

Summary
Background
Human platelet activation and aggregation is a complex process. To date, many therapies have been developed targeting proteins that mediate this process to prevent unwanted activation. However, the current standard of care for acute coronary syndromes still has limitations including bleeding risk.
Objective
The aim of the current study is to evaluate the PAR4 anionic cluster as a viable antiplatelet target using a polyclonal antibody (CAN12).
Methods
We used western blotting, aggregation, and secretion ex vivo to evaluate the ability of CAN12 to interact with PAR4 and inhibit platelet activation. The effects of CAN12 in vivo were evaluated with the Rose Bengal arterial thrombosis model and two models of hemostasis.
Results
We show that CAN12 is able to interact with human PAR4 and delay PAR4 cleavage. In addition, CAN12 inhibits thrombin induced human platelet aggregation and secretion in a dose dependent manner. We next determined that the specificity of CAN12 is agonist dependent. In vivo, we determined that CAN12 is able to inhibit arterial thrombosis and using two independent methods, we found that CAN12 does not influence hemostasis.
Conclusion
Targeting the extracellular anionic cluster on PAR4 is a viable novel strategy as an anti-platelet therapy.
Keywords: Protease-Activated Receptor 4 - human, Platelet Aggregation Inhibitors, Antibodies, Thrombosis, Hemostasis, G-Protein-Coupled Receptors
Protease activated receptors (PARs) are G-protein coupled receptors activated by cleavage of their N-terminus by serine proteases. There are four members of the PAR family: PAR1-4. PAR1, PAR3, and PAR4 are activated by thrombin, but PAR2 is activated by trypsin. Upon cleavage of the N-terminus, the new N-terminus acts as a tethered ligand for the receptor. PAR activation initiates multiple downstream signaling events in platelets including activation of the small GTPase RhoA, intracellular calcium release, secretion, and a decrease in accumulation of cAMP through Gq, G12/13, and Gi. The culmination of these events leads to platelet aggregation, and if uncontrollably activated, thrombosis.
The current standard for the treatment of acute coronary syndromes by antiplatelet therapy is dual treatment with aspirin and a thienopyridine. However, the current standard for treatment still has a number of limitations, most significantly, increased bleeding risk. This has led to a need for better anti-platelet therapies. Additional therapies, such as glycoprotein IIb/IIIa and PAR1 inhibitors have been investigated as alternate antiplatelet therapies, but have had limited success.
One innovative approach to antiplatelet therapies is targeting PAR4. Specifically, near the anionic cluster on PAR4, D57, D59, E62, and D65, which is C-terminal of the thrombin cleavage site at R47. The anionic cluster of PAR4 is crucial for thrombin interacting with the purified PAR4 exodomain and PAR4 expressed on cells. The anionic cluster slows the dissociation of PAR4 from thrombin in a way that cleavage can occur. We propose that if this region is blocked so that thrombin cannot as easily bind and cleave PAR4, human platelet aggregation can be delayed. Therefore, the anionic region on PAR4 could be a potential therapeutic target.
The current study examined a novel approach for antiplatelet therapy by targeting PAR4. We developed a goat polyclonal antibody that specifically targets the anionic cluster on PAR4. The antibody dose dependently inhibited thrombin induced human platelet aggregation and secretion. In addition, we demonstrated that the antibody is able to prevent carotid artery thrombosis when administered before or after initiation of an injury. Interestingly, using two independent methods, we demonstrated that treatment with the antibody does not influence hemostasis in mice. This study provides a new antiplatelet therapy target, the anionic region of PAR4, which could be a more efficacious approach for the treatment of acute coronary syndromes without the commonly associated increased bleeding risk.
Materials and Methods
Reagents and Antibodies
All cell culture reagents were purchased from Invitrogen. Human α-thrombin was purchased from Haematological Technologies (specific activity 3200–3400 U/mg) (Essex Junction, VT). PAR4 activating peptide (AYPGKF-NH2) and PAR1 activating peptide (SFLLRN-NH2) were synthesized at PolyPeptide Laboratories (San Diego, CA). Convulxin was purchased from Enzo Life Sciences Inc. (Farmingdale, NY). Collagen, ADP, and CHRONO-LUME were purchased from Chrono-log Corporation (Havertown, PA). Protease inhibitor cocktail tablets were purchases from Roche. Rose-Bengal sodium salt, fibrinogen from human plasma, red blood cell lysing buffer, and sepharose 2B were purchased from Sigma-Aldrich. The CAN12 antibody was prepared against the human PAR4 sequence (CANDSDTLELPD) by Bethyl Laboratories as a custom synthesis using a goat as the host. The antibody was affinity purified using a CANDSDTLTLPD peptide-specific immunosorbent. For control experiments, ChromPure goat IgG, whole molecule was purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). The secondary antibodies IRDYE 800CW donkey anti-mouse IgG, IRDYE 680RD donkey anti-goat IgG, and IR 680RD goat anti-rabbit IgG were purchased from LI-COR Biosciences (Lincoln, NE).
Animals
C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, Maine). F2RL3−/− mice (referred to as PAR4−/−) were obtained from the Mutant Mouse Regional Resource Center (MMRRC) (Chapel Hill, NC). All animal studies were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University School of Medicine.
Western blotting
HEK293 cells were purchased from American Type Culture Collection (Bethesda, MD) and were cultured in DMEM supplemented with 10% fetal bovine serum and 1% Pen/Strep. Cells were transfected with Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. HEK293 cells were transfected with 1 μg mouse PAR4 or human PAR4. After 48hrs, the cells were lysed in RIPA buffer (1% NP-40, 0.5% Deoxycholate, 0.1% SDS) on ice. Following lysis, the lysate was collected and quantitated using the Biorad DC protein assay. 100μg of total protein was loaded and resolved by SDS-PAGE and transferred onto nitrocellulose. The membranes were incubated with primary goat antibody to CAN12. To determine equal protein loading, the membrane was probed with rabbit antibody to α-actinin (1, 1000; Santa Cruz Biotechnology Inc.). The membrane was developed using the Odyssey Infrared Imaging System. The optical densities of proteins in the blot were quantified using software provided with the imaging system (Application Software version 3.0).
For examining the CAN12 binding region of PAR4, HEK293 cells were transfected with 4.0 μg human PAR4 or human PAR4-AAAA (D57A, D59A, E62A, D65A). The lysis and blotting procedure was the same as above.
Cleavage assays
HEK293 cells were transfected with HA-PAR1 (human) (4.0 μg) and V5-PAR4 (human) (0.1 μg) for 48 hours (described previously). Aliquots containing 1×106 cells were treated with PBS, IgG (2 μg/ml), or CAN12 (2 μg/ml) for 10 min and were activated with thrombin (100 nM) for 0, 2, or 30 min. The cells were then pelleted and lysed in RIPA buffer on ice. Following lysis, the entire sample was resolved by SDS-PAGE and transferred onto nitrocellulose for Western blotting with anti-V5-tag (1, 10,000; AbD Serotec.) and anti-α-actinin (1, 1000; Santa Cruz Biotechnology Inc.) as described above.
PRP, Gel Filtered Platelets, and Washed Platelets Preparation
Human platelets were obtained from healthy donors. These studies were approved by the Case Western Reserve University Institutional Review Board and informed consent was obtained from all donors. Whole blood was collected into the anticoagulant acid citrate dextrose (ACD) (2.5% sodium citrate, 71.4 mM citric acid, 2% D-glucose) and centrifuged at 200×g for 10 minutes at room temperature. The platelet rich plasma (PRP) was used for aggregation experiments within 3 hours or used to generate gel filtered platelets as previously described. Briefly, platelets were separated from plasma proteins using a sepharose 2B column in HEPES-Tyrode buffer pH 7.4 (10 mM HEPES, 12 mM NaHCO3, 130 mM NaCl, 5 mM KCl, 0.4 mM Na2HPO4, 1 mM MgCl2, 5 mM glucose, 0.1% bovine serum albumin). The number of platelets was quantitated using a Hemavet 950FS (Drew Scientific Inc, Waterbury, CT, USA).
Mice were anesthetized by intraperitoneal injection of pentobarbital (62 mg/kg). Blood was collected by heparinized capillary puncture of the retro-orbital venous sinus and immediately combined with ACD (1/5). The whole blood was centrifuged at 2300×g for 20 sec at room temperature to isolate the PRP. To pellet the platelets, the PRP was centrifuged at 2200×g for 3 minutes at room temperature. The platelets were resuspended in HEPES-Tyrode buffer pH 7.4 (10 mM HEPES, 12 mM NaHCO3, 130 mM NaCl, 5 mM KCl, 0.4 mM Na2HPO4, 1 mM MgCl2, 5 mM glucose, 0.33% human serum albumin). The number of platelets was quantitated using a Hemavet 950FS (Drew Scientific Inc, Waterbury, CT, USA).
Aggregation
Aggregation was measured using a lumi-aggregometer (Model 700, Chrono-log Corporation). The sample was stirred constantly at 1200 RPM at 37°C. Dense granule secretion was detected by ATP luminescence using CHROMO-LUME. For all experiments, PRP, gel filtered platelets, or washed platelets were treated with HEPES-Tyrode buffer, IgG (goat), or CAN12 for 10 minutes prior to activation with thrombin, AYPGKF, SFLLRN, ADP, collagen, or convulxin. Aggrolink8 version 1.3.98 was used for data acquisition. The IC50 was determined using GraphPad Prism.
Mouse Arterial Thrombosis Studies
The arterial thrombosis experiments were performed using the Rose Bengal carotid artery thrombosis model, as previously described. Saline, IgG (2 mg/kg), or CAN12 (1, 0.5, 0.25, 0.125 mg/kg) was injected by tail vein into C57BL/6 mice 10 minutes before initiation of injury. Alternatively, injury was initiated first and 15 minutes later IgG (2 μg/ml) or CAN12 (0.5 mg/kg) was injected via tail vein. The experiment was terminated at 90 min. After the experiment, blood was collected from the retro-orbital venous sinus and immediately combined with (1/5) volume of ACD as an anticoagulant. The platelets were counted using a Hemavet 950FS.
Mouse Tail Bleeding Assay
C57BL/6 mice were retro-orbitaly injected with IgG (2 mg/kg) or CAN12 (2 mg/kg); untreated PAR4−/− mice were used as a control. Ten minutes following injection, tail bleeding times were measured by clipping 3 mm from the tip of the tail of anesthetized mice. The tail was placed in prewarmed saline at 37°C, and the time to cessation of bleeding was measured or the experiment was terminated at 10 min. The quantity of blood loss was determined by lysing the red blood cells in red blood cell lysing buffer, measuring the absorbance of hemoglobin at 550 nm, and comparing to a standard curve.
Mouse Saphenous Vein Bleeding Assay
The method was modified from the previously described protocol by Buyue et al.. Mice were treated as described for tail bleeding assay. The saphenous vein was exposed in the right hind limb and pierced with a 23-G needle. Blood was removed around the site of injury using a Kimwipe. After the formation of a clot (~1.5 min), a 1 mm longitudinal incision was made using the initial site of injury for entry into the vessel. Bleeding was observed for 20 min and each time a subsequent clot formed it was disrupted by stroking a needle across the site of injury in the direction of blood flow. The average bleeding time was calculated by dividing the total bleeding time by the number of clots formed. In addition, the number of clots formed was recorded.
Results
CAN12 reduces the rate of PAR4 cleavage
The interaction between PAR4 and thrombin is mediated by two sites, the cleavage site (L43PAPR) and an anionic region (D57-D65). The anionic region has a single amino acid change between human and mouse PAR4 (CANDSDTLELPD vs. CANDSDTLELPA) suggesting a conserved function. Therefore, we developed a goat polyclonal PAR4 antibody (CAN12) that targets the anionic cluster of PAR4 (Fig. 1A). CAN12 interacted with both human (hPAR4) and mouse PAR4 (mPAR4) in transfected HEK293 cells (Fig. 1B, C). To determine if CAN12 was specifically interacting with the anionic region of PAR4, we transfected 293 cells with hPAR4 or hPAR4-AAAA; a construct in which each of the residues in the anionic region is mutated to alanine (D57A, D59A, E62A, D65A). CAN12 did not interact with the mutated anionic region (Fig. 1 D). We next determined if CAN12 was able to inhibit PAR4 cleavage by α-thrombin. To mirror the expression profile of PARs in human platelets, HEK293 cells were cotransfected with HA-PAR1 and V5-PAR4 and the cleavage of PAR4 was measured by the disappearance of the N-terminal V5-tag. CAN12 (2 μg/ml) treatment completely inhibited PAR4 cleavage by α-thrombin at 2 min compared to 44.5 ± 6.9% cleavage for the IgG control (2 μg/ml). Thirty minutes after CAN12 treatment, PAR4 cleavage was reduced to 39.4 ± 12.5% compared to 64.5 ± 15.4% for the IgG control (Fig. 1E, F). All together these results demonstrate that CAN12 interacts with mPAR4 and hPAR4 at the anionic region and is able to slow the rate of PAR4 cleavage.
Figure 1. CAN12 reduces the rate of PAR4 cleavage.

(A) The N-terminus of PAR4 with C54ANDSDTLELPD (CAN12) and the thrombin cleavage site (*) indicated. (B) HEK293 cells (293) or cells transfected with mouse PAR4 (1.0 μg) or human PAR4 (1.0 μg) blotted with CAN12 (1:100) and actinin (1:1000). (C) Full gel for CAN12 interacting with hPAR4 expressed in HEK293 cells. (D) HEK293 cells (293) or cells transfected with hPAR4 (4.0 μg) or hPAR4-AAAA (D57A, D59A, E62A, D65A) (4.0 μg) blotted with CAN12, PAR4 (C-10), and actinin. (E) HEK293 cells transfected with HA-hPAR1 (4.0 μg) and V5-hPAR4 (0.1 μg) pretreated with buffer (PAR1PAR4), IgG (2 μg/ml), or CAN12 (2 μg/ml) for 10 min at room temperature. The cells were then activated with thrombin (100nM) for 0, 2, or 30 min at 37°C. Cleavage of PAR4 was measured by loss of the N-terminal V5 epitope. (F) Quantitation of percent of uncleaved PAR4 compared to 0 min as 100%. n=4 **p<0.01 vs. time zero
The effect of CAN12 treatment on human platelet aggregation and secretion
We next sought to determine if CAN12 is able to inhibit thrombin induced platelet aggregation and secretion. Gel filtered platelets were pretreated with buffer, IgG (2 μg/ml), or CAN12 (0.2 ng/ml-2 μg/ml) for 10 min at 37°C. Platelets were stimulated with a threshold-dose of α-thrombin (0.5 nM) and the percent aggregation was measured. CAN12 inhibited thrombin induced aggregation in a dose dependent manner with an IC50 of 10 ng/ml (2–49 ng/ml, 95% CI) (Fig. 2A, B). In contrast, IgG did not influence aggregation at a 200 fold higher concentration. CAN12 also inhibited dense granule secretion as measured by ATP release in a dose dependent manner similar to aggregation (Fig. 2C).
Figure 2. CAN12 inhibits thrombin induced aggregation and dense granule secretion.
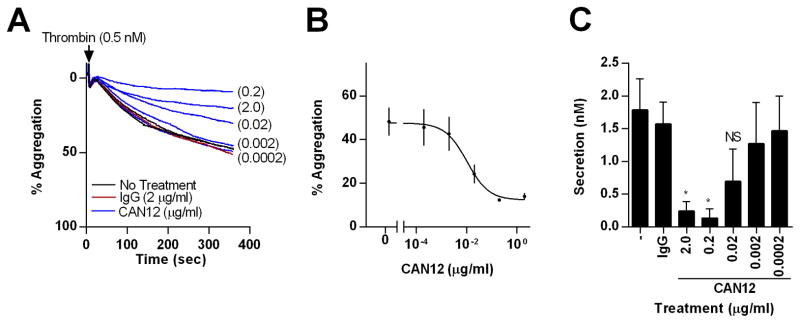
(A) Gel filtered platelets were isolated from healthy donors. Following treatment with goat IgG (2.0 μg/ml) or CAN12 (0.2 ng/ml – 2.0 μg/ml) for 10 min at 37°C, percent aggregation was determined for thrombin (0.5 nM). Representative curves shown. (B) Dose response curve for CAN12 inhibition of thrombin induced aggregation (0.5 nM). IC50=10 ng/ml (2–49 ng/ml, 95% CI) (C) Dense granule secretion as determined by ATP (nM). *p<0.05 vs. IgG, NS=not significant
We next examined the influence of CAN12 on platelet aggregation and secretion initiated by other agonists (AYPGKF, SFLLRN, ADP, and collagen). PRP was isolated from normal healthy donors and treated with buffer, IgG (2 μg/ml), or CAN12 (0.2 ng/ml-2 μg/ml) for 10 min at 37°C. High doses of CAN12 (0.02–2 μg/ml) inhibited aggregation by the PAR4 agonist peptide, AYPGKF (500 μM), but when the dose was decreased to 2 ng/ml the percent aggregation returned to 100% (Fig. 3A). This is likely due to the large antibody molecule sterically blocking access of AYPGKF to the proposed binding site in extracellular loop 2. In contrast, CAN12 had no effect on aggregation of platelets activated with SFLLRN (25 μM) (Fig. 3B). CAN12 dose dependently inhibited ADP (5 μM) induced aggregation (Fig. 3C). For collagen (1 μg/ml) activated platelets, CAN12 inhibited in a dose dependent manner (Fig. 3D). This could be due to PAR4 having a synergistic effect on collagen activation of GPVI. Interestingly, CAN12 had no effect on convulxin induced aggregation of platelets (Fig. 3E). Similar to aggregation, CAN12 inhibited dense granule secretion from platelets activated with AYPGKF (500 μM), ADP (5 μM), and collagen (1 μg/ml) (Fig. 4 A, C, D). However, CAN12 had no effect on secretion induced by SFLLRN (25 μM) and convulxin (5 nM) (Fig. 4 B, E).
Figure 3. CAN12 specificity on aggregation.

PRP was isolated from healthy donors. Following treatment with goat IgG (2.0 μg/ml) or CAN12 (dose response) for 10 min at 37°C, percent aggregation was determined for (A) AYPGKF (500 μM), (B) SFLLRN (25 μM), (C) ADP (5 μM), (D) collagen (1 μg/ml), (E) convulxin (5 nM). Representative curves and average percent aggregation shown. *p<0.05 vs. IgG, **p<0.01 vs. IgG, NS=not significant
Figure 4. CAN12 specificity on dense granule secretion.

PRP was isolated from healthy donors. Following treatment with goat IgG (2.0 μg/ml) or CAN12 (dose response) for 10 min at 37°C, secretion was determined for (A) AYPGKF (500 μM), (B) SFLLRN (25 μM), (C) ADP (5 μM), (D) collagen (1 μg/ml), (E) convulxin (5 nM). *p<0.05 vs. IgG, **p<0.01 vs. IgG, NS=not significant
CAN12 inhibits aggregation through PAR4
PAR4 and P2Y12 are known to heterodimerize and these interactions influence the function of each receptor. Therefore, we examined if CAN12’s influence on ADP-induced aggregation was dependent on PAR4 using platelets isolated from PAR4−/− mice. Washed platelets isolated from wild-type or PAR4−/− mice were pretreated with IgG (2 μg/ml) or CAN12 (2 μg/ml) for 10 min at 37°C. In wild-type mice, AYPGKF-induced (500 μM) platelet aggregation of CAN12 treated platelets was reduced to 9.3% from 37% for the IgG control (Fig. 5A). As expected, platelets from PAR4−/− mice did not respond to AYPGKF (Fig. 5B). Similarly, CAN12 reduced ADP-induced aggregation of platelets from wild-type mice from 21% to 8.3%. Importantly, ADP-induced aggregation was not influenced in platelets from PAR4−/− mice. CAN12 also appeared to have a slight effect on collagen aggregation; however, the results did not reach statistical significance (Fig. 5A). These data are in agreement with data from human platelets in which CAN12 reduced ADP-induced aggregation. Further, these data demonstrate that inhibitory effects of the antibody are dependent on the presence of PAR4.
Figure 5. CAN12 inhibits aggregation through PAR4.

Washed platelets were isolated from wild-type and PAR4−/− mice. Following treatment with goat IgG (2.0 μg/ml) or CAN12(2.0 μg/ml) for 10 min at 37°C percent aggregation was evaluated for (A) wild-type and (B) PAR4−/− platelets activated with AYPGFK (500 μM), ADP (5 μM) supplemented with 1 mg/ml fibrinogen, or collagen (1 μg/ml). **p<0.01 vs. IgG
CAN12 inhibits arterial thrombosis
Since CAN12 is able to interact with murine PAR4 (Fig. 1B) and affect mouse platelet activation (Fig. 5), our studies determined the ability of CAN12 to affect arterial thrombosis in vivo in the Rose Bengal model. The time to thrombosis was delayed to more than 90 minutes when CAN12, 1.0 mg/kg (~14 μg/ml plasma concentration), was injected 10 minutes prior to injury (Fig. 6A). We next wanted to determine the minimal dose of CAN12 required to influence the time to thrombosis. The intermediate doses of 0.5 mg/kg and 0.25 mg/kg had a time to thrombosis of 82 minutes and 60 min, respectively. At 0.125 mg/kg CAN12, the time to occlusion was 37 minutes; the same time as the controls (saline and IgG) (Fig 6A). We verified that the delay in thrombosis was not due to a decrease in the platelet number (Fig. 6B). Next we investigated whether CAN12 prolonged the time to thrombosis when administered after initiation of the injury. For these studies we used the lowest dose of CAN12 (0.5 mg/kg) that significantly prolonged the time to occlusion (see Fig. 6A). CAN12 delivered 15 minutes after injury was able to prolong the time to complete occlusion to 84 minutes (Fig. 6C). CAN12 also did not reduce platelet numbers when administered after the injury (Fig. 6D). Similarly, there was no difference in platelet number between IgG and CAN12 treatment when injury was not initiated (425 × 106 ± 56 platelets/ml vs. 462 × 106 ± 90 platelets/ml, respectively). Overall, CAN12 treatment is able to delay arterial thrombosis when delivered either before or after injury.
Figure 6. CAN12 inhibits arterial thrombosis.

(A) C57BL/6 mice were pretreated with saline, goat IgG (2 mg/kg), or CAN12 (1, 0.5, 0.25, 0.125 mg/kg) for 10 min and then subjected to the Rose Bengal carotid artery thrombosis model. Time to complete occlusion is indicated or the experiment was terminated at 90 min. (B) The concentration of platelets in the blood at termination of the experiment was determined. (C) 15 minutes after the initiation of carotid artery thrombosis, C57BL/6 mice were injected with goat IgG (2 mg/kg) or CAN12 (0.5 mg/kg) and the time to complete arterial occlusion was determined. The experiment was terminated at 90 min. (D) The concentration of platelets in the blood at termination of the experiment was determined. **p<0.01
CAN12 does not affect bleeding time
Finally, we wanted to examine if CAN12 treatment influences hemostasis using two assays. The first was the tail clip assay. C57BL/6 mice were injected with IgG (2 mg/kg) or a high dose of CAN12 (2 mg/kg) 10 minutes before the procedure. There was no difference in time to cessation of bleeding or total blood loss between IgG or CAN12 treated mice (Fig. 7A, B). PAR4−/− mice have a prolonged bleeding phenotype and were used as controls. An alternative method for examining the effect of CAN12 on hemostasis was the saphenous vein model. CAN12 (2 mg/kg) had no effect on the bleeding time or number of clot formations compared to the IgG (2 mg/kg) control (Fig. 7C, D). Similar to the tail clip model, PAR4−/− mice had a prolonged bleeding time and fewer clot formations. Using two independent methods, we demonstrated that CAN12 treatment does not delay hemostasis in mice.
Figure 7. CAN12 does not affect bleeding time.

(A) C57BL/6 mice or PAR4−/− mice were anesthetized and 3 mm of the tail was cut. The time to cessation of bleeding was determined or the experiment was terminated at 10 min. (B) The total amount of blood loss was determined by reading the absorbance of hemoglobin from lysed red blood cells and was compared to a standard curve. (C) C57BL/6 mice or PAR4−/− mice were anesthetized and the saphenous vein was exposed and pierced. Once, bleeding ceased, the clot was disrupted. The procedure was repeated for 20 min. The average time of bleeding and (D) the number of clot formations were determined. **p<0.01, NS=not significant
Discussion
In the current study, we have identified the anionic region of PAR4 as a potential therapeutic target using an inhibitory antibody. The antibody is directed toward the sequence C54ANDSDTLELPD, which has been identified to be important for PAR4’s interaction with thrombin using purified exodomains and cell lines. This region is conserved between murine and human PAR4. A co-crystal with a murine PAR4 derived peptide and murine thrombin shows that the anionic region of PAR4 makes direct contact with thrombin’s autolysis loop. The antibody CAN12 exploits these interactions to slow the rate of PAR4 cleavage (Fig. 1E and F) resulting in a decrease in PAR4 activation. These data are consistent with published results that demonstrate the importance of the anionic region for PAR4 activation by thrombin. By interfering with PAR4 activation, CAN12 inhibits thrombin-induced human platelet aggregation and thrombosis in the Rose Bengal thrombosis mouse model (Fig. 2 and 6). Importantly, CAN12 does not delay hemostasis in two mouse models. The studies in the current report demonstrate the feasibility of targeting PAR4 in general and, in particular, the anionic region of PAR4’s exodomain.
Human platelets express two subtypes of protease activated receptors, PAR1 and PAR4, which mediate thrombin-induced platelet activation. The interaction and subsequent activation of PAR1 and PAR4 by thrombin is mechanistically different. PAR1 contains a hirudin-like sequence that binds exosite I of thrombin, which likely allosterically induces thrombin into the protease conformation. The net effect is efficient activation of PAR1 by low concentrations of thrombin. In contrast, PAR4 relies on an anionic cluster (D57, D59, E62, D65), which slows the rate of thrombin dissociation and prolongs the interaction time between PAR4 and thrombin. However, this region does not interact with thrombin’s exosite I and likely does not contribute to thrombin allostery, which leads to inefficient PAR4 activation.. However the rate of PAR4 activation is enhanced when it is coexpressed with PAR1. We have recently demonstrated that PAR1-mediated enhancement of PAR4 cleavage is dependent on PAR1-PAR4 heterodimerization. We have previously shown that the anionic cluster contributes to PAR4’s interaction with thrombin both in the presence and absence of PAR1. Therefore, we targeted this region for developing a PAR4 antagonist. The inhibitory antibody, CAN12, was able to block thrombin-induced human platelet aggregation in a dose-dependent manner (Fig. 2). Since PAR1 and PAR4 form homodimers and heterodimers, the antibody, CAN12, is likely blocking PAR1 activation due to the interactions of PAR1 and PAR4 on the platelet surface as CAN12 does not influence PAR1 activation.
The primary focus of thrombin signaling has been directed at PAR1, which has led to the development of two PAR1 antagonists that have undergone clinical trials with mixed results. The most recent clinical trial for a PAR1 antagonist, the vorapaxar TRA-CER trial, had to be terminated due to intracranial hemorrhage. PAR4 has received much less attention than PAR1. However, there have been two PAR4 antagonists developed, the small molecule YD-3 and the peptide P4pal-10. CAN12 differs from these antagonists in two ways. First, it targets the extracellular anionic region of PAR4. Second, it does not delay hemostasis in mouse models. This effect is likely due to the fact that CAN12 slows the rate of PAR4 activation rather than completely inhibiting it. In the arterial thrombosis model in which there is a high flow rate in the vessel, the platelets with CAN12 bound will be rapidly cleared from the site of injury containing high thrombin concentrations, thus, reducing PAR4 mediated platelet activation at the site of injury. In contrast, in the tail clip and saphenous vein models both have a lower flow rate. This may allow the platelets with CAN12 bound to remain in proximity of higher thrombin concentrations at the site of injury for a prolonged length of time allowing for PAR4 to be cleaved by thrombin and enable platelet activation. This also explains why the bleeding times of the PAR4−/− mice (with zero PAR4 expression) are significantly prolonged in both hemostasis assays and the CAN12 treated mice are unaffected (Fig. 7).
The antibody, CAN12, also affects ADP and collagen-induced human platelet aggregation. The inhibition of ADP induced aggregation is likely through PAR4’s direct and indirect interactions with the ADP receptor, P2Y12. PAR4 and P2Y12 physically interact in heterodimeric complexes that directly influence thier signaling activities. In addition, the signaling from the two receptors indirectly influences the activity of one another. CAN12 was able to interfere with collagen-induced aggregation, but did not affect a different GPVI agonist, convulxin (Fig. 3). One theory is that CAN12 is indirectly affecting collagen-induced aggregation through inhibition of P2Y12 as previously demonstrated with the P2Y12 antagonist AR-C69931MX, which decreased platelet activation by collagen. A second explanation is that CAN12 may be sterically interfering with the α2β1 integrin since collagen activates both α2β1 integrin and GPVI, whereas convulxin specifically activates GPVI. A third option is the size discrepancy between collagen and convulxin. CAN12 could be blocking the larger collagen molecule (300 KDa) from interacting with its receptor, but the smaller convulxin (72 KDa) can still reach its receptor. While each of these theories is possible, the net effect is that targeting the extracellular domain of PAR4 inhibits human platelet aggregation ex vivo and thrombosis in vivo in mice. Importantly, our experiments with PAR4−/− platelets demonstrate the inhibition of ADP induced aggregation is dependent upon the presence of PAR4. Finally, targeting multiple pathways with a single agent can be beneficial. For example, the FDA approved anti-platelet therapy Ticagralor, a non-competitive P2Y12 antagonist, also influences multiple pathways, which likely contributes to its overall effectiveness.
The current study demonstrates that targeting the thrombin-binding site away from the cleavage site on PAR4 is a viable strategy for anti-platelet therapy. These studies provide justification for developing a humanized monoclonal CAN12 antibody, which is currently underway. The current study also justifies targeting PAR4 for anti-platelet therapies in general. To date, PAR4 has been considered a back-up receptor for PAR1 signaling and has been studied less. However, a recent study has shown that black populations have higher levels of microRNA’s that are associated with increased PAR4 reactivity; PAR1 was unaffected. In this light, PAR4 may be an attractive target for specific populations. Our studies that identify a novel target region on PAR4 provide insight toward developing future PAR antagonists based on the mechanism of thrombin binding to PAR4.
Acknowledgments
The authors would like to thank Drs. Matthew Bilodeau and Lalitha Nayak for helpful discussions. We also thank Dr. Alvin Schmaier for use of the thrombosis equipment. The work was supported by grants from the American Heart Association (AHA Beginning Grant In Aid, number) and (AHA Scientist Development Grant, 10SDG2600021), American Society for Hematology (ASH Scholar Award) and the NIH (HL098217) to M.T. Nieman, the Cardiovascular Training Grant 5T32HL105338 (M.M. Mumaw), and CWRU/UH Center for AIDS Research (NIH P30 AI036219).
Footnotes
Addendum
M. M. Mumaw, and M. T. Nieman conceived and designed the experiments. M. M. Mumaw, D. N. Noble, and M. de la Fuente performed the experiments. M. M. Mumaw, and M. T. Nieman analyzed the data. M. M. Mumaw and M. T. Nieman wrote the manuscript.
Disclosure of Conflict of Interest
The authors state that they have no conflicts of interest.
References
- 1.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–64. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 2.Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26:1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- 3.Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–68. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 4.Nanevicz T, Ishii M, Wang L, Chen M, Chen J, Turck CW, Cohen FE, Coughlin SR. Mechanisms of thrombin receptor agonist specificity. Chimeric receptors and complementary mutations identify an agonist recognition site. J Biol Chem. 1995;270:21619–25. doi: 10.1074/jbc.270.37.21619. [DOI] [PubMed] [Google Scholar]
- 5.Keularts IM, van Gorp RM, Feijge MA, Vuist WM, Heemskerk JW. alpha(2A)-adrenergic receptor stimulation potentiates calcium release in platelets by modulating cAMP levels. J Biol Chem. 2000;275:1763–72. doi: 10.1074/jbc.275.3.1763. [DOI] [PubMed] [Google Scholar]
- 6.Varga-Szabo D, Braun A, Nieswandt B. Calcium signaling in platelets. J Thromb Haemost. 2009;7:1057–66. doi: 10.1111/j.1538-7836.2009.03455.x. [DOI] [PubMed] [Google Scholar]
- 7.Woulfe DS. Platelet G protein-coupled receptors in hemostasis and thrombosis. J Thromb Haemost. 2005;3:2193–200. doi: 10.1111/j.1538-7836.2005.01338.x. [DOI] [PubMed] [Google Scholar]
- 8.Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, Cattaneo M, Kunapuli SP. Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood. 2002;99:3629–36. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Jin J, Kunapuli SP. Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood. 2006;107:947–54. doi: 10.1182/blood-2005-07-3040. [DOI] [PubMed] [Google Scholar]
- 10.Angiolillo DJ, Guzman LA, Bass TA. Current antiplatelet therapies: benefits and limitations. American heart journal. 2008;156:S3–9. doi: 10.1016/j.ahj.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Angiolillo DJ, Ferreiro JL. Antiplatelet and anticoagulant therapy for atherothrombotic disease: the role of current and emerging agents. American journal of cardiovascular drugs : drugs, devices, and other interventions. 2013;13:233–50. doi: 10.1007/s40256-013-0022-7. [DOI] [PubMed] [Google Scholar]
- 12.Schwarz M, Meade G, Stoll P, Ylanne J, Bassler N, Chen YC, Hagemeyer CE, Ahrens I, Moran N, Kenny D, Fitzgerald D, Bode C, Peter K. Conformation-specific blockade of the integrin GPIIb/IIIa: a novel antiplatelet strategy that selectively targets activated platelets. Circ Res. 2006;99:25–33. doi: 10.1161/01.RES.0000232317.84122.0c. [DOI] [PubMed] [Google Scholar]
- 13.Kastrati A, Neumann FJ, Schulz S, Massberg S, Byrne RA, Ferenc M, Laugwitz KL, Pache J, Ott I, Hausleiter J, Seyfarth M, Gick M, Antoniucci D, Schomig A, Berger PB, Mehilli J. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med. 2011;365:1980–9. doi: 10.1056/NEJMoa1109596. [DOI] [PubMed] [Google Scholar]
- 14.Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, Lokhnygina Y, Pei J, Leonardi S, Rorick TL, Kilian AM, Jennings LH, Ambrosio G, Bode C, Cequier A, Cornel JH, et al. Thrombin-Receptor Antagonist Vorapaxar in Acute Coronary Syndromes. The New England journal of medicine. 2011 [Google Scholar]
- 15.Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem J. 2003;376:733–40. doi: 10.1042/BJ20030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nieman MT. Protease-activated receptor 4 uses anionic residues to interact with alpha-thrombin in the absence or presence of protease-activated receptor 1. Biochemistry. 2008;47:13279–86. doi: 10.1021/bi801334s. [DOI] [PubMed] [Google Scholar]
- 17.Mathews II, Padmanabhan KP, Ganesh V, Tulinsky A, Ishii M, Chen J, Turck CW, Coughlin SR, Fenton JW., 2nd Crystallographic structures of thrombin complexed with thrombin receptor peptides: existence of expected and novel binding modes. Biochemistry. 1994;33:3266–79. doi: 10.1021/bi00177a018. [DOI] [PubMed] [Google Scholar]
- 18.Jacques SL, LeMasurier M, Sheridan PJ, Seeley SK, Kuliopulos A. Substrate-assisted catalysis of the PAR1 thrombin receptor. Enhancement of macromolecular association and cleavage. J Biol Chem. 2000;275:40671–8. doi: 10.1074/jbc.M004544200. [DOI] [PubMed] [Google Scholar]
- 19.Arachiche A, Mumaw MM, de la Fuente M, Nieman MT. Protease-activated Receptor 1 (PAR1) and PAR4 Heterodimers Are Required for PAR1-enhanced Cleavage of PAR4 by alpha-Thrombin. The Journal of biological chemistry. 2013;288:32553–62. doi: 10.1074/jbc.M113.472373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tangen O, Berman HJ, Marfey P, Gelfiltration A new technique for separation of blood platelets from plasma. Thrombosis et diathesis haemorrhagica. 1971;25:268–78. [PubMed] [Google Scholar]
- 21.Nieman MT, Warnock M, Hasan AA, Mahdi F, Lucchesi BR, Brown NJ, Murphey LJ, Schmaier AH. The preparation and characterization of novel peptide antagonists to thrombin and factor VIIa and activation of protease-activated receptor 1. J Pharmacol Exp Ther. 2004;311:492–501. doi: 10.1124/jpet.104.069229. [DOI] [PubMed] [Google Scholar]
- 22.Buyue Y, Whinna HC, Sheehan JP. The heparin-binding exosite of factor IXa is a critical regulator of plasma thrombin generation and venous thrombosis. Blood. 2008;112:3234–41. doi: 10.1182/blood-2008-01-136820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graff J, Klinkhardt U, Harder S. Pharmacodynamic profile of antiplatelet agents: marked differences between single versus costimulation with platelet activators. Thrombosis research. 2004;113:295–302. doi: 10.1016/j.thromres.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Khan A, Li D, Ibrahim S, Smyth E, Woulfe DS. The Physical Association of the P2Y12 Receptor with PAR4 Regulates Arrestin-mediated Akt Activation. Mol Pharmacol. 2014 doi: 10.1124/mol.114.091595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holinstat M, Voss B, Bilodeau ML, McLaughlin JN, Cleator J, Hamm HE. PAR4, but not PAR1, signals human platelet aggregation via Ca2+ mobilization and synergistic P2Y12 receptor activation. J Biol Chem. 2006;281:26665–74. doi: 10.1074/jbc.M602174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li D, D’Angelo L, Chavez M, Woulfe DS. Arrestin-2 differentially regulates PAR4 and ADP receptor signaling in platelets. J Biol Chem. 2011;286:3805–14. doi: 10.1074/jbc.M110.118018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature. 2001;413:74–8. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- 28.Bah A, Chen Z, Bush-Pelc LA, Mathews FS, Di Cera E. Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc Natl Acad Sci U S A. 2007;104:11603–8. doi: 10.1073/pnas.0704409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95:6642–6. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Jr, Tam C, Coughlin SR. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–4. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 31.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–87. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamath P, Huntington JA, Krishnaswamy S. Ligand binding shuttles thrombin along a continuum of zymogen- and proteinase-like states. J Biol Chem. 2010;285:28651–8. doi: 10.1074/jbc.M110.154914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–54. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- 34.de la Fuente M, Noble DN, Verma S, Nieman MT. Mapping human protease-activated receptor 4 (PAR4) homodimer interface to transmembrane helix 4. J Biol Chem. 2012;287:10414–23. doi: 10.1074/jbc.M112.341438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin H, Liu AP, Smith TH, Trejo J. Cofactoring and dimerization of proteinase-activated receptors. Pharmacol Rev. 2013;65:1198–213. doi: 10.1124/pr.111.004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, Lokhnygina Y, Pei J, Leonardi S, Rorick TL, Kilian AM, Jennings LH, Ambrosio G, Bode C, Cequier A, Cornel JH, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. The New England journal of medicine. 2012;366:20–33. doi: 10.1056/NEJMoa1109719. [DOI] [PubMed] [Google Scholar]
- 37.Wu CC, Huang SW, Hwang TL, Kuo SC, Lee FY, Teng CM. YD-3, a novel inhibitor of protease-induced platelet activation. British journal of pharmacology. 2000;130:1289–96. doi: 10.1038/sj.bjp.0703437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu CC, Hwang TL, Liao CH, Kuo SC, Lee FY, Lee CY, Teng CM. Selective inhibition of protease-activated receptor 4-dependent platelet activation by YD-3. Thrombosis and haemostasis. 2002;87:1026–33. [PubMed] [Google Scholar]
- 39.Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc Natl Acad Sci U S A. 2002;99:643–8. doi: 10.1073/pnas.022460899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuliopulos A, Covic L. Blocking receptors on the inside: pepducin-based intervention of PAR signaling and thrombosis. Life Sci. 2003;74:255–62. doi: 10.1016/j.lfs.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 41.Storey RF, Sanderson HM, White AE, May JA, Cameron KE, Heptinstall S. The central role of the P(2T) receptor in amplification of human platelet activation, aggregation, secretion and procoagulant activity. British journal of haematology. 2000;110:925–34. doi: 10.1046/j.1365-2141.2000.02208.x. [DOI] [PubMed] [Google Scholar]
- 42.Nieswandt B, Brakebusch C, Bergmeier W, Schulte V, Bouvard D, Mokhtari-Nejad R, Lindhout T, Heemskerk JW, Zirngibl H, Fassler R. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. The EMBO journal. 2001;20:2120–30. doi: 10.1093/emboj/20.9.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santoro SA, Walsh JJ, Staatz WD, Baranski KJ. Distinct determinants on collagen support alpha 2 beta 1 integrin-mediated platelet adhesion and platelet activation. Cell regulation. 1991;2:905–13. doi: 10.1091/mbc.2.11.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor? Blood. 2003;102:449–61. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- 45.Polgar J, Clemetson JM, Kehrel BE, Wiedemann M, Magnenat EM, Wells TN, Clemetson KJ. Platelet activation and signal transduction by convulxin, a C-type lectin from Crotalus durissus terrificus (tropical rattlesnake) venom via the p62/GPVI collagen receptor. The Journal of biological chemistry. 1997;272:13576–83. doi: 10.1074/jbc.272.21.13576. [DOI] [PubMed] [Google Scholar]
- 46.Nylander S, Femia EA, Scavone M, Berntsson P, Asztely AK, Nelander K, Lofgren L, Nilsson RG, Cattaneo M. Ticagrelor inhibits human platelet aggregation via adenosine in addition to P2Y12 antagonism. Journal of thrombosis and haemostasis : JTH. 2013;11:1867–76. doi: 10.1111/jth.12360. [DOI] [PubMed] [Google Scholar]
- 47.VANG JJ, Nilsson L, Berntsson P, Wissing BM, Giordanetto F, Tomlinson W, Greasley PJ. Ticagrelor binds to human P2Y(12) independently from ADP but antagonizes ADP-induced receptor signaling and platelet aggregation. Journal of thrombosis and haemostasis : JTH. 2009;7:1556–65. doi: 10.1111/j.1538-7836.2009.03527.x. [DOI] [PubMed] [Google Scholar]
- 48.Edelstein LC, Simon LM, Montoya RT, Holinstat M, Chen ES, Bergeron A, Kong X, Nagalla S, Mohandas N, Cohen DE, Dong JF, Shaw C, Bray PF. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat Med. 2013;19:1609–16. doi: 10.1038/nm.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]