

Abstract
Performing genetic studies in multiple human populations can identify disease risk alleles that are common in one population but rare in others1, with the potential to illuminate pathophysiology, health disparities, and the population genetic origins of disease alleles. We analyzed 9.2 million single nucleotide polymorphisms (SNPs) in each of 8,214 Mexicans and Latin Americans: 3,848 with type 2 diabetes (T2D) and 4,366 non-diabetic controls. In addition to replicating previous findings2–4, we identified a novel locus associated with T2D at genome-wide significance spanning the solute carriers SLC16A11 and SLC16A13 (P=3.9×10−13; odds ratio (OR)=1.29). The association was stronger in younger, leaner people with T2D, and replicated in independent samples (P=1.1×10−4; OR=1.20). The risk haplotype carries four amino acid substitutions, all in SLC16A11; it is present at ≈50% frequency in Native American samples and ≈10% in East Asian, but rare in European and African samples. Analysis of an archaic genome sequence indicated the risk haplotype introgressed into modern humans via admixture with Neandertals. The SLC16A11 mRNA is expressed in liver, and V5-tagged SLC16A11 protein localizes to the endoplasmic reticulum. Expression of SLC16A11 in heterologous cells alters lipid metabolism, most notably causing an increase in intracellular triacylglycerol levels. Despite T2D having been well studied by genome-wide association studies (GWAS) in other populations, analysis in Mexican and Latin American individuals identified SLC16A11 as a novel candidate gene for T2D with a possible role in triacylglycerol metabolism.
The Slim Initiative in Genomic Medicine for the Americas (SIGMA) Type 2 Diabetes Consortium set out to characterize the genetic basis of T2D in Mexican and Latin American populations, where the prevalence is roughly twice that of U.S. non-Hispanic whites5,6. This report considers 3,848 T2D cases and 4,366 controls (Table 1) genotyped using the Illumina OMNI 2.5 array that were unrelated to other samples, and that fall on a cline of Native American and European ancestry7 (Extended Data Fig. 1). Association analysis included 9.2 million variants that were imputed8,9 from the 1,000 Genomes Project Phase I release10 based on 1.38 million SNPs directly genotyped at high quality with minor allele frequency (MAF) >1%.
Table 1.
Study cohorts comprising the SIGMA T2D Project dataset with sample location, study design, numbers of cases and controls (including numbers before quality control (QC) checks), % male participants, age ± standard deviation (SD), age-of-onset in cases ± SD, body mass index ± SD, and fasting plasma glucose in controls ± SD.
| Study | Sample Location | Study Design | N (Before QC) | % Male | Age (years) | Age-of- onset (years) | Body mass index (kg/m2) | Fasting plasma glucose (mmol/l) | |
|---|---|---|---|---|---|---|---|---|---|
| UNAM/INCMNSZ Diabetes Study (UIDS) | Mexico City, Mexico | Prospective Cohort | Controls | 1,138 (1,195) | 41.1% | 55.3 ± 9.4 | – | 28.1 ± 4.0 | 4.8 ± 0.5 |
| T2D Cases | 815 (872) | 40.9% | 56.2 ± 12.3 | 44.2 ± 11.3 | 28.4 ± 4.5 | – | |||
| Diabetes in Mexico Study (DMS) | Mexico City, Mexico | Prospective Cohort | Controls | 472 (505) | 25.8% | 52.5 ± 7.7 | – | 28.0 ± 4.4 | 5.0 ± 0.4 |
| T2D Cases | 690 (762) | 33.0% | 55.8 ± 11.1 | 47.8 ± 10.6 | 29.0 ± 5.4 | – | |||
| Mexico City Diabetes Study (MCDS) | Mexico City, Mexico | Prospective Cohort | Controls | 613 (790) | 39.3% | 62.5 ± 7.7 | – | 29.4 ± 4.8 | 5.0 ± 0.5 |
| T2D Cases | 287 (358) | 41.1% | 64.2 ± 7.5 | 55.1 ± 9.7 | 29.9 ± 5.4 | – | |||
| Multiethnic Cohort (MEC) | Los Angeles, California, USA | Case- Control | Controls | 2,143 (2,464) | 48.3% | 59.3 ± 7.0 | – | 26.6 ± 3.9 | N/A |
| T2D Cases | 2,056 (2,279) | 47.9% | 59.2 ± 6.9 | N/A | 30.0 ± 5.4 | – |
Extended Data Figure 1. Principle component analysis (PCA) projection of SIGMA samples onto principal components calculated using data from samples collected by the Human Genome Diversity Project (HGDP).

PCA projection of SIGMA onto HGDP Yoruba, French, Karitiana and Han (Chinese) populations (a) before ancestry quality control filters were applied, with cohort centroids as indicated and (b) after all quality control filters were applied, with case and control centroids as indicated. Principal components 3 and 4 (c) before filtering samples on ancestry (a small number of samples in the MEC cohort show East Asian admixture) and (d) after all quality control filters were applied. Additional plots as in (b) but separating (e) cases and (f) controls.
The association of SNP genotype with T2D was evaluated using LTSOFT11, a method that increases power by jointly modeling case-control status with non-genetic risk factors. Our analysis utilized body mass index (BMI) and age to construct liability scores and also included adjustment for sex and ancestry via principal components7. The quantile-quantile (QQ) plot is well calibrated under the null (λGC = 1.05; Fig. 1a, red), indicating adequate control for confounders, with substantial excess signal at P<10−4.
Figure 1. Identification of a novel T2D risk haplotype carrying 5 SNPs in SLC16A11.

(a) QQ plot of association statistics in genome-wide scan shows calibration under the null and enrichment in the tail for all SNPs (red), and after removing SNPs within 1 Mb of previously published T2D associations (blue). Removal of sites within 1 Mb of 68 known loci and two novel loci results in a null distribution (black). (b) Regional plot of association at 17p13.1 that spans SLC16A11 and SLC16A13. (c) Analysis conditional on genotype at rs13342232 (the top associated variant) reduces signal to far below genome-wide significance across the surrounding region. Color indicates r2 to the most strongly associated site; recombination rate is shown, each based on the 1,000 Genomes ASN population. (d) Graphical depictions of SLC16A11 haplotypes constructed from the synonymous and four missense SNPs associated to T2D, with haplotype frequencies derived from the 1,000 Genomes Project and SIGMA samples. AFR, Africa (n=185); EUR, European (n=379); ASN, East Asian (n=286); MXL, Mexican samples from Los Angeles (n=66). Frequencies from SIGMA samples are calculated from genotypes and represent either the entire dataset (“All”) or only samples estimated to have ≥95% Native American ancestry (“≥95 N.A.”, n=290; Supplementary Note). Haplotypes with population frequency <1% are not depicted. (e) Predicted membrane topology of human SLC16A11 generated using TMHMM 2.0 and visualized with TeXtopo. Locations of SNPs carried by the T2D-associated haplotype are indicated. (f) Forest plot depicting odds ratio estimates at rs75493593 from the four SIGMA cohorts, the SIGMA pooled mega-analysis, the replication cohorts, replication-only meta-analysis, and the overall meta-analysis (including all replication cohorts and the SIGMA mega-analysis). Accompanying table lists ethnicity, cohort names, estimated odds ratio (OR) and 95% confidence interval (95% CI). Replication cohorts are the Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES), Multiethnic Cohort (MEC), and Singapore Chinese Health Study (SCHS). Further details provided in Supplementary Table 8.
We first examined SNPs previously reported to be associated to risk of T2D. Two such variants reached genome-wide significance: TCF7L2 (rs7903146; P=2.5×10−17; OR=1.41 [95% confidence interval 1.30–1.53]) and KCNQ1 (rs2237897; P=4.9×10−16; OR=0.74 [0.69–0.80]) (Extended Data Figs. 2, 3a), with effect sizes and frequencies consistent with previous studies3,4,12. At KCNQ1, we identified a signal3 of association that shows limited linkage disequilibrium both to rs2237897 (r2=0.056) and to rs231362 (r2=0.028) (previously seen in Europeans), suggesting a third allele at this locus (rs139647931; after conditioning, P=5.3×10−8; OR=0.78 [0.70–0.86]; Extended Data Fig. 3b; Supplementary Note).
Extended Data Figure 2. Regional plot for signal at TCF7L2.

Point color indicates r2 to the most strongly associated site (rs7903146) and recombination rate is also shown, both based on the 1,000 Genomes ASN population.
Extended Data Figure 3. Conditional analyses reveal multiple independent signals at INS-IGF2 and KCNQ1.

Regional plots are shown for the interval spanning INS-IGF2 and KCNQ1 (a) without conditioning, (b) conditional on rs2237897 at KCNQ1, (c) conditional on rs2237897 and rs139647931 (both at KCNQ1), and (d) conditional on rs2237897 and rs139647931 (both at KCNQ1) and rs11564732 (the top associated variant in the INS-IGF2-TH region). The top SNPs in 11p15.5 and KCNQ1 are ~700 kb away from each other, but despite this proximity, there is a strong residual signal of association at INS-IGF2 after analysis conditional on genotype at KCNQ1. Point color indicates r2 to rs11564732 and recombination rate is also shown, both based on the 1,000 Genomes ASN population.
More generally, of SNPs previously associated with T2D at genome-wide significance, 56 of 68 are directionally consistent with the initial report (P=3.1×10−8; Supplementary Table 1). Nonetheless, a QQ plot excluding all SNPs within 1 Mb of the 68 T2D associations remains strikingly non-null (Fig. 1a, blue).
This excess signal of association is entirely attributable to two regions of the genome: chromosome 11p15.5 and 17p13.1 (Fig. 1a, black). The genome-wide significant association at 11p15.5 spans insulin, IGF2, and other genes (Extended Data Fig. 3a). The strongest association lies in the 3′-UTR of IGF2 and the non-coding INS-IGF2 transcript (rs11564732, P=2.6×10−8; OR=0.77 [0.70–0.84]; Supplementary Table 2). The associated SNPs are ~700 kb from the genome-wide significant signal in KCNQ1 (above), and analysis conditional on the two significant KCNQ1 SNPs reduced the INS-IGF2 association signal to just below genome-wide significance (P=7.5×10−7, Extended Data Fig. 3c). Conditioning on the two KCNQ1 SNPs and the INS-IGF2 SNP reduces the signal to background (Extended Data Fig. 3d). Further analysis is needed to determine whether the INS-IGF2 signal is reproducible and independent of that at KCNQ1.
The strongest novel association is at 17p13.1 spanning SLC16A11 and SLC16A13 (Fig. 1b), both poorly characterized members of the monocarboxylic acid transporter family of solute carriers13. The strongest signal of association includes a silent mutation as well as four missense SNPs, all in SLC16A11 (Fig. 1d, e). These five variants are (a) in strong LD (r2 ≥ 0.85 in 1,000 Genomes samples from the Americas) and co-segregate on a single haplotype, (b) common in samples of Mexican and Latin American ancestry, and (c) show equivalent levels of association to T2D (P=2.4×10−12 to P=3.9×10−13; OR=1.29 [1.20–1.38]; Supplementary Tables 3, 4, and 5). Analysis conditional on any of these variants leaves no genome-wide significant signal (Fig. 1c, Extended Data Fig. 4). Computational prediction with SIFT14 (which considers each site independently) labels one of the missense SNPs (rs13342692, D127G) as damaging and the other three “tolerated” (Supplementary Table 6).
Extended Data Figure 4. Regional plots for SLC16A11 conditional on associated missense variants of that gene.

Association signal at chromosome 17p13 (a) without conditioning, or conditional on the four missense SNPs in SLC16A11: (b) rs117767867, (c) rs13342692, (d) rs75418188, and (e) rs75493593. Point color indicates r2 to the most strongly associated SNP (rs13342232) and recombination rate is also shown, both based on the 1,000 Genomes ASN population.
Individuals with T2D that carry the risk haplotype develop T2D 2.1 years earlier (P=3.1×10−4), and at 0.9 kg/m2 lower BMI (P=5.2×10−4) than non-carriers (Extended Data Fig. 5). The odds ratio for the risk haplotype estimated using young cases (≤ 45 years) was higher than in older cases (OR=1.48 versus 1.11; Pheterogeneity=1.7×10−3). We tested the haplotype for association with related metabolic quantitative traits in the fasting state in a subset of SIGMA participants (n=1,505–3,855). No associations surpass nominal significance (P<0.05; Supplementary Table 7).
Extended Data Figure 5. Cases with risk haplotype develop T2D younger and at a lower BMI than non-carriers.
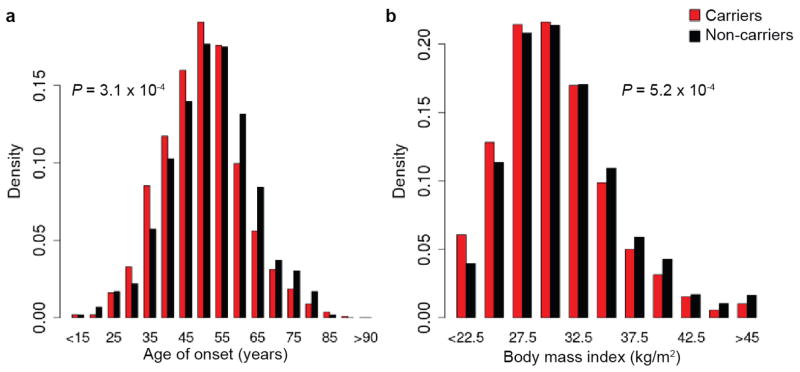
(a) Distribution of age-of-onset in T2D cases based on genotype at rs13342232, binned every 5 years with upper bounds indicated (carriers n=1,126; non-carriers n=594). (b) Distribution of BMI in T2D cases for carriers and non-carriers of rs13342232, binned every 2.5 kg/m2 with upper bounds indicated (carriers n=2,161; non-carriers n=1,647). P-values from two-sample t-test between T2D risk haplotype carriers and T2D non-carriers.
Given that large GWAS have been performed for T2D in samples of European and Asian ancestry, it may seem surprising that associated variants at SLC16A11/13 were not previously identified. Using data generated by the 1,000 Genomes Project and the current study, we observed that the risk haplotype (henceforth referred to as “5 SNP” haplotype) is rare or absent in samples from Europe and Africa, has intermediate frequency (≈10%) in samples from East Asia, and up to ≈50% frequency in samples from the Americas (Fig. 1d; Extended Data Fig. 6a). A second haplotype carrying one of the four missense SNPs (D127G) and the synonymous variant (termed the “2 SNP” haplotype) is very common in samples from Africa but rare elsewhere, including in the Americas (Fig. 1d). The low frequency of the 5 SNP haplotype in Africa and Europe may explain why this association was not found in previous studies.
Extended Data Figure 6. Frequency distribution of the risk haplotype and dendrogram depicting clustering with Neandertal haplotypes.

(a) Allele frequency of missense SNP rs117767867 (tag for risk haplotype) in the 1,000 Genomes Phase I dataset. (b) Dendrogram generated from haplotypes across the 73 kb Neandertal introgressed region. Nodes for modern human haplotypes are labeled in red or blue with the 1,000 Genomes population in which the corresponding haplotype resides. Archaic Neandertal sequences are labeled in black and include the low coverage Neandertal sequence15 (labeled Vindija), and the unpublished Neandertal sequence that is homozygous for the 5 SNP risk haplotype18 (Altai). H1 includes haplotypes from MXL and FIN, and H2 and H3 both include haplotypes from CLM, MXL, CHB, and ASW. Modern human sequences included are all 1,000 Genomes Phase I samples that are homozygous for the 5 SNP risk haplotype (n=15), and 16 non-risk haplotypes—four haplotypes (from two randomly selected individuals) from each of the CLM, MXL, CHB, and FIN 1,000 Genomes populations (the populations with carriers of the 5 SNP haplotype). The red subtree depicts the Neandertal clade, with all risk haplotypes clustering with the Altai and Vindija sequences. In blue are all other modern human haplotypes. The dendrogram was generated by the R function hclust using a complete linkage clustering algorithm on a distance matrix measuring the fraction of SNPs called in the 1000 genomes project at which a pair of haplotypes differs (the Y-axis represents this distance). Since haplotypes are unavailable for the archaic samples, we picked a random allele to compute the distance matrix.
We attempted to replicate this association in ~22,000 samples from a variety of ancestry groups. A proxy for the 5 SNP haplotype of SLC16A11 showed strong association with T2D (Preplicaton=1.1×10−4; ORreplication=1.20 [1.09–1.31]; Pcombined=5.4×10−15; ORcombined=1.25 [1.18–1.32]; Fig. 1f; Supplementary Table 8). The association was clearly observed in East Asian samples, a population which lacks admixture of Amerindian and European populations and shows little genetic substructure. This result argues against population stratification as an explanation for the finding in Latino populations.
We estimated the difference in disease prevalence attributable to a risk factor with OR=1.20 (1.09–1.31), 26% frequency in Mexican Americans (as in the SIGMA control samples), and 2% in European Americans. Approximately 20% (9.2%-29%) of the difference in prevalence could be explained by such a risk factor (Online Methods).
Two population genetic features of the 5 SNP haplotype struck us as discordant. The haplotype sequence is highly divergent, with an estimated time to most recent common ancestor (TMRCA) of 799k years to a European haplotype (Supplementary Table 9 and Supplementary Note). This long precedes the “out of Africa” bottleneck. And yet, the haplotype is not observed in Africa and is rare throughout Europe (Fig. 1d).
This combination of age and geographic distribution could be consistent with admixture from Neandertals into modern humans. Neither the published Neandertal genome15 nor the Denisova genome16 contained the variants observed on the 5 SNP haplotype. However, an unpublished genome of a Neandertal from Denisova Cave17,18 is homozygous across 5 kb for the 5 SNP haplotype at SLC16A11, including all four missense SNPs. Over a span of 73 kb this Neandertal sequence is nearly identical to that of individuals from the 1000 Genomes Project who are homozygous for the 5 SNP haplotype (Supplementary Note).
Two lines of evidence suggest that the 5 SNP haplotype entered modern humans through archaic admixture. First, the Neandertal sequence is more closely related to the 5 SNP haplotype than to random non-risk haplotypes (mean TMRCA=250k years versus 677k years; Supplementary Tables 10 and 11, Supplementary Note), forming a clade (Extended Data Fig. 6b), with a coalescence time that postdates the range of estimated split times between modern humans and Neandertals16,19. Second, the genetic length of the 73 kb haplotype is longer than would be expected if it had undergone recombination for ~9,000 generations since the split with Neandertals (P=3.9×10−5; Supplementary Note). These two features indicate that the 5 SNP haplotype is not only similar to the Neandertal sequence, but was likely introduced into modern humans relatively recently through archaic admixture. We note that while this particular Neandertal-derived haplotype is common in the Americas, Latin Americans have the same proportion of Neandertal ancestry genome-wide as other Eurasian populations (~2%)16.
With an absence of multiple independently segregating functional mutations in the same gene, we lack formal genetic proof that SLC16A11 is the gene responsible for association to T2D at 17p13.1. Nonetheless, as the associated haplotype encodes four missense SNPs in a single gene (Supplementary Table 12), we set out to begin characterizing the function of SLC16A11.
We examined the tissue distribution of SLC16A11 mRNA expression using Nanostring and ~55,000 curated microarray samples. In both datasets, we observed SLC16A11 expression in liver, salivary gland, and thyroid (Extended Data Figs. 7 and 8). We used immunofluorescence to determine the subcellular localization of V5-tagged SLC16A11 introduced into HeLa cells. SLC16A11-V5 co-localizes with the endoplasmic reticulum membrane protein Calnexin, but shows minimal overlap with plasma membrane, Golgi apparatus and mitochondria (Fig. 2a). Distinct patterns were seen for other SLC16 family members, which are known to have diverse cellular functions20: SLC16A13-V5 localizes to the Golgi apparatus and SLC16A1-V5 appears at the plasma membrane21 (Extended Data Fig. 9, data not shown).
Extended Data Figure 7. Analysis of gene expression for SLC16A11, SLC16A13, and SLC16A1 in 30 human tissues.

Data measured using nCounter is shown as mean, normalized mRNA counts per 200ng RNA +/− SEM. Threshold for background (non-specific) binding is indicated by the red line. Sample size for each tissue (n): pancreas (5), adipose, brain, colon, liver, skeletal muscle, and thyroid (3), adrenal, fetal brain, breast, heart, kidney, lung, placenta, prostate, small intestine, spleen, testes, thymus, and trachea (2), bladder, cervix, esophagus, fetal liver, ovary, salivary gland, fetal skeletal muscle, skin, umbilical cord, and uterus (1).
Extended Data Figure 8. Microarray-based analysis of SLC16A11 expression in human tissues.

(a) Results from the “55k screen”, a survey of gene expression in 55,269 samples profiled on the Affymetrix U133 plus 2.0 array, are shown as the fraction of samples of a given tissue in which SLC16A11 is expressed. Sample size for each tissue (n): adipose (394), adrenal (69), brain (1990), breast (4104), heart (178), kidney (675), liver (721), lung (1442), pancreas (150), placenta (107), prostate (578), salivary gland (26), skeletal muscle (793), skin (947), testis (102), thyroid (108). (b) Histograms show the expression level distribution of SLC16A11 and other well-studied liver genes in 721 liver samples from the “55k screen.” INS is shown as reference for a gene not expressed in liver. Based on negative controls a normalized log2 expression of 4 is considered baseline and log2 expression values greater than 6 are considered expressed.
Figure 2. SLC16A11 localizes to the endoplasmic reticulum and alters lipid metabolism in HeLa cells.
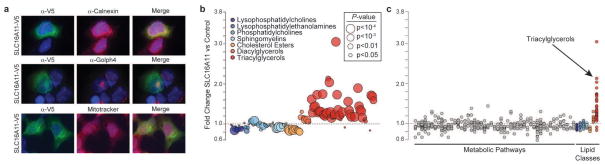
(a) Localization of SLC16A11 to the endoplasmic reticulum. HeLa cells expressing C-terminus, V5-tagged SLC16A11 were immunostained for SLC16 expression (α-V5) along with markers for the endoplasmic reticulum (α-Calnexin), cis-Golgi apparatus (α-Golph4), or mitochondria (MitoTracker). Imaging of each protein was optimized for clarity of localization rather than comparison of expression level across proteins. (b) Changes in intracellular lipid metabolites following expression of SLC16A11-V5 in HeLa cells. The fold-change in cells expressing SLC16A11 relative to cells expressing control proteins is plotted for individual lipid metabolites, with lipid classes indicated by point color and P-values (of the Wilcox rank sum test) by point size. (c) Fold change plotted for both polar and lipid metabolites, grouped according to metabolic pathway or class. Each point within a pathway or class shows the fold-change of a single metabolite within that pathway or class. Pathway names and statistical analyses are shown in Extended Data Fig. 10 and Supplementary Table 14.
Extended Data Figure 9. SLC16A13 localizes to Golgi apparatus.

HeLa cells transiently expressing C-terminus, V5-tagged (a) SLC16A13 or (b) BFP were immunostained for SLC16A13 or BFP expression (α-V5) along with specific markers for the endoplasmic reticulum (α-Calnexin), cis-Golgi apparatus (α-Golph4) and mitochondria (MitoTracker). Due to heterogeneity in expression levels of overexpressed proteins and endogenous organelle markers, imaging of each protein was optimized for clarity of localization and varied across images; therefore, images are not representative of relative expression levels of each protein as compared to the other proteins.
As SLC16 family members are solute carriers, we expressed SLC16A11 (or control proteins) in HeLa cells (which do not express SLC16A11 at appreciable levels) and profiled ~300 polar and lipid metabolites. Expression of SLC16A11 resulted in substantial increases in triacylglycerol (TAG) levels (P=7.6×10−12), with smaller increases in intracellular diacylglycerols (P=7.8×10−3) and decreases in lysophosphatidylcholine (P=2.0×10−3), cholesterol ester (P=9.8×10−4) and sphingomyelin (P=3.9×10−3) lipids (Fig. 2b, c, Supplementary Tables 13 and 14). As TAG synthesis takes place in the endoplasmic reticulum in the liver22, these results suggest that SLC16A11 may play a role in hepatic lipid metabolism. We note that serum levels of specific TAGs have been prospectively associated with future risk of T2D23 and accumulation of intracellular lipids has been implicated in insulin resistance in human populations24,25.
In summary, GWAS in Mexican and Latin American samples identified a haplotype containing four missense SNPs, all in SLC16A11, that is much more common in individuals with Native American ancestry than in other populations. Each haplotype copy is associated with a ~20% increased risk of T2D. With these properties, the haplotype would be expected to contribute to the higher burden of T2D in Mexican and Latin American populations26. The haplotype derives from Neandertal introgression, providing an example of Neandertal admixture affecting physiology and disease susceptibility today. Our data suggest the hypothesis for future studies that SLC16A11 may influence diabetes risk through effects on lipid metabolism in the liver. Our results also indicate that genetic mapping in understudied populations can identify previously undiscovered aspects of disease pathophysiology1.
Methods Summary
DNA samples were prepared using strict quality control procedures and genotyped using the Illumina HumanOmni2.5 array. Stringent sample and SNP quality (including ancestry) filters were applied on the resulting genotypes. Following imputation8,9, SNPs were quality filtered (MAF ≥ 1% and info score ≥ .6) and association testing was performed via LTSOFT11 with T2D status, BMI, and age modeling liability and adjusting for sex and top 2 principal components as fixed effect covariates. P-values were corrected for genomic control (λGC=1.046). Odds ratios (ORs) are from logistic regression in PLINK27 using BMI, age, sex, and top 2 PCs as covariates. Proportion of Native American ancestry was estimated using ADMIXTURE28 (K=3) run including unadmixed individuals from several populations.
Odds ratios for young (≤45 years) and older age of onset cases were calculated using logistic regression in each group compared to two randomly selected non-overlapping sets of controls. Significance testing used a Z-score calculated from these ORs.
Population prevalence was modeled using OR to approximate relative risk in a log-additive effect model29. Relative change in population prevalences is reported based on removing a locus with relative risk of 1.20 and the indicated frequency.
Gene expression analyses were performed on data collected using Nanostring and a compendium of publicly available Affymetrix U133 Plus 2.0 microarrays. The subcellular localization of SLC16A11-V5 and metabolic profiling studies were performed following expression of C-terminus, V5-tagged SLC16A11 in HeLa cells. Metabolite values were normalized to the total metabolite signal obtained for each sample. Measurements were obtained in replicate from each of three independent experiments, with data combined after subtracting the mean of the log-transformed values. The Wilcox rank sum test was used to test for differences in individual metabolite levels in cells expressing SLC16A11 compared to controls; the Wilcoxon signed rank test was used to assess differences in lipid classes.
Supplementary Material
Extended Data Figure 10. Pathway and class-based metabolic changes induced by SLC16A11 expression.

Changes in metabolite levels in HeLa cells expressing SLC16A11-V5 compared to control-transfected cells are plotted in groups according to metabolic pathway or class. Pathways shown include all KEGG pathways from the human reference set for which metabolites were measured as well as eight additional classes of metabolites covering carnitines and lipid sub-types. Each point within a pathway or class shows the fold-change of a single metabolite within that pathway or class. For each pathway or class with at least six measured metabolites, enrichment was computed as described in Online Methods. Asterisks indicate pathways with P ≤ 0.05 and FDR ≤ 0.25. Supplementary Table 14 shows additional details from the enrichment analysis.
Acknowledgments
We thank Mark Daly, Vamsi Mootha, Eric Lander and Karol Estrada for comments on the manuscript, Benjamin Voight, Ayellet Segre, Joseph Pickrell and the Scientific Advisory Board of the SIGMA Project (especially Carlos Bustamante) for useful discussions, and Aravind Subramanian and Victor Rusu for assistance with expression analyses. This work was conducted as part of the Slim Initiative for Genomic Medicine, a joint U.S.-Mexico project funded by the Carlos Slim Health Institute. The UNAM/INCMNSZ diabetes study was supported by Consejo Nacional de Ciencia y Tecnología grants 138826, 128877, CONACyT- SALUD 2009-01-115250, and a grant from Dirección General de Asuntos del Personal Académico, UNAM, IT 214711. The Diabetes in Mexico Study was supported by Consejo Nacional de Ciencia y Tecnología grant 86867 and by Instituto Carlos Slim de la Salud, A.C. The Mexico City Diabetes Study was supported by National Institutes of Health (NIH) grant R01HL24799 and by the Consejo Nacional de Ciencia y Tenologia grants: 2092, M9303, F677-M9407, 251M, and 2005-C01-14502, SALUD 2010-2-151165. The Multiethnic Cohort was supported by NIH grants CA164973, CA054281, and CA063464. The Singapore Chinese Health Study was funded by the National Medical Research Council of Singapore under its individual research grant scheme and by NIH grants R01 CA55069, R35 CA53890, R01 CA80205, and R01 CA144034. The Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) project was supported by NIH grant U01DK085526. The San Antonio Mexican American Family Studies (SAMAFS) were supported by R01 DK042273, R01 DK047482, R01 DK053889, R01 DK057295, P01 HL045522, and a Veterans Administration Epidemiologic grant to R.A.D. A.L.W. is supported by National Institutes of Health Ruth L. Kirschstein National Research Service Award number F32 HG005944.
The SIGMA (Slim Initiative in Genomic Medicine for the Americas) Type 2 Diabetes Genetics Consortium
Writing Team
Amy L. Williams1,2, Suzanne B. R. Jacobs1, Hortensia Moreno-Macías3, Alicia Huerta-Chagoya4,5, Claire Churchhouse1, Carla Márquez-Luna6, Humberto García-Ortíz6, María José Gómez-Vázquez4,7, Noël P. Burtt1, Carlos A. Aguilar-Salinas4, Clicerio González-Villalpando8,*, Jose C. Florez1,9,10,*, Lorena Orozco6,*, Christopher A. Haiman11,*, Teresa Tusié-Luna4,5,*, and David Altshuler1,2,9,10,12,13,14,*
Analysis Team
Amy L. Williams1,2, Carla Márquez-Luna6, Alicia Huerta-Chagoya4,5, Stephan Ripke1,15, María José Gómez-Vázquez4,7, Alisa K. Manning1, Hortensia Moreno-Macías3, Humberto García-Ortíz6, Benjamin Neale1,15, Noël P. Burtt1, Carlos A. Aguilar-Salinas4, David Reich1,2, Daniel O. Stram11, Juan Carlos Fernández-López6, Sandra Romero-Hidalgo6, David Altshuler1,2,9,10,12,13,14, Jose C. Florez1,8,10, Teresa Tusié-Luna4,5, Nick Patterson1, and Christopher A. Haiman11
Clinical Research, Study Design and Metabolic Phenotyping
Diabetes in Mexico Study: Irma Aguilar-Delfín6, Angélica Martínez-Hernández6, Federico Centeno-Cruz6, Elvia Mendoza-Caamal6, Cristina Revilla-Monsalve16, Sergio Islas-Andrade16, Emilio Córdova6, Eunice Rodríguez-Arellano17, Xavier Soberón6, and Lorena Orozco6
Massachusetts General Hospital: Jose C. Florez1,9,10
Mexico City Diabetes Study: Clicerio González-Villalpando8 and María Elena González-Villalpando8
Multiethnic Cohort Study: Christopher A. Haiman11, Brian E. Henderson11, Kristine Monroe11, Lynne Wilkens18, Laurence N. Kolonel18, and Loic Le Marchand18
UNAM/INCMNSZ Diabetes Study: Laura Riba5, María Luisa Ordóñez-Sánchez4, Rosario Rodríguez-Guillén4, Ivette Cruz-Bautista4, Maribel Rodríguez-Torres4, Linda Liliana Muñoz-Hernández4, Tamara Sáenz4, Donají Gómez4, and Ulices Alvirde4
Sample QC and Whole Genome Genotyping
Noël P. Burtt1, Robert C. Onofrio19, Wendy M. Brodeur19, Diane Gage19, Jacquelyn Murphy1, Jennifer Franklin19, Scott Mahan19, Kristin Ardlie19, Andrew T. Crenshaw19, and Wendy Winckler19
Neandertal analysis team
Kay Prüfer20, Michael V. Shunkov21, Susanna Sawyer20, Udo Stenzel20, Janet Kelso20, Monkol Lek1,15, Sriram Sankararaman1,2, Amy L. Williams1,2, Nick Patterson1, Daniel G. MacArthur1,15, David Reich1,2, Anatoli P. Derevianko21, and Svante Pääbo20
Functional analysis and metabolite profiling
Suzanne B. R. Jacobs1, Claire Churchhouse1, Shuba Gopal22, James A. Grammatikos22, Ian C. Smith23, Kevin H. Bullock22, Amy A. Deik22, Amanda L. Souza22, Kerry A. Pierce22, Clary B. Clish22, and David Altshuler1,2,9,10,12,13,14
Replication genotyping and analysis
Broad Institute of Harvard and MIT: Timothy Fennell19, Yossi Farjoun19, Broad Genomics Platform19, and Stacey Gabriel19
Singapore Chinese Health Study: Daniel O. Stram11, Myron D. Gross24, Mark A. Pereira24, Mark Seielstad25, Woon-Puay Koh26,27, and E-Shyong Tai,26,27,28
T2D-GENES Consortium: Jason Flannick1,9, Pierre Fontanillas1, Andrew Morris29, Tanya M. Teslovich30, Noël P. Burtt1, Gil Atzmon31, John Blangero32, Don W. Bowden33, John Chambers34,35,36, Yoon Shin Cho37, Ravindranath Duggirala32, Benjamin Glaser38,39, Craig Hanis40, Jaspal Kooner35,36,41, Markku Laakso42, Jong-Young Lee43, E-Shyong Tai26,27,28, Yik Ying Teo44,45,46,47,48, James G. Wilson49, and the T2D-GENES Consortium
Multiethnic Cohort Study: Christopher A. Haiman11, Brian E. Henderson11, Kristine Monroe11, Lynne Wilkens18, Laurence N. Kolonel18, and Loic Le Marchand18
Texas Biomedical Research Institute and University of Texas Health Science Center at San Antonio: Sobha Puppala32, Vidya S. Farook32, Farook Thameem50, Hanna E. Abboud50, Ralph A. DeFronzo51, Christopher P. Jenkinson51, Donna M. Lehman52, Joanne E. Curran32, John Blangero32, and Ravindranath Duggirala32
Scientific and Project Management
Steering Committee
David Altshuler1,2,9,10,12,13,14, Jose C. Florez1,9,10, Christopher A. Haiman11, Brian E. Henderson11, Carlos A. Aguilar-Salinas4, Clicerio González-Villalpando8, Lorena Orozco6, and Teresa Tusié-Luna4,5
Footnotes
Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, Massachusetts, USA
Department of Genetics, Harvard Medical School, Boston, Massachusetts, USA
Universidad Autonoma Metropolitana, Mexico City, Mexico
Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, Mexico
Instituto de Investigaciones Biomédicas, UNAM. Unidad de Biología Molecular y Medicina Genómica, UNAM/INCMNSZ, Mexico City, Mexico
Instituto Nacional de Medicina Genómica, Mexico City, Mexico
Universidad Autónoma de Nuevo León, San Nicolás de los Garza, Nuevo León 66451, México
Centro de Estudios en Diabetes, Unidad de Investigacion en Diabetes y Riesgo Cardiovascular, Centro de Investigacion en Salud Poblacional, Instituto Nacional de Salud Publica, Mexico City, Mexico
Center for Human Genetic Research and Diabetes Research Center (Diabetes Unit), Massachusetts General Hospital, Boston, Massachusetts, USA
Department of Medicine, Harvard Medical School, Boston, Massachusetts, USA
Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, California,, USA
Center for Human Genetic Research, Massachusetts General Hospital, Boston, Massachusetts, USA
Department of Molecular Biology, Harvard Medical School, Boston, Massachusetts, USA
Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA
Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA
Instituto Mexicano del Seguro Social SXXI, Mexico City, Mexico
Instituto de Seguridad y Servicios Sociales para los Trabajadores del Estado, Mexico City, Mexico
Epidemiology Program, University of Hawaii Cancer Center, Honolulu, Hawaii, USA
The Genomics Platform, The Broad Institute of Harvard and MIT, Cambridge, Massachusetts, USA
Department of Evolutionary Genetics, Max Planck Institute for Evolutionary Anthropology, D-04103 Leipzig, Germany
Palaeolithic Department, Institute of Archaeology and Ethnography, Russian Academy of Sciences, Siberian Branch, 630090 Novosibirsk, Russia
The Metabolite Profiling Platform, The Broad Institute of Harvard and MIT, Cambridge, Massachusetts, USA
Cancer Biology Program, The Broad Institute of Harvard and MIT, Cambridge, Massachusetts, USA
University of Minnesota, Minneapolis, Minnesota, USA
University of California San Francisco, San Francisco, California, USA
Duke-National University of Singapore Graduate Medical School, Singapore, Singapore
Saw Swee Hock School of Public Health, National University of Singapore, Singapore
Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK
Department of Biostatistics, Center for Statistical Genetics, University of Michigan, Ann Arbor, Michigan, USA
Department of Medicine, Department of Genetics, Albert Einstein College of Medicine, Bronx, New York, USA
Department of Genetics, Texas Biomedical Research Institute, San Antonio, Texas, USA
Center for Genomics and Personalized Medicine Research, Center for Diabetes Research, Department of Biochemistry, Department of Internal Medicine, Wake Forest School of Medicine, Winston-Salem, North Carolina, USA
Department of Epidemiology and Biostatistics, Imperial College London, London, UK
Imperial College Healthcare NHS Trust, London, UK.
Ealing Hospital National Health Service (NHS) Trust, Middlesex, UK
Department of Biomedical Science, Hallym University, Chuncheon, Gangwon-do, Korea
Endocrinology and Metabolism Service, Hadassah-Hebrew University Medical School, Jerusalem, Israel
Israel Diabetes Research Group (IDRG), Israel
Human Genetics Center, University of Texas Health Science Center at Houston, Houston, Texas, USA
National Heart and Lung Institute (NHLI), Imperial College London, Hammersmith Hospital, London, UK
Department of Medicine, University of Eastern Finland, Kuopio Campus and Kuopio University Hospital, Kuopio, Finland
Center for Genome Science, National Institute of Health, Osong Health Technology Administration Complex, Chungcheongbuk-do, Cheongwon-gun, Gangoe-myeon, Yeonje-ri, Korea
Department of Epidemiology and Public Health, National University of Singapore, Singapore, Singapore
Centre for Molecular Epidemiology, National University of Singapore, Singapore, Singapore
Genome Institute of Singapore, Agency for Science, Technology and Research, Singapore, Singapore
Graduate School for Integrative Science and Engineering, National University of Singapore, Singapore, Singapore
Department of Statistics and Applied Probability, National University of Singapore, Singapore, Singapore.
Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, Mississippi, USA
Division of Nephrology, Department of Medicine, University of Texas Health Science Center at San Antonio, San Antonio, Texas
Division of Diabetes, Department of Medicine, University of Texas Health Science Center at San Antonio, San Antonio, Texas
Division of Clinical Epidemiology, Department of Medicine, University of Texas Health Science Center at San Antonio, San Antonio, Texas
Broad Institute of Harvard and MIT, Cambridge, Massachusetts, USA
denotes co-corresponding authors
The authors declare no competing financial interests.
References
- 1.Rosenberg NA, et al. Genome-wide association studies in diverse populations. Nature reviews. Genetics. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grant SFA, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature genetics. 2006;38:320–323. doi: 10.1038/ng1732. doi: http://www.nature.com/ng/journal/v38/n3/suppinfo/ng1732_S1.html. [DOI] [PubMed] [Google Scholar]
- 3.Unoki H, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nature genetics. 2008;40:1098–1102. doi: 10.1038/ng.208. doi: http://www.nature.com/ng/journal/v40/n9/suppinfo/ng.208_S1.html. [DOI] [PubMed] [Google Scholar]
- 4.Yasuda K, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nature genetics. 2008;40:1092–1097. doi: 10.1038/ng.207. doi: http://www.nature.com/ng/journal/v40/n9/suppinfo/ng.207_S1.html. [DOI] [PubMed] [Google Scholar]
- 5.Villalpando S, et al. Prevalence and distribution of type 2 diabetes mellitus in Mexican adult population: a probabilistic survey. Salud Pública de México. 2010;52:S19–S26. doi: 10.1590/s0036-36342010000700005. [DOI] [PubMed] [Google Scholar]
- 6.U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; Atlanta, GA: 2011. [Google Scholar]
- 7.Patterson N, Price AL, Reich D. Population Structure and Eigenanalysis. PLoS genetics. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howie BN, Donnelly P, Marchini J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLoS genetics. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams Amy L, Patterson N, Glessner J, Hakonarson H, Reich D. Phasing of Many Thousands of Genotyped Samples. American journal of human genetics. 2012;91:238–251. doi: 10.1016/j.ajhg.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. doi: http://www.nature.com/nature/journal/v491/n7422/abs/nature11632.html#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaitlen N, et al. Informed Conditioning on Clinical Covariates Increases Power in Case-Control Association Studies. PLoS genetics. 2012;8:e1003032. doi: 10.1371/journal.pgen.1003032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voight BF, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nature genetics. 2010;42:579–589. doi: 10.1038/ng.609. doi: http://www.nature.com/ng/journal/v42/n7/suppinfo/ng.609_S1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halestrap AP. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life. 2012;64:1–9. doi: 10.1002/iub.573. [DOI] [PubMed] [Google Scholar]
- 14.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic acids research. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green RE, et al. A Draft Sequence of the Neandertal Genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer M, et al. A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science. 2012 doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mednikova MB. A proximal pedal phalanx of a Paleolithic hominin from denisova cave, Altai. Archaeology, Ethnology and Anthropology of Eurasia. 2011;39:129–138. doi: 10.1016/j.aeae.2011.06.017. [DOI] [Google Scholar]
- 18.A high-quality Neandertal genome sequence. < http://www.eva.mpg.de/neandertal/> (
- 19.Hublin JJ. The origin of Neandertals. Proceedings of the National Academy of Sciences. 2009;106:16022–16027. doi: 10.1073/pnas.0904119106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halestrap AP, Wilson MC. The monocarboxylate transporter family—Role and regulation. IUBMB Life. 2012;64:109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- 21.Garcia CK, Goldstein JL, Pathak RK, Anderson RGW, Brown MS. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: Implications for the Cori cycle. Cell. 1994;76:865–873. doi: 10.1016/0092-8674(94)90361-1. [DOI] [PubMed] [Google Scholar]
- 22.Fu S, Watkins Steven M, Hotamisligil Gökhan S. The Role of Endoplasmic Reticulum in Hepatic Lipid Homeostasis and Stress Signaling. Cell metabolism. 2012;15:623–634. doi: 10.1016/j.cmet.2012.03.007. doi: http://dx.doi.org/10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 23.Rhee EP, et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. The Journal of clinical investigation. 2011;121:1402–1411. doi: 10.1172/jci44442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savage DB, Semple RK. Recent insights into fatty liver, metabolic dyslipidaemia and their links to insulin resistance. Current Opinion in Lipidology. 2010;21:329–336. doi: 10.1097/MOL.0b013e32833b7782. 310.1097/MOL.1090b1013e32833b37782. [DOI] [PubMed] [Google Scholar]
- 25.Samuel Varman T, Shulman Gerald I. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Florez J, et al. Strong association of socioeconomic status with genetic ancestry in Latinos: implications for admixture studies of type 2 diabetes. Diabetologia. 2009;52:1528–1536. doi: 10.1007/s00125-009-1412-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell S, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome research. 2009 doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Risch N, et al. 12th International Congress of Human Genetics/61st Annual Meeting of The American Society of Human Genetics.. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.