

Abstract
Recent studies indicate that the cardioprotective effects of ischemic preconditioning (IPC) against sustained ischemia/reperfusion (IR) can be replicated by angiotensin II (Ang II). However, it is not clear whether IPC and Ang II-induced preconditioning (APC) act through similar mechanisms or synergize to enhance cardioprotection. In this study, Langendorff-perfused rat hearts were subjected to IPC, APC or their combination (IPC/APC) followed by IR. IPC and less potently APC, significantly increased the percent recovery of the left ventricular developed-pressure, the first derivative of developed pressure and the rate pressure product compared to control. Furthermore, the post-ischemic recovery of the heart was significantly higher for IPC/APC compared to IPC or APC. The improvements in cardiac function by IPC, APC and IPC/APC were associated with similar reductions in LDH release and infarct size. However, a significant improvement in mitochondrial respiration was observed with IPC/APC. The post-ischemic recovery observed with APC and IPC/APC was inhibited by treatment with losartan, an Ang II type-1 receptor blocker, during the preconditioning phase but not by chelerythrine, a pan-PKC inhibitor. Both drugs, however, abolished the enhanced mitochondrial respiration by IPC/APC. Altogether, these results indicate that APC and IPC interact through mechanisms that enhance cardioprotection by affecting cardiac function and mitochondrial respiration.
Keywords: Preconditioning, Angiotensin II, Mitochondria, Cardioprotection, Ischemia-Reperfusion
Introduction
Ischemic preconditioning (ICP) is an adaptive response of the heart to protect the myocardium against ischemic/reperfusion (IR). The process is induced when the heart is exposed to brief and repetitive periods of ischemia making the heart resistant to the damage induced by a subsequent severe ischemia. Since its discovery by Murry et al.1 in 1986, IPC has gained enormous clinical interest because of its potential to reduce myocardial injury and improve ventricular recovery following IR injury in patients.2 The cellular pathways involved in IPC appear to be numerous and complex,2-3 although it is clear that leading roles are played by protein kinase C (PKC-ε and PKC-δ),4-5 mitochondria-derived reactive oxygen species (ROS),6-7 redox-sensitive MAPKs,8 mitochondrial permeability transition pore (mPTP)9-10 and mitochondrial KATP (mitKATP) channels11. It has been postulated that the activation of these mechanisms by IPC reduce the mitochondrial damage induced by calcium loading, ROS and mPTP opening. The preservation of mitochondria delays the cellular ATP depletion, and reduces necrosis and mitochondrial oxygen consumption following an ischemic insult.12 Adenosine and bradykinin, both of which are released by the myocardium during ischemia, are two recognized mediators of IPC.3
Recent studies13-17 also suggest that angiotensin II (Ang II), a well-known inducer of oxidative stress and PKC in the cardiovascular system,18 is involved in cardiac preconditioning. This notion, however, is at variance with the reported detrimental effects of the hormone on the myocardium during IR injury. Indeed, renin inhibitors19, angiotensin converting enzyme inhibitors20 or Ang II blockers20-21 have shown protection against IR injury. Differences in the experimental approach used in these studies are likely responsible for the contrasting effects of the hormone on IR injury. Nevertheless, evidence indicates that the preconditioning actions of Ang II involve the Ang II-type 1 receptor (AT-1R), G-protein independent signaling22-23 and/or AT-2 receptor stimulation.24 Activation of these receptor-dependent pathways results in improvements of post-ischemic ventricular recovery,13,15-16 reduced myocardial infarction13,15-16,22-23 and cardiomyocyte apoptosis.25 The cardioprotective effects of Ang II have been associated with PKC activation26 and ROS generation because they are sensitive to the antioxidants N-acetylcysteine and tempol, and the NAD(P)H oxidase inhibitor apocynin.26-28 Indeed, Ang II has been reported to increase the mRNA expression of the NAD(P)H subunits, gp91phox, p22phox and RAC-1 in rat hearts.25-28 These findings suggest that contrary to IPC that rely mostly on mitochondrial-derived ROS for cardioprotection, the beneficial effects of Ang II are mediated via NAD(P)H oxidase-induced ROS generation. However, recent studies demonstrated that mitKATP channels,26 lipid peroxidation and MAPK activation29 are also involved in Ang II-induced preconditioning (APC) in the heart. For these reasons, it has been proposed that the cardioprotective effects of Ang II in the heart are mediated by mitochondria-derived ROS through NAD(P)H oxidase and MAPK activation.26,28-29 Taking into consideration that APC like IPC, appears to converge on mitochondria, we hypothesized that the combination of these treatments could synergize to improve post-ischemic cardioprotection. However, this aspect of the Ang II-induced cardioprotection has not been evaluated.
In this work, we compared the effects of IPC, APC and their combination (IPC/APC) on heart contractility and mitochondrial function to establish whether concomitant application of IPC and APC interact to induce enhanced cardioprotection against IR. Cardioprotection was evaluated by the post-ischemic recovery of the left ventricular developed pressure (LVDP), the first derivative of the developed pressure (+dP/dt), the rate pressure product (RPP), lactate dehydrogenase (LDH) release, infarct size, mitochondrial respiration and mPTP opening. Our results confirm the preconditioning effects of Ang II and suggest the interaction of independent pathways for IPC and APC to improve cardiac function. Convergent mechanisms, however, appear to operate to promote mitochondrial respiration and to reduce myocardial damage.
Methods
Male Sprague-Dawley rats weighing 140-160g were purchased from Charles River (Wilmington, MA, USA). All experiments were performed according to protocols approved by the University Animal Care and Use Committee and conform to the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Animal groups
Animals were randomly assigned to the following four groups: (i) Control group, hearts were perfused for 40 min without any interventions followed by sustained IR, (ii) IPC group, hearts were perfused for 20 min (pre-equilibration period) and four cycles of short (5 min each) IR for a total of 40 min followed by sustained IR, (iii) APC group, hearts were perfused for 20 min (pre-equilibration period) and four cycles of short (5 min each) Ang II (10 nM)/washout for a total of 40 min followed by sustained IR, and (iv) IPC/APC group, hearts were perfused for 20 min (pre-equilibration period) and four cycles of short (5 min each) ischemia/Ang II reperfusion for a total of 40 min followed by sustained IR. A 5 min washout period prior to the global ischemia similar to the other groups was included in this group. IR was induced by switching off the pump to induce global ischemia for the indicated period of time after which the flow rate was restored to pre-ischemic levels. In all experiments, sustained IR was induced by 30-min of global ischemia followed by 90-min reperfusion. A diagram for the perfusion protocols is illustrated in Figure 1.
Figure 1.

Schematic representation of the perfusion protocols. See “Methods” for details.
To evaluate whether AT-1 receptors and PKC were involved in Ang II-mediated actions, losartan (10 μM) and chelerythrine (pan-PKC inhibitor, 5 μM) were infused prior to (15 and 30 min, respectively) and during APC and IPC/APC preconditioning cycles. The drugs were not present during reperfusion. At the concentrations used, losartan and chelerythrine abolished the Ang II-induced increase in coronary resistance (from 2.85±0.16 to 0.24 ± 0.43 (P<0.05) and to 0.52 ± 0.18 (P<0.05) mmHg × mL/min, n=10), respectively. These findings indicate the effectiveness of these inhibitors to reduce Ang II action in the experimental conditions used.
The concentration of Ang II used (10 nM) was selected following preliminary studies at higher concentrations that induced marked alterations in contractility and heart rhythm. Four hearts out of sixteen in the APC group with marked rhythm alterations did not recover after the 30 min global ischemia and were discarded.
Langendorff-mode perfusion
Isolated rat hearts were perfused by the Langendorff method as described previously30. Briefly, isolated hearts were perfused at constant flow (10-12 mL/g heart weight per min) with Krebs-Henseleit solution containing (in mM): 118 NaCl, 4.8 KCl, 1.2 KH2PO4, 1.25 CaCl2, 1.2 MgSO4, 11 glucose, and 25 NaHCO3 equilibrated at pH 7.4 with 5% CO2/95% O2. Left ventricular pressure was monitored throughout using a water-filled latex balloon inserted in the left ventricle and connected to a pressure transducer, and coronary perfusion pressure was monitored by the transducer connected through a filled line to a port in the cannula housing directly above the heart. An initial left ventricular end-diastolic pressure (LVEDP) was adjusted to 4-6 mmHg. Data acquisition to determine heart rate (HR), LVDP, +dP/dt, and ischemic contracture was performed with the Labscribe2 Data Acquisition Software (iWorx 308T, Dover, NH, USA). For each determination, 10 contraction-relaxation cycles spread in the last 5 min period prior to the termination of pre-ischemia or 60 min of reperfusion were evaluated. The rate pressure product for each heart was calculated as RPP = HR × LVDP. Samples of the coronary effluent were collected at the indicated time intervals to determine LDH activity as previously described.30
Isolation of mitochondria
At the end of the corresponding perfusion protocol, the ventricles were cut, weighed and homogenized with a Polytron homogenizer in 3 mL of ice-cold sucrose buffer containing 300 mM sucrose, 10 mM Tris-HCl and 2 mM EGTA, pH 7.4. The homogenate was then diluted to 40 mL of buffer containing 5 mg/mL BSA. Mitochondria were isolated29 by the centrifugation of the homogenate at 2,000×g for 3 min, and centrifugation of the supernatant at 10,000×g for 6 min. The pellet was then resuspended and washed twice with BSA-free sucrose buffer followed by centrifugation at 10,000×g for 6 min. The final pellet was resuspended in 200 μL of sucrose buffer and used for determination of mitochondrial respiration rates and mPTP opening.
Determination of mitochondrial respiration rates
Mitochondrial respiration rates31 were determined at 30°C using a YSI Oxygraph (Yellow Springs, OH, USA) model 5300 and a Clark-type oxygen electrode. Mitochondria were suspended in a buffer containing (in mM): 125 KCl, 20 MOPS, 10 Tris, 0.5 EGTA and 2 KH2PO4 supplemented with the substrates for complex I of the electron transfer chain, 2.5 mM 2-oxoglutarate and 1 mM L-malate. Oxygen consumption rates were measured in the absence (state 2) and presence of 1 mM ADP (state 3), and normalized to mg of mitochondrial protein. The respiratory control index (RCI) was calculated as the state 3/state 2 ratio. Solubility of oxygen in the buffer was 230 nmol of oxygen/mL.32
Determination of mitochondrial PTP opening
The extent of mPTP opening was determined by the swelling of de-energized mitochondria in the presence and absence of Ca2+ by the decrease in light scattering at 520 nm as described previously.31 Mitochondria were incubated at 25°C in 1 mL of a buffer containing (mM): 150 KSCN, 20 MOPS, 10 Tris and 2 nitrilotriacetic acid. The buffer was supplemented with 0.5 μM rotenone, 0.5 μM antimycin and 2 μM A23187. The reaction was started by the addition of 40 μL of the mitochondrial suspension containing approximately 220 μg of protein and swelling was initiated by addition of 200 μM CaCl2.
Infarct size assessment
A subset of hearts (n=3 per group) underwent the corresponding perfusion protocols for each group to determine infarct area described previously by Liu et al.26 Briefly, at the end of reperfusion, the ventricles were isolated, weighed and frozen for 90 min at -20°C. The ventricles were then cut into 1.5-mm-thick slices and stained with 1% triphenyltetrazolium chloride (TTC) in phosphate buffer (pH=7.4) at 37°C followed by fixation with 10% formalin at room temperature. The slices were then mounted on glass plates of 1.5 mm thickness and digitally photographed. The digital images were analyzed for the infarcted area (TTC negative) using the NIH ImageJ software. Results were expressed as infarcted area/total area × 100.
Statistical analysis
Data are presented as mean SEM of 9-14 experiments per group. Statistical differences between groups were analyzed using two-tailed, unpaired Student's t-test and ANOVA followed by Tukey-Kramer post-hoc tests. Differences were considered significant when p ≤ 0.05.
Results
Angiotensin II-induced preconditioning is additive to that of IPC
Table 1 depicts cardiac function parameters evaluated in the four groups studied during pre-ischemia (5 min before global ischemia) and post-ischemia (60 min after the initiation of reperfusion). As expected, both IPC and APC promoted post-ischemic ventricular recovery when compared with the control group. IPC increased the percent recovery of LVDP, +dP/dt and RPP by 2.92, 3.66 and 4.16-fold (P<0.05 for all), respectively. APC also stimulated the percent recovery of LVDP (2.23-fold, P<0.05), +dP/dt (66%, P<0.05) and RPP (91%, P<0.05). When combined, IPC and APC further improved cardioprotection compared to control hearts and induced increases of 3.61, 4.83 and 4.4-fold of LVDP, +dP/dt and RPP (P<0.05 for all), respectively. Compared with APC, the combination of IPC/APC significantly increased the percent recovery of these parameters (LVDP: 62%, +dP/dt: 2.9-fold and RPP: 2.3-fold (P<0.05 for all). Additive effects of IPC and APC are suggested by the finding that the absolute changes in LVDP, +dP/dt and RPP induced by IPC and APC together with their corresponding basal values, yield totals (LVDP: 21.6±3 mmHg, +dP/dt: 484±78 mmHg/sec and RPP: 6479±939 beats × mmHg × min-1) which are not statistically different from those observed with IPC/APC (Table 1). These findings demonstrate the Ang II-induced preconditioning and support the presence of independent pathways for improving the post-ischemic ventricular recovery by APC and IPC.
Table 1. Effects of IPC and APC on functional recovery of Langendorff-perfused rat hearts following global ischemia and reperfusion.
| Pre-ischemia | ||||
|---|---|---|---|---|
| Parameters | Control | IPC | APC | IPC/APC |
| HR, beats/min | 301±10 | 216±16* | 324±9 | 260±10*† |
| LVDP, mmHg | 55±3 | 40±3* | 59±3 | 43±2*& |
| +dP/dt, mmHg/sec | 1357±70 | 848±134* | 1431±68 | 1129±143† |
| RPP, beats × mmHg × min-1 | 16555±986 | 8640±934* | 19116±1021 | 11180±617*† |
| Post-ischemia | ||||
| HR, beats/min | 266±33 | 306±16 | 291±19 | 296±15 |
| LVDP, mmHg % of Pre-ischemia |
8±1 13±2 |
14±2* 38±5* |
15±2* 29±5* |
20±2* 47±5*& |
| +dP/dt, mmHg/sec % of Pre-ischemia |
159±29 12±2 |
362±52* 44±7* |
279±26* 20±3* |
570±45*†& 58±4*& |
| RPP, beats × mmHg × min-1 % of Pre-ischemia |
2128±335 12±2 |
4284±517* 50±7* |
4365±421* 23±4* |
5920±642*†& 53±2*& |
See Materials and Methods for details. Pre-ischemia values were determined 5 min before global ischemia whereas post-ischemia values were taken at 60 min of reperfusion. The values shown are the means ± SEM of 9-14 experiments per group.
P<0.05 vs Control;
P<0.05 vs IPC;
P<0.05 vs APC.
Angiotensin II reduces LDH release and exerts additive effects to that of IPC
To corroborate that IPC and APC induce cardioprotection, LDH release, a measure of damage to the myocardium, was determined in the coronary effluent. Figure 2 illustrates the release of LDH as a function of time in the four conditions evaluated. Both IPC and APC significantly reduced LDH release from the heart at reperfusion. At variance with IPC that reduced LDH release 17% (P=N.S.) in the first minute of reperfusion, APC decreased this parameter by 48% (P<0.05) in the same period of time. Thereafter both treatments exerted comparable reductions in LDH release. Of note, the combination of IPC and APC suppressed LDH release to a larger extent than that induced by IPC alone. Indeed, significant differences (∼43%, P<0.05) were observed at all time points evaluated. This suggests that the combined IPC/APC treatment was more effective in preventing cell damage than either treatment alone.
Figure 2.

LDH activity in cardiac effluents collected during the reperfusion as a function of time. The time intervals (in minutes) used for the sample collection are indicated in the inset. The values shown are the means ± SEM of 9-14 experiments per group. *P<0.05 vs Control and &P<0.05 vs IPC.
Angiotensin II stimulates mitochondrial respiration and significantly increases the respiratory control ratio
To examine whether the cardioprotection induced by Ang II is associated with improved mitochondrial function, respiration rates at complex I were determined in the absence and presence of 1 mM ADP. Results in Figure 3A indicate that state 2 was not significantly affected by IPC, APC or IPC/APC. In the presence of ADP (state 3) however, respiration rates for IPC were markedly enhanced 51% (P<0.05) when compared with controls. Likewise, APC stimulated state 3 over the controls by 30%, although it did not reach statistical significance (P=0.07). Moreover, the combination of the treatments (IPC/APC group) further stimulated state 3 over that observed in the IPC group 31% (P<0.05) and that of APC group 53% (P<0.05). Of note, RCI, an important functional parameter of mitochondrial function and an indicator of coupling electron transport and oxidative phosphorylation was not affected by either IPC or APC alone, but significantly increased (89%, P<0.05) in the IPC/APC group when compared with controls (Figure 2B). These results provide strong evidence that the combination of IPC and APC exerts a synergistic effect on the mitochondrial respiratory coupling and function.
Figure 3.
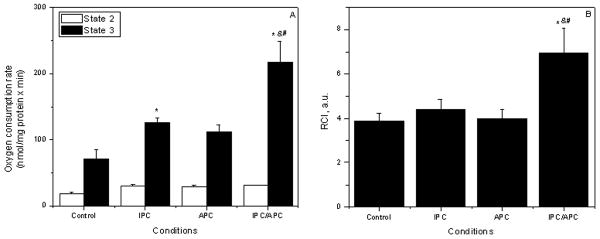
Effects of APC and IPC on mitochondrial oxygen consumption. A, State 2 and State 3; B, respiratory control index (RCI). Oxygen consumption rates are given in nmol O/mg protein × min. The values shown are the means ± SEM of 9-14 experiments per group. *P<0.05 vs Control, &P<0.05 vs IPC; #P<0.05 vs APC.
Angiotensin II inhibits mPTP opening
The extent of mPTP opening was determined to establish if the improvement in mitochondrial respiration and RCI were associated with inhibition of pore opening. Figure 4A depicts the Ca2+-induced swelling of mitochondria represented as the decrease in light scattering at 520nm in the four groups evaluated. Mitochondria isolated from hearts subjected to IPC and to a lower extent APC, showed a reduced Ca2+-induced swelling and hence, lower mPTP opening than controls. Likewise, IPC/APC also strongly reduced mPTP opening compared to control. Quantification of the rate of swelling of mitochondria induced by the addition of CaCl2 (Figure 4B) indicated a 28% (P<0.05) and 14% (P=N.S.) lower swelling in IPC and APC, respectively. Of note, IPC in combination with APC exerted maximum inhibition and reduced the extent of mPTP opening by 37% (P<0.05) compared to control. These results suggest that both IPC and APC attenuates mPTP opening induced by IR, and that IPC in combination with APC is more effective compared to either treatment alone in this regard.
Figure 4.

Ca2+-induced mitochondrial swelling as an indicator of mPTP opening following IR. Measurement of the PTP under de-energized conditions was performed by monitoring the Ca2+-induced decrease in light scattering (A520). A, Original traces are shown for one mitochondrial preparation derived from hearts of Control, IPC, APC or IPC/APC groups. Matrix swelling was induced by 200 μM or 1 mM (maximal swelling) Ca2+. B, Rate of swelling of mitochondrial swelling induced by 200 μM CaCl2. Data are shown as a change of optical density (ΔOD)/min per mg of protein. The values shown are the means ± SEM of 9-14 experiments per group. *P<0.05 vs Control.
APC reduced infarct size without affecting the ischemic contracture
Analysis of infarct size in the four groups evaluated indicated that both IPC and APC reduced infarction areas by ∼60%, (P<0.05) (Table 2). Likewise, the combined treatment (IPC/APC group) also reduced infarct size by 66% (P<0.05) compared to control hearts. In addition, IPC but not APC, decreased the maximum contracture during ischemia 34% (from 14.7±0.9 to 9.6±0.9 mmHg, P<0.05) consistent with previous reports.33 This effect was not observed when both treatments were combined. The onset of the ischemia-induced contracture was not significantly affected by any treatment. These findings suggest that IPC, APC or their combination are equally effective in reducing infarct area and that this effect is not necessarily related to a reduced ischemic-induced contracture.
Table 2. Effects of IPC and APC on infarct size and ischemic contracture of Langendorff-perfused rat hearts subjected to global ischemia and reperfusion.
| Control | IPC | APC | IPC/APC | |
|---|---|---|---|---|
| Infarct size, % | 53±4 | 22±6* | 20±3* | 18±2* |
| Ischemic contracture, mmHg | 14.7±0.9 | 9.6±0.9* | 15±1.2 | 12.2±0.8† |
| Time-contracture, min | 12.3±0.9 | 9.7±0.7 | 13.7±0.5 | 9.3±1 |
See Materials and Methods for details. The values shown are the means ± SEM of 6 experiments per group for infarct size and 9-14 experiments for ischemic contracture.
P<0.05 vs Control;
P<0.05 vs IPC.
Effect of losartan and chelerythrine on APC and IPC/APC-induced cardioprotection
To evaluate a role for AT-1R and PKC in the preconditioning effects of Ang II, APC and IPC/APC, hearts were pre-treated with losartan (10 μM) or chelerythrine (5 μM). Table 3 shows that losartan significantly (P<0.05) inhibited LVDP, +dP/dt and RPP in both APC (∼40%) and IPC/APC (∼30%) during the pre-ischemic and post-ischemic periods. However, compared with controls or IPC (Table 1), the post-ischemic increase in LVDP, +dP/dt and RPP by APC or IPC/APC, were significantly inhibited by losartan (>83%, P<0.05). This indicates that the effects of Ang II are mediated by AT-1R.
Table 3.
Effect of losartan and chelerythrine on cardiac function in Langendorff-perfused rat hearts.
| Pre-ischemia | ||||||
|---|---|---|---|---|---|---|
| Parameters | APC | IPC/APC | APC+ L | IPC/APC+ L | APC+C | IPC/APC+ C |
| HR, beats/min | 318±8 | 265±12* | 302±12 | 284±8 | 305±16 | 252±6┴ |
| LVDP, mmHg | 57±3 | 43±2* | 35±3* | 34±1& | 46±4* | 45±2 |
| +dP/dt, mmHg/sec | 1381±62 | 1018±40* | 903±67* | 789±16& | 1140±84* | 969±22 |
| RPP, beats × mmHg × min-1 | 18126±963 | 11395±699* | 10570±817* | 9656±320 | 14030±925* | 11340±403 |
| Post-ischemia | ||||||
| HR, beats/min | 296±15 | 297±12 | 288±9 | 310±7 | 325±14 | 266±13 |
| LVDP, mmHg % of Pre-ischemia |
15±1 29±4 |
22±2* 51±3* |
9±1* 26±3 |
14±1& 43±4 |
14±3 30±4 |
21±2 48±3┴ |
| +dP/dt, mmHg/sec % of Pre-ischemia |
295±23 23±2 |
591±40* 59±3 |
187±48* 20±4 |
383±36& 49±5 |
352±73 29±5 |
554±55 58±5┴ |
| RPP, beats × mmHg × min-1 % of Pre-ischemia |
4440±421 25±3 |
6534±576* 57±2* |
2592±442* 24±3 |
4340±421& 45±4& |
4550±768 32±5 |
5586±375 49±2┴ |
See Materials and Methods for details. Pre-ischemia values were determined 5 min before global ischemia whereas post-ischemia values were taken at 60 min of reperfusion. The values shown are the means ± SEM of 9-12 experiments per group. L= losartan and C = chelerythrine.
P<0.05 vs APC,
P<0.05 vs IPC/APC and
P<0.05 vs APC+C.
Table 3 also shows that chelerythrine significantly inhibited LVDP, +dP/dt and RPP by ∼20% (P<0.05) in the APC group during the pre-ischemia. No effect of the drug was observed in the post-ischemic period. Likewise, no effect of chelerythrine was observed in the IPC/APC group in both pre- and post-ischemic periods. These experiments are consistent with the idea that cardiac function can be improved by APC or IPC/APC through PKC-independent pathways.
The RCI increase by IPC/APC is losartan- and chelerythrine-sensitive
Table 4 depicts the results of experiments where the effects of losartan and chelerythrine were tested on mitochondrial respiration. Both losartan and chelerythrine significantly reduced the increase in state 2 and RCI observed with IPC/APC. State 3 was inhibited 38% (P<0.05) by losartan and 33% (P<0.05) by chelerythrine. Likewise, the RCI was inhibited 76% (P<0.05) by AT-1R blockade and 100% (P<0.05) by the PKC inhibitor. It is worth noting that chelerythrine significantly increased state 2 by 89% in APC (P<0.05) and 44% in IPC/APC group (P<0.05). These findings indicate that APC acting through AT-1R, synergize with IPC to modulate mitochondrial function through a PKC-dependent mechanism.
Discussion
The results of the present study confirm the preconditioning effects of Ang II on post-ischemic recovery of cardiac function. Four cycles of Ang II exposure improves post-ischemic ventricular recovery (LVDP and +dP/dt), increases RPP, decreases LDH activity in the coronary effluent, improves mitochondrial respiration (state 3) and reduces infarct size. The percent recovery of ventricular function by APC is lower in magnitude than that observed with IPC based on LVDP, +dP/dt and RPP measurements. However, this effect is partly explained by the fact that IPC depress cardiac function during the pre-ischemia whereas APC does not. When this factor is taken into consideration, the post-ischemic ventricular recoveries are similar for these two groups with the exception of +dP/dt that had a significant absolute increase in IPC over the APC group. These findings are consistent with similar protective effects of IPC and APC on LDH release, mitochondrial respiration and infarct size. These improvements appear to be independent of ischemic contracture changes.
The preconditioning actions of Ang II are sensitive to losartan indicating that they are mediated by AT-1R. These findings agree with previous reports13-16,22 indicating the importance of AT-1R-dependent signaling in the improvements of post-ischemic ventricular recovery with APC. An important mechanism of Ang II action in cells is through AT-1R-dependent, stimulation of PKC activity. This occurs mainly in response to the activation of AT-1R downstream effectors such as phospholipase C, inositol-1,4,5-triphosphate and diacylglycerol generation18 but could also involve redox-sensitive modification.34 PKC is an effector in the RAS/Raf/MEK/ERK pathway and has been implicated in the activation of p38MAPK, c-jun N-terminal kinase (JNK), and Akt/PKB signaling pathways by mitochondrial ROS released in response to NAD(P)H oxidase-mediated ROS generation.18,27-29 Besides, PKC can interact, directly or indirectly, with components of mitochondrial membranes such as mPTP, mKATP, BAX/BAD and Bcl-2 to determine the survival of myocytes.34 For these reasons, it was postulated that both IPC and APC share common pathways for cardioprotection involving central roles for PKC and ROS.
Our studies with chelerythrine indicate that cardiac function in APC depends partly on PKC activity during the pre-ischemia. Indeed, about 20% of cardiac function during this period was sensitive to pre-treatment with the PKC inhibitor. This agrees with a role of PKC as a modulator of muscle cell contraction following AT-1R activation.18 In the post-ischemic period, however, the sensitivity to chelerythrine was lost. Likewise, no effect of chelerythrine was observed in the pre-ischemic or the post-ischemic periods in the IPC/APC group. These results were unexpected, because the use of PKC inhibitors such as polymyxin B during the preconditioning phase have been reported to block the cardioprotective effects of Ang II13,16 and in our experimental conditions, chelerythrine abolished the increase in Ang II-induced coronary resistance and inhibited mitochondrial function in IPC/APC. The reason for the absence of chelerythrine sensitivity in cardiac function in IPC/APC and the post-ischemia of APC is unknown, but could be related to the use of multi-cycle preconditioning.35 Alternatively, cardioprotection in chelerythrine-treated hearts could involve redundant, PKC-independent pathways activated during the preconditioning phase or PKC-dependent pathways activated in the reperfusion. Indeed, evidence is available indicating that activation of prosurvival kinase pathways8,36 (i.e. MEK-1/2-ERK-1/2, PI3-Akt-NO-PKG) during the preconditioning or reperfusion phase could lead to cardioprotection. It is worth noting that chelerythrine (10 μM) has been shown to inhibit the protective effects of IPC in rat hearts37 when added at reperfusion, indicating that the post-ischemic phase is also important for cardioprotection. Therefore, additional experiments are necessary to clarify the role of PKC and the pro-survival kinase pathway in the experimental conditions evaluated in this work.
Our data suggest that APC and IPC induce improvements in the post-ischemic ventricular recovery using distinct signaling pathways because significant increases in the absolute values of +dP/dt and RPP were evident when the IPC/APC treatment was compared to APC or IPC alone. The absolute LVDP values also increased in the IPC/APC group compared with APC. In addition, the percent recoveries of LVDP, +dP/dt and RPP in the IPC/APC group were significantly higher than those observed for APC. It is noteworthy that losartan reduced post-ischemic ventricular function in APC and IPC/APC to values not different from control and IPC, respectively, supporting the existence of separate mechanisms for cardiac function improvement by APC and IPC.
The additive effects of IPC and APC on ventricular function reported here suggest that extra-mitochondrial effects of Ang II add cardioprotective effects to IPC. Several mechanisms could contribute to these actions of APC. These include among others, Ang II-dependent, ROS generation from NAD(P)H oxidase activity26-29 and redox-sensitive MAPKs activation.28-29 However, the improvement in post-ischemic ventricular contractility by APC could also involve changes in myocardial contractility38-40 that could take place in response to alterations in myofilament Ca2+ sensitivity.40-41 Indeed, a recent study42 identified the β-arrestin-dependent mechanism as the AT-1R-dependent pathway responsible for the increase in Ca2+ sensitivity and maximum tension of cardiac myofilaments. Besides, the rising intracellular pH at reperfusion could promote an enhanced contractility in Ang II-preconditioned hearts. For these reasons, distinct and additive mechanisms could be activated by IPC and APC to promote the increased post-ischemic ventricular contractility observed by IPC/APC.
Finally, it must be indicated that the additive effects of combined IPC and APC in cardiac function were not present when cardiac damage by either LDH release and infarct size was evaluated. These findings suggest that IPC and APC-dependent mechanisms converge on final pathways that reduce myocardial cell damage. It is likely that mitochondria are part of these converging pathways against reperfusion-mediated cell death because both IPC and APC reportedly affect these organelles through ROS-mediated mitKATP channels and mPTP regulation.27,29 Indeed, IPC/APC significantly increased mitochondrial state 3 and RCI when compared with IPC or APC alone. It is noteworthy that such interaction takes place through an AT-1R-dependent mechanism and requires PKC activation during the preconditioning phase. This suggests that early activation of PKC (PKC-ε) by APC is central to improving the coupling and/or efficiency of mitochondrial respiration by IPC/APC. It should be noted however, that this effect supports, but is not essential for the enhancement in ventricular recovery demonstrated by IPC/APC because chelerythrine did not affect the improvements in cardiac function by this treatment. To our knowledge this is the first time that a synergism between Ang II and IPC has been reported at the level of the mitochondria.
The precise mechanisms mediating the modulation of mitochondrial respiration by IPC/APC are not known but could involve the redox-sensitive MAPK signaling (i.e. p38 and SAPK/JNKs) that is activated secondary to mitKATP channel and ROS generation.27-29 Alternatively, mitochondrial respiration could be improved through activation of PKC-ε, which inhibits mPTP and increases the activity of cytochrome c oxidase (COX).43 It has been proposed that interaction of PKC-ε with COX regulates the electron transfer chain and improves electron flow from cytochrome c to molecular oxygen.43 A link between Ang II and PKC-ε has been suggested by the involvement of this kinase in the stimulation of cell hypertrophy by Ang II in neonatal cardiomyocytes.44 Therefore, it is possible that beneficial effects of both IPC and APC converge on PKC-ε to inhibit mPTP. Although our data do not show significant inhibition of mPTP in APC, there was a tendency toward reduced pore opening in both APC and IPC/APC compared with control and IPC, respectively. Our finding that the increased RCI observed with IPC/APC was abolished with chelerythrine supports a central role of PKC (PKC-ε) in the control of mPTP and hence, of ADP-dependent respiration. Future studies are required to clarify the relevance of these mechanisms to the improvements in mitochondrial respiration (state 3 and RCI) observed with the combination of IPC and APC.
Conclusion
The results of the present study confirm that APC and IPC improved cardiac recovery after IR injury. Both treatments improved post-ischemic ventricular recovery, reduced LDH release and decreased infarct size. When combined, IPC and APC lead to enhanced ventricular recovery suggesting additive effects of these treatments. However, additive effects of IPC and APC were not observed for LDH release or infarct size. The enhancement of post-ischemic ventricular recovery by IPC/APC was associated with an AT-1R-dependent and PKC-mediated increase in mitochondrial RCI. Altogether, these findings suggest the interaction of independent pathways for IPC and APC that improve cardiac function and converge on mitochondria to reduce myocardial damage. This interaction could be a protective mechanism to enhance cardioprotection in the setting of cardiac renin-angiotensin system upregulation and ischemic heart disease.
Acknowledgments
This study was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health through Research Grant SC1HL118669 (SJ), and in part, by the RCMI National Center for Research Resources NIH Grant G12RR-03051.
Footnotes
Conflict of interest: The authors confirm that there are no conflicts of interest.
References
- 1.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 2.Fernandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning and post conditioning. Pharmacol Rev. 2007;59:418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 3.Sadat U. Signaling pathways of cardioprotective ischemic preconditioning. Internal J Surg. 2009;7:490–498. doi: 10.1016/j.ijsu.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Budas GR, Churchill EN, Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemic-reperfusion injury. Pharmacol Res. 2007;55:523–536. doi: 10.1016/j.phrs.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Lancaster TS, Jefferson SL, Korzick DH. Local delivery of a PKC-activating peptide limits ischemia reperfusion injury in the aged female heart. Am J Physiol Regul Comp Physiol. 2011;301:R1242–R1249. doi: 10.1152/ajpregu.00851.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Guang S, Liu GS, Heusch G, Cohen MV, Downey JM. Opening of mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- 7.Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ Res. 2001;88:802–809. doi: 10.1161/hh0801.089342. [DOI] [PubMed] [Google Scholar]
- 8.Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 9.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exptl Ther. 2009;330:670–678. doi: 10.1124/jpet.109.153213. [DOI] [PubMed] [Google Scholar]
- 10.Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res. 2012;111:1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- 11.O'Rourke B. Myocardial KATP channels in preconditioning. Circ Res. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- 12.Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301:H1723–H1741. doi: 10.1152/ajpheart.00553.2011. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Tsuchida A, Cohen MV, Downey JM. Pretreatment with angiotensin II activates protein kinase C and limits myocardial infarction in isolated rabbit hearts. J Mol Cell Cardiol. 1995;27:883–892. doi: 10.1016/0022-2828(95)90038-1. [DOI] [PubMed] [Google Scholar]
- 14.Diaz RJ, Wilson GJ. Selective blockade of AT1 angiotensin receptors abolishes ischemic preconditioning in isolated rabbit hearts. J Mol Cell Cardiol. 1997;29:129–139. doi: 10.1006/jmcc.1996.0258. [DOI] [PubMed] [Google Scholar]
- 15.Sharma A, Singh M. Role of angiotensin in cardioprotective effect of ischemic preconditioning. J Cardiovasc Pharmacol. 1999;33:772–778. doi: 10.1097/00005344-199905000-00014. [DOI] [PubMed] [Google Scholar]
- 16.Sharma A, Singh M. Possible mechanism of cardioprotective effect of angiotensin preconditioning in isolated rat heart. Eur J Pharmacol. 2000;406:85–92. doi: 10.1016/s0014-2999(00)00582-3. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira AJ, Santos RA, Almeida AP. Angiotensin: cardioprotective effect in myocardial ischemia/reperfusion. Hypertension. 2001;38:665–668. doi: 10.1161/01.hyp.38.3.665. [DOI] [PubMed] [Google Scholar]
- 18.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 19.Shi X, Yan C, Nadtochiy SM, Abe J, Brookes PS, Berks BC. P90 ribosomal S6 kinase regulates activity of the renin-angiotensin system: A pathogenic mechanism for ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51:272–275. doi: 10.1016/j.yjmcc.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu B, Sun Y, Sievers RE, Browne AE, Pulukurthy S, Sudhirk, Lee RJ, Chou TM, Chatterjee k, Parmley WW. Comparative effects of pretreatment with captopril and losartan on cardiovascular protection in a rat model of ischemia-reperfusion. J Am Coll Cardiol. 2000;35:787–795. doi: 10.1016/s0735-1097(99)00592-6. [DOI] [PubMed] [Google Scholar]
- 21.Flynn JD, Akers WS. Effects of the angiotensin II subtype 1 receptor antagonist losartan on functional recovery of isolated rat hearts undergoing global myocardial ischemia-reperfusion. Pharmacotherapy. 2003;23:1401–1410. doi: 10.1592/phco.23.14.1401.31947. [DOI] [PubMed] [Google Scholar]
- 22.Nakano A, Miura T, Ura N, Suzuki K, Shimamoto K. Role of angiotensin II type 1 receptor in preconditioning against infarction. Coronary Artery Dis. 1997;8:343–350. doi: 10.1097/00019501-199706000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Hostrup A, Christensen GL, Bentzen BH, Liang B, Aplin M, Grunnet M, Hansen JL, Jespersen T. Functionally selective AT1 receptor activation reduces ischemia reperfusion injury. Cell Physiol Biochem. 2012;30:642–652. doi: 10.1159/000341445. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y, Menon V, Jugdutt BI. Cardioprotection after angiotensin II type blockade involves angiotensin II type 2 receptor expression and activation of protein kinase C-ε in acutely reperfused myocardial infarction in the dog: Effect of UP269 and losartan on AT-1 and AT-2 receptor expression and IP3 receptor and PKC-ε proteins. J RAAS. 2000;1:184–195. doi: 10.3317/jraas.2000.024. [DOI] [PubMed] [Google Scholar]
- 25.Das S, Engelman RM, Maulik N, Das DK. Angiotensin preconditioning of the heart. Cell Biochem Biophys. 2006;44:103–110. doi: 10.1385/CBB:44:1:103. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Tsuchida A, Cohen MV, Downey JM. Pretreatment with angiotensin II activates protein kinase C and limits myocardial infarction in isolated rabbit hearts. J Mol Cell Cardiol. 1995;27:883–892. doi: 10.1016/0022-2828(95)90038-1. [DOI] [PubMed] [Google Scholar]
- 27.Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- 28.Zhang GX, Lu XM, Kimura S, Nishiyama A. Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation. Cardiovasc Res. 2007;76:204–212. doi: 10.1016/j.cardiores.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 29.Daiber A. Redox signaling (cross talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Bioch Bioph Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 30.Javadov S, Choi A, Rajapurohitam V, Zeidan A, Basnakian AG, Karmazyn M. NHE-1 inhibition-induced cardioprotection against ischemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc Res. 2008;77:416–424. doi: 10.1093/cvr/cvm039. [DOI] [PubMed] [Google Scholar]
- 31.Barreto-Torres G, Parodi-Rullan R, Javadov S. The role of PPAR in metformin-induced attenuation of mitocondrial dysfunction in acute cardiac ischemia/reperfusion in rats. Int J Mol Sci. 2012;13:7694–7709. doi: 10.3390/ijms13067694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chappell JB. The oxidation of citrate, isocitrate and cis-aconitate by isolated mitochondria. Biochem J. 1964;90:225–237. doi: 10.1042/bj0900225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laclau MN, Boudina S, Thambo JB, Tariosse L, Gouverneur G, Bonoron-Adele S, Saks VA, Garlid KD, Dos Santos P. Cardioprotection by ischemic preconditioning preserves mitochondrial function and functional coupling between adenine nucleotide translocase and creatine kinase. J Mol Cell Cardiol. 2001;33:947–956. doi: 10.1006/jmcc.2001.1357. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. 2010;24(3):225–234. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandhu R, Diaz RJ, Mao GD, Wilson GJ. Differences in protection and susceptibility to blockade with single-cycle versus multi-cycle transient ischemia. Circulation. 1997;96:984–995. doi: 10.1161/01.cir.96.3.984. [DOI] [PubMed] [Google Scholar]
- 36.Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol. 2005;288:H971–H976. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- 37.Hausenloy DJ, Wynne AM, Yellon DM. Ischemic preconditioning targets the reperfusion phase. Basic Res Cardiol. 2007;102(5):445–452. doi: 10.1007/s00395-007-0656-1. [DOI] [PubMed] [Google Scholar]
- 38.Ishihata A, Endoh M. Pharmacological characteristics of the positive inotropic effect of angiotensin II in the rabbit ventricular myocardium. Br J Pharmacol. 1993;108(4):999–1005. doi: 10.1111/j.1476-5381.1993.tb13497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salas MA, Vila-Petroff MG, Palomeque J, Aiello EA, Mattiazzi A. Positive inotropic and negative lusitropic effect of angiotensin II: intracellular mechanisms and second messengers. J Mol Cell Cardiol. 2001;33(11):1957–1971. doi: 10.1006/jmcc.2001.1460. [DOI] [PubMed] [Google Scholar]
- 40.Ikenouchi H, Barry WH, Bridge JH, Weinberg EO, Apstein CS, Lorell BH. Effects of angiotensin II on intracellular Ca2+ and pH in isolated beating rabbit hearts and myocytes loaded with the indicator indo-1. J Physiol. 1994;480(Pt 2):203–215. doi: 10.1113/jphysiol.1994.sp020353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita S, Endoh M. Influence of a Na+-H+exchange inhibitor ethyisopropylamiloride, a Na+-Ca2+exchange inhibitor KB-R7943 and their combination on the increases in contractility and Ca2+transient induced by angiotensin II in isolated adult ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol. 1999;360(5):575–584. doi: 10.1007/s002109900123. [DOI] [PubMed] [Google Scholar]
- 42.Monasky MM, Taglieri DM, Henze M, Warren CM, Utter MS, Soergel DG, Violin JD, Solaro JR. The β-arrestin-biased ligand TRV 120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am J Physiol Heart Circ Physiol. 2013;305:H856–H866. doi: 10.1152/ajpheart.00327.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ogbi M, Johnson JA. Protein kinase Cε interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte preconditioning. Biochem J. 2006;393:191–199. doi: 10.1042/BJ20050757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Z, Wang W, Geng J, Wang L, Su G, Zhang Y, Ge Z, Kang W. Protein kinase C epsilon-dependent extracellular signal-regulated kinase 5 phosphorylation and nuclear translocation involved in cardiomyocyte hypertrophy with angiotensin II stimulation. J Cell Biochem. 2010;109:653–662. doi: 10.1002/jcb.22441. [DOI] [PubMed] [Google Scholar]