

Abstract
Objective
To test the hypothesis that homocysteine (Hcy) impairs angiogenic outgrowth through an inducible nitric oxide synthase (iNOS)-dependent mechanism.
Methods
Adult C57Bl/6 mouse choroid explants were used in angiogenic outgrowth assays. Mouse microvascular endothelial cells were studied in culture during scrape induced migration and dispersed cell locomotion experiments. Activity of iNOS was manipulated with pharmacology (1400W) siRNA, and by use of choroid explants from iNOS knockout mice (iNOS −/−).
Results
Hcy (20 μM) significantly decreased the area of endothelial outgrowth without altering the number of cells in the choroid explant angiogenic assay, resulting in more densely packed outgrowth. Hcy prevented the outward orientation of actin filaments and decreased the number of actin projections along the leading edge of outgrowth. Hcy also slowed outgrowth from the edge of a scraped endothelial monolayer and in cultures of dispersed cells, Hcy impaired cell locomotion without affecting proliferation. Inhibition of iNOS activity rescued the effect of Hcy on area of explant outgrowth, cell density, number of projections, cell locomotion, and rate of outgrowth following scraping.
Conclusions
Hcy impairs microvascular endothelial outgrowth, but not proliferation, by disrupting cell locomotion through an iNOS-dependent mechanism.
Keywords: homocysteine, angiogenesis, inducible nitric oxide synthase, actin, endothelium
INTRODUCTION
Angiogenesis is essential throughout life for processes including organ development, tumor growth, weight loss or gain, responses to exercise training, and wound healing [9,11,23]. Angiogenesis is the sprouting of new capillaries from pre-existing vessels, which requires endothelial cell proliferation and directional motility. Motility involves the intricate coordination of cytoskeletal elements including filamentous actin (F-actin) [4]. Disruption of the angiogenic response gives rise to, or exacerbates, many clinical disorders because of the fundamental importance of microvessels for nutrient delivery to tissues [11]. While much is known about the process of angiogenesis, the mechanisms by which it is impaired are poorly understood in many disease states.
Homocysteine (Hcy) is an aminothiol formed during methionine metabolism. Hyperhomocysteinemia (HHcy), a condition of elevated blood levels of Hcy, is a graded risk factor for cardio- and neurovascular disease [16]. HHcy impairs angiogenesis in response to hindlimb ischemia [15], decreases microvascular growth in the chick embryo [29], and slows repair of scrape-wounded endothelium [12]. Resolution of these pathways could aid in treatment of HHcy-related diseases and inform the development of prevention-based therapies.
Though the mechanisms by which Hcy disrupts angiogenesis have yet to be resolved, the pathway is likely to include redox and/or nitrative stress [15]. In previous studies, we have shown that Hcy increases iNOS protein expression in a time and dose dependent manner in brain microvascular cells [25]. Specifically, we demonstrated that HHcy increases endothelial nitrative stress by an increase in the expression of inducible nitric oxide synthase (iNOS) [7,25]. Increased expression of iNOS leads to oxidant-induced disruption of F-actin [2]. F-actin dynamics are a necessary component of cell motility, propelling cell movement and producing exploratory projections that lead expanding outgrowth [33]. Whether Hcy-mediated increases in iNOS activity play a role in disrupting F-actin organization and endothelial outgrowth during angiogenesis is not known.
In this study, we investigated molecular mechanisms by which Hcy modulates angiogenesis and endothelial cell migration. We tested the hypothesis that iNOS activity is responsible for key features of the aberrant phenotypical responses to Hcy during angiogenic outgrowth.
MATERIALS AND METHODS
All procedures were approved by the Institutional Animal Care and Use Committee of Idaho State University and of the University of Tennessee Health Science Center and performed in accord with the National Institutes of Health Guide for the Care and Use of Laboratory Animals [1].
Pharmaceutical Treatments
D,L-homocysteine (Sigma-Aldrich, St Louis, MO) was dissolved in sterile PBS as a stock solution and dissolved in culture media to 20 μM for treatment. A concentration of 20 μM Hcy is within the ‘mild HHcy’ classification (15–30 μM) in humans [10] and has been verified in previous work by our lab to inhibit wound healing and increase iNOS levels in endothelial cells [12,25]. The selective iNOS antagonist, 1400W (N-[[3-(aminomethyl) phenyl] methyl]-ethanimidamide dihydrochloride; Cayman Chemical), was dissolved in sterile PBS as a stock solution and dissolved in culture media to 10 μM for treatment. VEGF165aa mouse recombinant protein (VEGF, vascular endothelial growth factor, Cat# GF140, Millipore, Billerica, MA) was dissolved in sterile water as a stock solution and diluted in culture media to a working concentration of 385 pM (15 ng/ml).
Choroid Tissue Explants
C57Bl/6 male mice (3.38 ± 0.4 months of age, n=20) were group housed on a 12 hrs light, 12 hrs dark cycle and provided food and water ad libitum in the Idaho State University animal care facilities. Male iNOS knockout mice (iNOS−/−; 3–4 months of age, n=6) were bred at the University of Tennessee animal care facilities and similarly housed. Animals were euthanized by cervical dislocation under isoflurane anesthesia. Eyes from iNOS knockout animals were cyroprotected in complete media following standard procedures for cryopreservation of living cells and shipped overnight on dry ice from the University of Tennessee to Idaho State University. Extensive pilot studies validated this method for reliable preservation of tissue function for choroid vascular outgrowth experiments following cryorecovery. To harvest choroid explants, an incision was made across the cornea with a scalpel. The lens, vitreous and retina were removed by placing forceps close to the optic disk while pressing and pulling toward the incision. The remaining sclera, choroid, and retinal pigment epithelium were released from the eye socket by cutting the optic nerve. The tissue was rinsed in sterile PBS and laid flat in a glass petri dish. A biopsy punch with diameter of 1.5 mm was used to excise the explant tissue centered on the optic disk; this ensured explants of microvascular beds with consistent geometry and anatomic origin. Explants were placed in separate wells of a 12-well culture plate (Thermo Fisher Scientific, Rochester, NY), embedded in 600 μl of collagen-media solution (collagen type I, rat tail, Millipore; DMEM-H, Invitrogen, Grand Island, NY), with penicillin-streptomycin-amphotericin B (Invitrogen). NaOH was included to obtain the optimal pH of the collagen-media solution for solidification (pH ~7.0). Matrices with explants were maintained at 37°C for 2 hrs in the cell culture incubator to solidify (5% CO2 and 95% room air, passive humidity from pan of water). A global treatment of VEGF was established by adding media and growth factor on top of the solidified collagen [500 μL DMEM-H, 10% bovine calf serum (Hyclone FetalClone III; Fisher Scientific), penicillin-streptomycin-amphotericin B (Invitrogen), and 385 pM VEGF (15 ng/ml)]. This media was exchanged every 48 hrs. Explant cultures were harvested for analysis on day 4 of culture.
Cell Culture
Mouse microvascular endothelial cells [bEnd.3 [27]] were purchased from American Type Culture Collection (ATCC, Manassas, VA). The cells were cultured at 37°C in a humidified incubator with 5% CO2/95% room air in 75 cm2 plastic culture flasks (Thermo Scientific, Rochester, NY). Culture media comprised DMEM-H, 10% bovine calf serum (Hyclone, Logan, UT), and penicillin-streptomycin-amphotericin B (Invitrogen). For cell locomotion experiments, cells were plated at low density (<20% confluence) as dispersed, individual cells and immediately treated with Hcy, Hcy+VEGF, or 1400+Hcy+VEGF as indicated for 24 hrs. Human coronary artery smooth muscle cells (HCASMCs) and human coronary artery endothelial cells (HCAECs; Lonza, Walkersville, MD, USA) were cultured in growth medium (Genlantis; PM311500 for HCASMCs and PM112500 for HCAECs) under the same conditions.
Scrape Outgrowth Assay
Cells were studied three days post-confluence to allow the development of a stable monolayer with the full complement of adhesion molecules and morphology as previously described [5,6]. After three days of indicated pharmaceutical treatment, a scrape wound was induced with a 20 μl pipette tip (tip diameter 1 mm). The well was subsequently rinsed three times to remove scraped cells and debris. At wounding and 48 hrs after wounding, the area of the wound was measured. The difference in wound area between initial wounding and 48 hrs post-wounding was divided by the time elapsed to determine the average rate of outgrowth into the denuded area. A time course of 48 hrs allowed sufficient time for migration but ensured that none of the wounds had completely closed.
Time Lapse Imaging of Cell Locomotion
Cells were plated in 8-well coverslip chambers (Thermo Scientific; Nunc Lab-Tek) as dispersed individual cells at ~10–20% confluence. Culture conditions were maintained using a Tokai Hit stage-top incubator at 37°C, 5% CO2 (balance air) and passive humidity from a surrounding heated water moat. Time-lapse images were obtained every 15 mins over a 16 hr period with an Olympus FV1000 confocal laser scanning microscope. Single cell start and end positions were generated by tracking movement of the cell nucleus over time using the open-source MATLAB digitizing software DLTdv5 [21]. A MATLAB (version 7.10.0) program, written in our lab by co-author CHC, quantified the net distance travelled (start position to end position) per unit time.
Small interfering RNA (siRNA)
For experiments involving siRNA, confluent cells were transfected with iNOS specific siRNA (1 μM, Ambion Silencer Select-NOS2 #4390815, Life Technologies) for 24 hrs. Cells used in the scrape outgrowth assay were wounded at the time of transfection. After 24 hrs of transfection, cells were washed with PBS and incubated with the indicated pharmaceuticals.
Cell Proliferation
Mouse microvascular endothelial cells were plated at 20–30% confluence and 2 hrs later the media was changed to remove any cells that did not adhere. Cell number was quantified by counting the number of cells in a field of view at the center of each well (10X objective). A cell count was made 2 hrs after plating, indicated as time zero, and 24 hrs later. Proliferation was defined as the number of new cells (cell count at 24 hrs minus cell count at time 0). Cells for a given experiment were seeded at the same time to ensure that the starting densities were equal. Indicated treatment for cells began at time zero.
Immunohistochemistry
Explants were fixed with 4% paraformaldehyde (PFA) at room temperature for 30 mins. Cells were fixed with 4% PFA for 15 mins. Both were washed 3 times for 10 mins each with PBS, permeabilized with 0.1% Triton Χ-100 in PBS at room temperature, and incubated in 0.1% Triton Χ-100 for 45 mins. Cells were incubated in 0.1% Triton Χ-100 for 15 mins. Nuclei were labeled with Hoechst 33258 (Sigma Aldrich, 1 ng/ml) for 2 hrs (explants) or 20 mins (cells). F-actin was labeled with rhodamine phalloidin (6.6 μM) for 12 hrs (explants) or 30 mins (cells) in PBS. Isolectin (GS-IB4 Alexafluor 488, I21411, Life Technologies, 1:1000) was used as an endothelial cell marker. Rabbit anti-iNOS (ab15323, Abcam, 1:250) and rabbit anti-nitrotyrosine (A21285; Invitrogen, 1:1000) were used to label iNOS and 3-nitrotyrosine respectively.
Density of nuclei and organization of F-actin were measured with ImageJ [31] and the Java plug-in OrientationJ [32]. Cell number was quantified using ImageJ to count nuclei labeled with Hoechst 33258. F-actin in the explant outgrowth was labeled with rhodamine phalloidin; images were processed using OrientationJ to calculate the mean vector of actin orientation at the leading edge of outgrowth.
F-actin images were acquired using fluorescence microscopy (Leica, DMLFS) with a digital camera (MicroPublisher 3.3 RTV; Qimaging; Qcapture software). Phalloidin-labeled outgrowth was also visually examined with a fluorescent microscope to count the number of actin projections beyond the leading edge of growth. Images of isolectin B4 and iNOS labeling were acquired with an Olympus FV1000 inverted laser scanning confocal microscope. Images of isolectin B4 and iNOS shown in figures are Z-stacked, 3D projections.
Enzyme Linked Immunosorbent Assay (ELISA)
Cells grown to confluence and treated with the indicated pharmaceuticals for 3 days were fixed in PFA for 15 mins. Cells were rinsed three times for five mins with TBS (Tris-buffered saline), permeabilized with 1% Triton X-100 in TBS, and blocked with 2% bovine serum albumin in TBS with 0.05% Tween (TBST). Samples were then incubated with the indicated primary antibody in blocking solution overnight at 4° C and washed 3 times for 10 mins with TBST. For detection, cells were incubated with alkaline phosphatase-conjugated goat anti-rabbit IgG (Pierce) in blocking solution for 30 mins, and, after washing as indicated above, detected using a colorimetric ELISA substrate (BluePhos; KPL, Gaithersburg, MD, USA). Absorbance was measured at 620 nm using a plate reader (Synergy HT, BioTek).
Statistical Analyses
Alpha was set at 0.05. In initial experiments, right and left eyes of mice were paired so that one eye received the VEGF treatment and the other eye from the same animal received the experimental treatment condition. Significant differences between individual mice were not observed. Whether the left or right eye received any given treatment condition was systematically designed to ensure that right and left eyes had equal representation (± 1) in each treatment condition. When comparing only two groups, a student’s paired t-test was used. One-way analysis of variance (ANOVA) was used to compare three or more groups with Tukey or Dunnett post-hoc analysis. Analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). Data are presented as mean ± standard error of the mean.
RESULTS
Hcy Impairs Choroid Endothelial Outgrowth by an iNOS Dependent Mechanism
Explants exhibited little to no outgrowth without VEGF stimulation: vehicle 1.34 ± 0.71 mm2 vs. VEGF 8.76 ± 1.04 mm2 (p < 0.05, n=9/group). All further experiments were conducted on a VEGF background. Figure 1 shows representative images of all treatment conditions with differential interference contrast (DIC) (Figure 1 A–E) and corresponding fluorescence images where explants were labeled with the endothelial cell marker isolectin B4 (Figure 1 F–J). Outgrowth territories in all treatment groups were composed of endothelial cells (Figure 1 K), which formed a sprout-like network that extended out from the explant tissue disc (Figure 1 L). Endothelial cell specificity of the isolectin B4 was confirmed by the absence of labeling on a confluent monolayer of coronary artery smooth muscle cells (Figure 1 M) and the presence of labeling on a monolayer of coronary artery endothelial cells (Figure 1 N). Hcy treatment reduced the area of outgrowth (Figure 1 O) without reducing the number of cells (Figure 1 P), resulting in a smaller outgrowth territory with more densely packed cells (Figure 1 O and Q). The iNOS inhibitor 1400W rescued the effects of Hcy on total outgrowth area (Figure 1 O) without affecting the number of cells (Figure 1 P), resulting in an outgrowth area and density that was not different from controls (Figure O and Q). Similarly, explants harvested from iNOS −/− mice and treated with Hcy did not have impaired outgrowth or an increased density of cells within the resulting outgrowth as compared to the VEGF control (Figure 2).
Figure 1. Choroid Explant Tissue VEGF Stimulated Outgrowth.

(A–E) DIC images of explant outgrowth across treatments after four days of cell culture incubation with the corresponding fluorescence images of the same outgrowth labeled with isolectin B4 (F–J). (K) Representative image of explant outgrowth labeled with isolectin 488 (green) and the nuclear label, Hoechst (blue). Panel (L) is a representative image labeled with isolectin 488 (green) demonstrating the network of sprout-like endothelial cells projecting outward from the explant tissue that was measured as outgrowth across all treatment groups after 4 days of cell culture incubation. (M) Monolayer of vascular smooth muscle and endothelial cells (N) labeled with isolectin (green) and Hoechst (blue) demonstrating specificity of the labeling procedure. (O) Area of outgrowth for choroid explants treated as indicated for 4 days. * p < 0.05 from VEGF group, n=6. (P) Total cell count measured by the product of nuclei density and area of growth, n=6. (Q) Nuclei density of outgrowth, represented as nuclei per mm2, * p < 0.05 from VEGF group, n=6. E; explant, O; Outgrowth.
Figure 2. iNOS −/− Choroid Explant Outgrowth.

(A–B) DIC images of iNOS −/− explant outgrowth across treatments after four days of cell culture incubation with the corresponding fluorescence images of the same outgrowth labeled with isolectin B4 (C–D). (E) Area of outgrowth for iNOS −/− choroid explants treated as indicated for 4 days. * p < 0.05 from VEGF group, n=6. (P) Total cell count measured by the product of nuclei density and area of growth, n=6. (Q) Nuclei density of outgrowth, represented as nuclei per mm2, * p < 0.05 from VEGF group, n=6. E; explant, O; Outgrowth.
Hcy Disrupts F-actin Orientation at the Leading Edge by an iNOS Independent Mechanism
Actin organization was evaluated by measuring the mean vector of F-actin alignment at the leading edge of endothelial outgrowth. A vector aligned outward from the explant center (like spokes from a wheel axle) was defined as 0° and a vector aligned perpendicular to this was defined as 90° as shown in Figure 3 A. In VEGF-treated explants, endothelial sprouting and migration was consistently oriented outward from the explant, with F-actin aligning close to 0° (Figure 3 B and C). Hcy disrupted this organization and produced F-actin morphology nearly perpendicular to that of the VEGF-only group (Figure 3D). Inhibition of iNOS activity had no discernible effect on these measures of F-actin orientation under VEGF or VEGF+Hcy-treated conditions (Figure3 B, E, and F).
Figure 3. Measures of F-actin Orientation in Explant Outgrowth.

(A) Illustration of F-actin orientation measurement. (B) Quantification of orientation across treatment groups after 4 days of outgrowth. (C–F) Representative images of actin orientation in indicated treatment groups. Quantification of orientation was made using the ImageJ plug in; OrientationJ. Sections of outgrowth at the edge of the explant labeled with rhodamine phalloidin were imaged. A line that ran from the center of the explant tissue and bisected the center of the region of interest (black circle with white outline) was set at zero degrees. This line, representing direct outgrowth, is represented as a white line with a closed arrow head in panels A (labeled with 0°), and C–F. The red lines in panels C–F indicate the mean relative vector of F-actin orientation at the edge of outgrowth. This quantifies the deviation, in degrees, from direct outgrowth. * significant difference from VEGF group, VEGF group n=6, VEGF+Hcy group n=11, VEGF+1400W n=3, and VEGF+Hcy+1400W n=6. E; explant, O; Outgrowth.
Hcy Reduces the Number of Projections at the Leading Edge of Outgrowth by an iNOS Dependent Mechanism
Under control conditions (VEGF-only stimulated outgrowth), the leading edge of outgrowth formed numerous multicellular actin-filled projections that extended beyond the primary edge of growth (Figure 4 A and B). Hcy treatment reduced the number of these projections (Figure 4 A and C), consistent with reduced outgrowth area. Inhibiting iNOS with 1400W restored the occurrence of projections (Figure 4 A, D and E), indicating that iNOS is involved in forming or maintaining this outgrowth behavior.
Figure 4. Actin Extensions at Leading Edge of Outgrowth.

(A) After four days, explants were labeled with phalloidin and a visually inspected using a fluorescent microscope to count the number of actin extensions present at the leading edge of outgrowth per explant, * p < 0.05 from VEGF, VEGF group n=6, VEGF+Hcy group n=9, VEGF+1400W group n=3, VEGF+Hcy+1400W n=6. (B–E) Representative images of F-actin extensions at leading edge, labeled with phalloidin. White arrows indicate extensions at leading edge of outgrowth.
Hcy Increases iNOS Expression in Endothelium
Hcy treatment increased levels of iNOS expression within the explant outgrowth (Figure 5 A–J). A monolayer of microvascular endothelial cells also displayed an increase in iNOS expression with Hcy treatment (Figure 5 K). Treatment with 1400W decreased levels of iNOS in both the explant outgrowth and the endothelial cell line (Figure 5 D and K). Treatment with 1400W also reduced the formation of 3-nitrotyrosine (3-NT) under all conditions (Figure 5 L).
Figure 5. iNOS Expression in Endothelium with Hcy Treatment.

(A) Outgrowth after 4 days of treatment (VEGF 385 pM) labeled with a non-specific rabbit IgG antibody and anti-rabbit Alexa Fluor 647 (red). Fluorescence image (A) of the same area of growth shown in the DIC image (F). (B–E) iNOS rabbit IgG antibody labeling (red) with corresponding isolectin labeling (green) (G–J) of the same area of growth. For imaging of both the isolectin and the iNOS labeling, laser power, offset, and intensity levels were held constant across conditions. Outgrowth after 4 days of treatment (VEGF 385 pM, Hcy 20 μM, and 1400W 10 μM). (K) iNOS expression in a monolayer of microvascular endothelial cells after indicated treatment, VEGF group n=28, VEGF+Hcy group n=18, VEGF+1400W group n=13, VEGF+Hcy+1400W group n=29, * p < 0.05 from VEGF. (L) Quantification of 3-NT after treatment with 1400W, VEGF group n=4, VEGF+Hcy group n=5, VEGF+1400W group n=5, VEGF+Hcy+1400W group n=12, * p < 0.05 from VEGF group.
Hcy Impairs Endothelial Cell Locomotion by an iNOS-dependent Mechanism
Microvascular endothelial cells (bEnd.3) cultured at low density were used to study the behavior of single cells to respective treatments. Hcy reduced cell locomotion as defined by the net distance travelled by each cell in a given period of time during 16 hrs time-lapse laser scanning confocal microscopy (Figure 6 A). Locomotion was restored to control levels with inhibition of iNOS activity. Treatment conditions did not affect the proliferation of microvascular endothelial cells (Figure 6 B).
Figure 6. Effect of Hcy and iNOS Activity Time Lapse of Cell Locomotion.
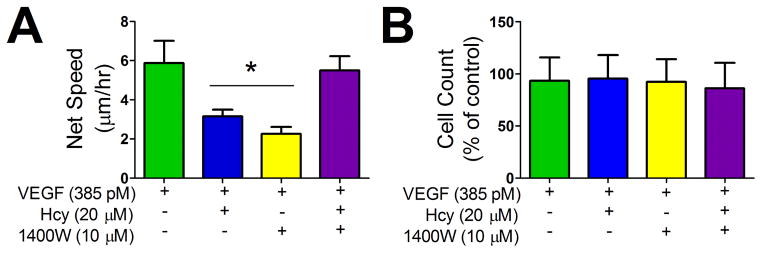
(A) Net speed of microvascular endothelial cells quantified as start position to end position over the time of the experiment. Time lapse image acquisition occurred every 15 mins over a 16 hour period. * p < 0.05 vs. VEGF group, n=6 wells per group over 3 experiments. (B) Effect of treatment groups on dispersed microvascular endothelial cell proliferation, VEGF n=19 wells, VEGF+Hcy, VEGF+Hcy+1400W, and VEGF+1400W n=22 wells.
Hcy Inhibits Endothelial Monolayer Outgrowth by an iNOS-dependent Mechanism
Hcy reduced the outgrowth rate of endothelial cells following the creation of a denuded area by scraping a monolayer of endothelial cells (Figure 7). Outgrowth rate was restored to control levels following inhibition of iNOS activity by both 1400W and transfection with iNOS-specific siRNA.
Figure 7. Effect of Hcy and iNOS Activity on Monolayer Endothelial Outgrowth.

(A–D) siRNA dose response, Hoechst nuclear stain (blue) and iNOS labeling (red). Panel A is a representative image of a monolayer of cells with the empty siRNA vector and panel B is a representative image of the transfection agent, lipofectin, alone. (E–H) siRNA time response after an initial 24 hr transfection period. A final dose of 1μM of siRNA was used for wound healing experiments which did not exceed 96 hrs from transfection. (I–J) Confluent microvascular endothelial cells were scrape-wounded and the initial wound area was measured. Wound area was measured again at 48 hrs. The difference between initial wound area and final wound area was divided by time to quantify the rate of outgrowth into the denuded area between groups. (I) Rate of outgrowth into the denuded area with VEGF 385 pM, Hcy 20 μM, and 1400W 10 μM as treatments as indicated, n=12, * p < 0.05 vs. VEGF group. (J) Wound healing rate with VEGF 385 pM, Hcy 20 μM, and 1 μM siRNA or an empty siRNA vector as indicated. VEGF+Empty siRNA group n=6, VEGF+Hcy+Empty siRNA group n=5, VEGF+siRNA group n=5, VEGF+Hcy+siRNA group n=6, * p < 0.05 vs. VEGF+Empty siRNA group.
DISCUSSION
Angiogenesis requires both proliferation and locomotion but it is unknown whether Hcy influences one or both of these processes when it inhibits angiogenesis. In our primary tissue explant assay, Hcy reduced endothelial outgrowth area without reducing cell number (denser, smaller outgrowth) (Figure 1). Because total cell count was not affected by Hcy, we hypothesized that cell locomotion was impaired without significantly altering cell proliferation. We have previously found that while Hcy impaired the rate of wound repair in microvascular endothelial cell culture, proliferation was not affected [12]. To confirm the effect of Hcy on cell locomotion directly, we used mouse microvascular endothelial cells dispersed in culture in conjunction with time lapse laser scanning confocal microscopy. The results demonstrated a 1400W-sensitive mechanism by which Hcy impairs endothelial cell locomotion without affecting proliferation (Figure 6). We brought these results back to an endothelial syncytium outgrowth model by testing this pathway in a scrape-induced outgrowth assay. Again, outgrowth inhibition by Hcy was rescued by iNOS inhibition with 1400W (Figure 7). We further demonstrated the specific importance of iNOS in mediating the effects of Hcy on endothelial outgrowth in the scrape assay by utilizing siRNA as a second approach to inhibit iNOS (Figure 7 E and F). Our results are consistent with previous reports of the effects of Hcy on endothelial migration. The inhibition of migration of bovine aortic endothelial cells with Hcy treatment has been shown in vitro along with complementary decreases in the angiogenic response in vivo [15,28].
We confirmed that Hcy treatment increased iNOS levels in the choroid explants as well as in a monolayer of endothelial cells (Figure 5). Treatment with 1400W reduced iNOS levels across treatments both in the ex vivo tissue model and in cell culture. 1400W is a highly selective, slow, tight binding, competitive inhibitor of iNOS activity [18]. While 1400W is recognized as an inhibitor of iNOS activity to the exclusion of the other NOS isoforms [17,35], the mechanism of expression decrease is not known and appears to be dependent on cell type [30,34]. Increased iNOS activity and the production of 3-NT are highly correlated and increases in iNOS activity are the major mechanism of 3-NT formation [8,14]. To verify 1400W treatment inhibited iNOS activity, we quantified 3-NT formation across treatment conditions. As expected, 1400W reduced the production of 3-NT (Figure 5 L).
A decrease in iNOS levels, either by siRNA or by treatment with 1400W, rescued Hcy impaired rate of outgrowth, providing additional evidence that Hcy’s affect on cell migration is dependent on increased iNOS levels (Figure 5 and Figure 7). Whether increased iNOS levels, independent of increases in iNOS activity, or an increase in iNOS activity are the ultimate effectors of Hcy impaired migration is not known.
Although 1400W is widely used as a selective inhibitor of iNOS activity, pharmaceutical treatments can have off target effects. To address this issue we used the choroids from iNOS−/− mice to further verify the involvement of iNOS in Hcy induced decrease in explant outgrowth. Choroid explants from treated iNOS−/− mice were resistant to Hcy; they grew out normally and did not show any signs of impaired outgrowth relative to control (Figure 2).
Production of 3-NT in a monolayer of endothelial cells was not significantly different between control and Hcy treated groups under these conditions, while iNOS levels were increased with Hcy treatment (Figure 5). This would suggest that the effects of Hcy on cell migration that are mediated by iNOS are independent of gross cellular protein nitrosylation modifications but does not exclude the possible importance of individual protein nitrosylation events that could be beyond the sensitivity of these measurements.
Cell locomotion involves the dynamic regulation of cytoskeletal architecture [4,22]. Our results show that Hcy caused a re-orientation of F-actin, making it perpendicular to the vector of outgrowth (sideways instead of straight out; Figure 3B and D). In contrast, the VEGF-only group developed F-actin filaments that oriented directly out from the explant (Figure 3B and C). Adding to the directional differences in outgrowth morphology was the observation of F-actin filled projections at the leading edge of outgrowth in the VEGF-only group that were blocked by Hcy and rescued with inhibition of iNOS (Figure 4A and C). These results indicate that Hcy disrupts not only the orientation but also the morphology of probing actin micro-domains during migration, which are a key feature of angiogenesis [19,26,33]. The loss of these micro-domains contributes to impaired outward growth of new vascular endothelium from the native microvascular network in response to VEGF [19]. Our results indicating iNOS dependent formation of actin projections at the leading edge of growth (Figure 4) are supported by the close physical interaction between iNOS and alpha-actinin [13], which tethers enzymes to actin polymers, and the established role of NO in regulating actin dynamics [24].
Nitrosylation of the cytoskeleton in lamellipodia is implicated in modulation of the functional dynamics as cells migrate [20,24]. Though reactive oxygen species (ROS) are necessary for actin polymerization, some have suggested that excess production of reactive oxygen species may damage actin, resulting in a loss of lamellipodia formation [3]. Whether nitrative stress, oxidative stress, or a combination of the two are the ultimate effector(s) of iNOS-dependent actin filament disruption and reduced outgrowth is currently undetermined. Overall, the results of our explant experiments, document evidence for the role of iNOS in Hcy-mediated disruption of endothelial subcellular actin dynamics during VEGF-stimulated growth. Determining whether the effect of Hcy on actin dynamics during endothelial outgrowth is directly related to the observed impaired migration, however, will require further exploration.
While iNOS was a significant mediator of the effects of Hcy in our studies, there are likely multiple mechanisms by which Hcy disrupts angiogenesis. Specifically, our data indicate that the influence of Hcy on the overall orientation of actin near the leading edge of outgrowth may be iNOS independent because 1400W treatment did not rescue this phenotype (Figure 3). The role of iNOS was different depending on the cellular microenvirnment. Constitutive iNOS was important for individual cell locomotion (Figure 6) but dispensable for the outgrowth of primary endothelial tissue explants (Figure 1 O). In individual cells, where there is a lack of contacting neighbors, Hcy appears to elevate iNOS to levels that are detrimental to cellular locomotion as quantified by the net rate of travel. Blocking basal iNOS activity in control cells had a similar effect, indicating that a basal level of iNOS activity is required for the locomotion of individual cells while too much or too little are similarly detrimental (Figure 6). The relation was different in the explant model where cells have additional constraints established by the microenvironment of neighboring cells and/or the extracellular matrix. In this context, blocking iNOS activity in control explants did not alter the outgrowth of endothelium, which is consistent with the idea that basal iNOS activity was not necessary for outgrowth of endothelium from the intact tissue into boarding matrix. Hcy-impaired outgrowth and decreased locomotion in dispersed cells are both sensitive to fluctuations in iNOS activity, however, because the impairment of both was rescued with 1400W treatment. These results emphasize the importance of subtle regulation of iNOS activity in migrating cells.
PERSPECTIVES
The mechanisms that control and regulate angiogenesis dictate the outcomes of tissue injury, metabolic stress, and repair in health and disease. Here, we present the results of initial studies dissecting the mechanisms by which Hcy disrupts endothelial cell migration and cellular F-actin architecture during VEGF-stimulated angiogenesis. These results describe one of the potential mechanisms for the elevated risk and severity of cardio- and neurovascular disease in patients with HHcy.
Acknowledgments
Funding: NIH R15 HL106548 to S.E.B. and NIH R21 AG041934 to F-F. L.
We thank Dr. Yueqiang Xue for technical assistance.
Abbreviations
- 1400W
N-[[3-(aminomethyl) phenyl]methyl]-ethanimidamide dihydrochloride
- eNOS
endothelial nitric oxide synthase
- F-actin
filamentous actin
- HHcy
hyperhomocysteinemia
- Hcy
homocysteine
- iNOS
inducible nitric oxide synthase
- VEGF
vascular endothelial growth factor
- DMEM-H
Dulbecco’s Modified Eagle’s Medium high glucose
- 3-NT
3-nitrotyrosine
Footnotes
Disclosure/Conflict of Interest:
None
References
- 1.Guide for the Care and Use of Laboratory Animals. National Academy of Sciences; 2011. [Google Scholar]
- 2.Banan A, Fields JZ, Zhang Y, Keshavarzian A. iNOS upregulation mediates oxidant-induced disruption of F-actin and barrier of intestinal monolayers. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1234–1246. doi: 10.1152/ajpgi.2001.280.6.G1234. [DOI] [PubMed] [Google Scholar]
- 3.Barth BM, Stewart-Smeets S, Kuhn TB. Proinflammatory cytokines provoke oxidative damage to actin in neuronal cells mediated by Rac1 and NADPH oxidase. Mol Cell Neurosci. 2009;41:274–285. doi: 10.1016/j.mcn.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bayless KJ, Johnson GA. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J Vasc Res. 2011;48:369–385. doi: 10.1159/000324751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beard RS, Jr, Reynolds JJ, Bearden SE. Hyperhomocysteinemia increases permeability of the blood-brain barrier by NMDA receptor-dependent regulation of adherens and tight junctions. Blood. 2011;118:2007–2014. doi: 10.1182/blood-2011-02-338269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beard RS, Jr, Reynolds JJ, Bearden SE. Metabotropic glutamate receptor 5 mediates phosphorylation of vascular endothelial cadherin and nuclear localization of beta-catenin in response to homocysteine. Vascul Pharmacol. 2012;56:159–167. doi: 10.1016/j.vph.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bearden SE, Beard RS, Jr, Pfau JC. Extracellular transsulfuration generates hydrogen sulfide from homocysteine and protects endothelium from redox stress. Am J Physiol Heart Circ Physiol. 2010;299:H1568–1576. doi: 10.1152/ajpheart.00555.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 9.Bloor CM. Angiogenesis during exercise and training. Angiogenesis. 2005;8:263–271. doi: 10.1007/s10456-005-9013-x. [DOI] [PubMed] [Google Scholar]
- 10.Carmel R, Jacobsen DW. Cambridge University Press, editor. Homocysteine in Helath and Disease. 2001. [Google Scholar]
- 11.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 12.Chen CH, Beard RS, Bearden SE. Homocysteine impairs endothelial wound healing by activating metabotropic glutamate receptor 5. Microcirculation. 2012;19:285–295. doi: 10.1111/j.1549-8719.2012.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daniliuc S, Bitterman H, Rahat MA, Kinarty A, Rosenzweig D, Lahat N. Hypoxia inactivates inducible nitric oxide synthase in mouse macrophages by disrupting its interaction with alpha-actinin 4. J Immunol. 2003;171:3225–3232. doi: 10.4049/jimmunol.171.6.3225. [DOI] [PubMed] [Google Scholar]
- 14.Drew B, Leeuwenburgh C. Aging and the role of reactive nitrogen species. Annals of the New York Academy of Sciences. 2002;959:66–81. doi: 10.1111/j.1749-6632.2002.tb02084.x. [DOI] [PubMed] [Google Scholar]
- 15.Duan J, Murohara T, Ikeda H, Sasaki K, Shintani S, Akita T, Shimada T, Imaizumi T. Hyperhomocysteinemia impairs angiogenesis in response to hindlimb ischemia. Arterioscler Thromb Vasc Biol. 2000;20:2579–2585. doi: 10.1161/01.atv.20.12.2579. [DOI] [PubMed] [Google Scholar]
- 16.Faraci FM. Hyperhomocysteinemia: a million ways to lose control. Arterioscler Thromb Vasc Biol. 2003;23:371–373. doi: 10.1161/01.ATV.0000063607.56590.7F. [DOI] [PubMed] [Google Scholar]
- 17.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nature chemical biology. 2008;4:700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 19.Gu W, Gu C, Jiang W, Wester P. Neurotransmitter synthesis in poststroke cortical neurogenesis in adult rats. Stem cell research. 2010;4:148–154. doi: 10.1016/j.scr.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Harris LK, McCormick J, Cartwright JE, Whitley GS, Dash PR. S-nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp Cell Res. 2008;314:1765–1776. doi: 10.1016/j.yexcr.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 21.Hedrick TL. Software techniques for two- and three-dimensional kinematic measurements of biological and biomimetic systems. Bioinspiration & biomimetics. 2008;3:034001. doi: 10.1088/1748-3182/3/3/034001. [DOI] [PubMed] [Google Scholar]
- 22.Isenberg JS, Martin-Manso G, Maxhimer JB, Roberts DD. Regulation of nitric oxide signalling by thrombospondin 1: implications for anti-angiogenic therapies. Nature reviews. Cancer. 2009;9:182–194. doi: 10.1038/nrc2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lijnen HR. Angiogenesis and obesity. Cardiovasc Res. 2008;78:286–293. doi: 10.1093/cvr/cvm007. [DOI] [PubMed] [Google Scholar]
- 24.Lindsay SL, Ramsey S, Aitchison M, Renne T, Evans TJ. Modulation of lamellipodial structure and dynamics by NO-dependent phosphorylation of VASP Ser239. J Cell Sci. 2007;120:3011–3021. doi: 10.1242/jcs.003061. [DOI] [PubMed] [Google Scholar]
- 25.Mayo JN, Beard RS, Jr, Price TO, Chen CH, Erickson MA, Ercal N, Banks WA, Bearden SE. Nitrative stress in cerebral endothelium is mediated by mGluR5 in hyperhomocysteinemia. J Cereb Blood Flow Metab. 2012;32:825–834. doi: 10.1038/jcbfm.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain research reviews. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Montesano R, Pepper MS, Mohle-Steinlein U, Risau W, Wagner EF, Orci L. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell. 1990;62:435–445. doi: 10.1016/0092-8674(90)90009-4. [DOI] [PubMed] [Google Scholar]
- 28.Nagai Y, Tasaki H, Takatsu H, Nihei S, Yamashita K, Toyokawa T, Nakashima Y. Homocysteine inhibits angiogenesis in vitro and in vivo. Biochem Biophys Res Commun. 2001;281:726–731. doi: 10.1006/bbrc.2001.4400. [DOI] [PubMed] [Google Scholar]
- 29.Oosterbaan AM, Steegers EA, Ursem NT. The effects of homocysteine and folic acid on angiogenesis and VEGF expression during chicken vascular development. Microvasc Res. 2011;83:98–104. doi: 10.1016/j.mvr.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Patel P, Qi WN, Allen DM, Chen LE, Seaber AV, Stamler JS, Urbaniak JR. Inhibition of iNOS with 1400W improves contractile function and alters nos gene and protein expression in reperfused skeletal muscle. Microsurgery. 2004;24:324–331. doi: 10.1002/micr.20029. [DOI] [PubMed] [Google Scholar]
- 31.Rasband WS. Image J. U.S. National Institutes of Health; Bethesda, Maryland, USA: 1997–2012. http://rsb.info.nih.gov/ij/index.html. [Google Scholar]
- 32.Rezakhaniha R, Agianniotis A, Schrauwen JT, Griffa A, Sage D, Bouten CV, van de Vosse FN, Unser M, Stergiopulos N. Experimental investigation of collagen waviness and orientation in the arterial adventitia using confocal laser scanning microscopy. Biomech Model Mechanobiol. 2012;11:461–473. doi: 10.1007/s10237-011-0325-z. [DOI] [PubMed] [Google Scholar]
- 33.Ridley AJ. Life at the leading edge. Cell. 2011;145:1012–1022. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Uno K, Iuchi Y, Fujii J, Sugata H, Iijima K, Kato K, Shimosegawa T, Yoshimura T. In vivo study on cross talk between inducible nitric-oxide synthase and cyclooxygenase in rat gastric mucosa: effect of cyclooxygenase activity on nitric oxide production. J Pharmacol Exp Ther. 2004;309:995–1002. doi: 10.1124/jpet.103.061283. [DOI] [PubMed] [Google Scholar]
- 35.Zhu Y, Nikolic D, Van Breemen RB, Silverman RB. Mechanism of inactivation of inducible nitric oxide synthase by amidines. Irreversible enzyme inactivation without inactivator modification. Journal of the American Chemical Society. 2005;127:858–868. doi: 10.1021/ja0445645. [DOI] [PubMed] [Google Scholar]