived Quantities | ||||
|---|---|---|---|---|---|
|
| |||||
| Days until ART started | log10(vr) | r | log10(vmax) | R0 | Estimated days to VL=50 copies/ml |
| 3 | −6.57 | 1.12 | 4.22 | 6.10 | 17.0 |
| 3 | −0.27 | 0.27 | 4.83 | 1.84 | 16.8 |
| 3 | −2.18 | 0.85 | 4.88 | 4.51 | 10.5 |
| 3 | −4.34 | 0.80 | 5.22 | 4.20 | 17.5 |
| 7 | 0.72 | 0.40 | 4.70 | 2.53 | 5.6 |
| 7 | −1.23 | 0.94 | 3.62 | 8.12 | 7.2 |
| 7 | 0.76 | 0.49 | 3.79 | 2.75 | 4.4 |
| 7 | −0.60 | 0.69 | 5.01 | 3.78 | 7.7 |
| 10 | −1.33 | 0.96 | 5.26 | 4.95 | 7.3 |
| 10 | −1.17 | 0.89 | 4.00 | 3.74 | 7.4 |
| 10 | 1.25 | 0.24 | 5.71 | 1.84 | 4.2 |
| 10 | 0.12 | 0.38 | 2.50 | 2.24 | 10.1 |
| 14 | 1.38 | 0.19 | 3.39 | 1.56 | 3.9 |
| 14 | 1.23 | 0.36 | 4.22 | 2.34 | 3.0 |
| 14 | 1.07 | 0.52 | 4.41 | 3.02 | 2.7 |
| 14 | 0.24 | 0.51 | 5.00 | 2.37 | 6.6 |
Acknowledgments
We thank M. Pensiero, M. Marovich, C. Dieffenbach, W. Wagner, C. Gittens, J. Yalley-Ogunro, M. Nowak, R. Siliciano, D. Persaud, L. Picker, F. Stephens, R. Hamel, K. Kelly, and L. Dunne for generous advice, assistance, and reagents. The SIVmac239 peptides were obtained from the NIH AIDS Research and Reference Reagent Program. We acknowledge support from the U.S. Military Research and Material Command and the U.S. Military HIV Research Program through its cooperative agreement with the Henry M. Jackson Foundation (W81XWH-07-2-0067, W81XWH-11-2-0174); the National Institutes of Health (AI060354, AI078526, AI084794, AI095985, AI096040, AI100645); and the Ragon Institute of MGH, MIT, and Harvard. The views expressed in this manuscript are those of the authors and do not represent the official views of the Department of the Army or the Department of Defense.
Footnotes
Author Contributions
J.B.W., R.G., M.L.R., J.H.K., N.L.M., and D.H.B. designed the studies and interpreted the data. J.B.W. and S.S. led the virologic assays. P.P., J.L., M.S., L.P., C.C., J.S., S.B., J.Y.S., A.L.B., L.E.P., E.N.B., and K.M.S. led the study operations and the immunologic assays. A.L.H. and D.I.S.R. led the mathematical modeling and statistical analysis. M.G.L. led the clinical care of the rhesus monkeys. B.L., J.H., J.H., and R.G. developed the antiretroviral drug cocktail. J.B.W. and D.H.B. wrote the paper with all co-authors.
The authors declare no competing financial interests.
References
- 1.Finzi D, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 2.Zhang L, et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N Engl J Med. 1999;340:1605–1613. doi: 10.1056/NEJM199905273402101. [DOI] [PubMed] [Google Scholar]
- 3.Chun TW, Davey RT, Jr, Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. 1999;401:874–875. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- 4.Chun TW, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 5.Chun TW, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Persaud D, Zhou Y, Siliciano JM, Siliciano RF. Latency in human immunodeficiency virus type 1 infection: no easy answers. J Virol. 2003;77:1659–1665. doi: 10.1128/JVI.77.3.1659-1665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho YC, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, et al. Low-dose mucosal simian immunodeficiency virus infection restricts early replication kinetics and transmitted virus variants in rhesus monkeys. J Virol. 2010;84:10406–10412. doi: 10.1128/JVI.01155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barouch DH, et al. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature. 2012;482:89–93. doi: 10.1038/nature10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, et al. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature. 2009;457:87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cline AN, Bess JW, Piatak M, Jr, Lifson JD. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol. 2005;34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 12.Nowak MA, et al. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J Virol. 1997;71:7518–7525. doi: 10.1128/jvi.71.10.7518-7525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmer S, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003;41:4531–4536. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitney JB, et al. T-cell vaccination reduces simian immunodeficiency virus levels in semen. J Virol. 2009;83:10840–10843. doi: 10.1128/JVI.01202-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenbloom DI, Hill AL, Rabi SA, Siliciano RF, Nowak MA. Antiretroviral dynamics determines HIV evolution and predicts therapy outcome. Nat Med. 2012;18:1378–1385. doi: 10.1038/nm.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Archin NM, et al. Immediate antiviral therapy appears to restrict resting CD4+ cell HIV-1 infection without accelerating the decay of latent infection. Proc Natl Acad Sci U S A. 2012;109:9523–9528. doi: 10.1073/pnas.1120248109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haase AT. Targeting early infection to prevent HIV-1 mucosal transmission. Nature. 2010;464:217–223. doi: 10.1038/nature08757. [DOI] [PubMed] [Google Scholar]
- 18.Ananworanich J, et al. Impact of multi-targeted antiretroviral treatment on gut T cell depletion and HIV reservoir seeding during acute HIV infection. PLoS ONE. 2012;7:e33948. doi: 10.1371/journal.pone.0033948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wyl V, et al. Early antiretroviral therapy during primary HIV-1 infection results in a transient reduction of the viral setpoint upon treatment interruption. PLoS ONE. 2011;6:e27463. doi: 10.1371/journal.pone.0027463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hocqueloux L, et al. Long-term antiretroviral therapy initiated during primary HIV-1 infection is key to achieving both low HIV reservoirs and normal T cell counts. The Journal of antimicrobial chemotherapy. 2013;68:1169–1178. doi: 10.1093/jac/dks533. [DOI] [PubMed] [Google Scholar]
- 21.Saez-Cirion A, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013;9:e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steingrover R, et al. HIV-1 viral rebound dynamics after a single treatment interruption depends on time of initiation of highly active antiretroviral therapy. AIDS. 2008;22:1583–1588. doi: 10.1097/QAD.0b013e328305bd77. [DOI] [PubMed] [Google Scholar]
- 23.Tsai CC, et al. Effectiveness of postinoculation (R)-9-(2-phosphonylmethoxypropyl) adenine treatment for prevention of persistent simian immunodeficiency virus SIVmne infection depends critically on timing of initiation and duration of treatment. J Virol. 1998;72:4265–4273. doi: 10.1128/jvi.72.5.4265-4273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai CC, et al. Prevention of SIV infection in macaques by (R)-9-(2-phosphonylmethoxypropyl)adenine. Science. 1995;270:1197–1199. doi: 10.1126/science.270.5239.1197. [DOI] [PubMed] [Google Scholar]
- 25.Saez-Cirion A, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS pathogens. 2013;9:e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stohr W, et al. Duration of HIV-1 viral suppression on cessation of antiretroviral therapy in primary infection correlates with time on therapy. PLoS ONE. 2013;8:e78287. doi: 10.1371/journal.pone.0078287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenberg ES, et al. Safety and immunogenicity of therapeutic DNA vaccination in individuals treated with antiretroviral therapy during acute/early HIV-1 infection. PLoS ONE. 2010;5:e10555. doi: 10.1371/journal.pone.0010555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Persaud D, et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N Engl J Med. 2013;369:1828–1835. doi: 10.1056/NEJMoa1302976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Li H, Iampietro MJ, Barouch DH. Accelerated heterologous adenovirus prime-boost SIV vaccine in neonatal rhesus monkeys. J Virol. 2012;86:7829–7835. doi: 10.1128/JVI.00512-12. [DOI] [PMC free article] [PubMed] [Google Scholar]