

Summary
Background
Our previous studies have demonstrated that platelet-specific gene delivery to hematopoietic stem cells can induce sustained therapeutic levels of platelet-FVIII expression in hemophilia A (HA) mice.
Objective
In this study, we aimed to enhance platelet-FVIII expression while minimizing potential toxicities.
Methods
A novel lentiviral vector (LV), which harbors dual genes, the FVIII gene driven by the αIIb promoter (2bF8) and a drug-resistance gene, the MGMTP140K cassette, was constructed. Platelet-FVIII expression in HA mice was introduced by transduction of HSCs and transplantation. The recipients were treated with O6-benzylguanine followed by 1,3-bis-2 chloroethyl-1-nitrosourea monthly for 3 or 4 times. Animals were analyzed by PCR, qPCR, FVIII:C assays, and inhibitor assays. Phenotypic correction was assessed by tail clipping tests and ROTEM analysis.
Results
Even using a low MOI (multiplicity of infection) of 1 and a non-myeloablative conditioning regimen, after in vivo selection, the levels of platelet-FVIII expression in recipients increased to 4.33 ± 5.48 mU per 108 platelets (n = 16), which were 19.7-fold higher than the levels obtained from the recipients before treatment. qPCR results confirmed that 2bF8/MGMT-LV-transduced cells were effectively enriched after drug-selective treatment. Fifteen of 16 treated animals survived tail clipping. Blood loss and whole blood clotting time were normalized in the treated recipients. Notably, no anti-FVIII antibodies were detected in the treated animals even after rhF8 challenge.
Conclusion
we have established an effective in vivo selective system that allows us to enrich 2bF8LV-transduced cells, enhancing platelet-FVIII expression while reducing the potential toxicities associated with platelet gene therapy.
Keywords: Blood platelet, Factor VIII, Genetic therapy, Hemophilia A, Immune tolerance
Introduction
Hemophilia A (HA) is considered to be an ideal target for gene therapy. [1–3] Classical approaches for gene therapy of HA, which rely on correcting the level of plasma factor VIII (FVIII), have resulted in phenotypic correction in animal models, [4–16] but have not yet been beneficial in clinical trials. [17–19] Our previous studies have demonstrated that targeting FVIII expression to platelets using a platelet-specific αIIb promoter (2bF8) results in FVIII storage in platelet α-granules and that platelet-derived FVIII corrects the bleeding diathesis in HA mice with or without inhibitory antibodies (inhibitors) using either a transgenic model [20;21] or lentiviral gene delivery to hematopoietic stem cells (HSCs). [22–25]
The challenges of HSC gene therapy may include the toxicities of HSC transplantation preconditioning regimens, the inability to genetically modify sufficient numbers of target cells, the risk of insertional mutagenesis, and the immune response to the foreign protein. Although our previous studies have demonstrated that sustained therapeutic levels of platelet-FVIII were achieved in 2bF8-transduced mice using our non-selectable 2bF8 lentiviral vector (2bF8LV), the transduction efficiency was only about 10% even using a myeloablative conditioning regimen and an approximate MOI (multiplicity of infection) of 10. [22;23] While using a higher MOI to introduce 2bF8LV to HSCs may enhance the transduction efficiency, it may also augment the potential genotoxicities associated with lentivirus integration. Since the risks involved in myeloablative conditioning are high, it is likely that in vivo representation of transduced autologous cells in patients will be substantially less than required to correct the bleeding phenotype when a lower intensity level of non-myeloablative conditioning regimen is employed.
Thus, developing a protocol by which therapeutic results can be achieved while the potential toxicities associated with platelet gene therapy can be minimized will be desired.
Human trials for the severe combined immunodeficiency disorders have demonstrated that efficient gene transfer and myeloablation is not required when there is a powerful selective advantage to the genetically modified cells. [26;27] We hypothesize that incorporating a drug-resistance gene into the 2bF8LV will allow for post-transduction selection of 2bF8-transduced HSCs which will result in the increase of the platelet-FVIII expression while reducing the potential risks of insertional mutagenesis and the toxicities associated with the intense transplantation pre-conditioning.
In the current study, we investigated the efficacy of in vivo selection of the 2bF8-transduced HSCs using the O6-methylguanine-DNA-methyltransferase (MGMT)-based selection system in a HA mouse model. The question we addressed in the current study is whether we can achieve sustained therapeutic levels of platelet-FVIII while reducing the MOI, the stringency of the transplantation preconditioning regimen, and the immune response in this novel 2bF8/MGMT platelet gene therapy.
Materials and Methods
Mice
FVIIInull mice used in this study were in a 129/SV x C57BL/6 mixed genetic background with a targeted disruption of exon 17 of the FVIII gene. [28] Isoflurane or ketamine was used for anesthesia. Animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin.
Vector construction, virus production and purification
A 1.066-kb MGMTP140K expression cassette driven by the murine stem cell virus promoter was removed from Hu889B3WPT-mscvMGMT [29] and inserted into the pWPT-2bF8 vector [22] to generate a new lentiviral construct, pWPT-2bF8/MGMT (Fig. 1A). The 2bF8/MGMT lentivirus (2bF8/MGMT-LV) was produced and purified as previously described. [22;30]
Fig. 1. 2bF8 transgene analysis.
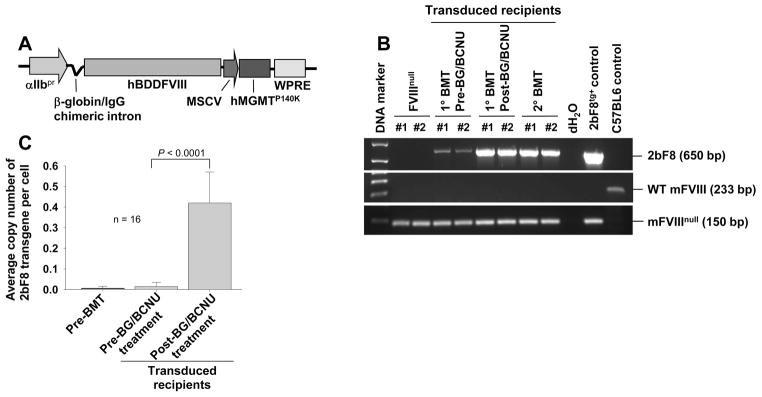
(A) Diagram of the pWPT-2bF8/MGMT construct. FVIII is driven by the platelet-specific αIIb promoter. MGMTp140k was driven by the MSCV promoter. (B) PCR detection of the 2bF8 transgene. DNA was purified from peripheral white blood cells. A 0.65 kb fragment from the 2bF8 expression cassette was amplified. Wild type (WT) mFVIII and mFVIIInull PCRs were used as controls to confirm that 2bF8/MGMT-transduced recipients were on the FVIIInull background. (C) qPCR determination of the average copy number of the 2bF8/MGMT transgene per cell in transduced recipients. Peripheral blood cell-derived DNA was analyzed for the 2bF8 transgene and normalized to the ApoB gene. These results demonstrate that 2bF8/MGMT genetically modified hematopoietic cells were viable and selectable by BG/BCNU treatments.
HSCs transduction and transplantation
FVIIInull HSCs were isolated using the anti-Sca-l+ MicroBead Kit (Miltenyl Biotec, Auburn, CA, USA) following the protocol provided by the manufacturer. The transduction of Sca-1+ cells with 2bF8/MGMT-LV was performed using the protocol described in our previous reports. [22;23] The cells were transduced with 2bF8/MGMT-LV at MOI of ~1 for the principal studies. An additional transduction experiment using an MOI of 10 was performed for comparison. Six- to eight-week-old FVIIInull mice were conditioned with a non-myeloablative conditioning regimen, 660 or 440 cGy total body irradiation (TBI). 1 × 106 Sca-1+ cells were transplanted into each animal. Animals were analyzed starting at 3 weeks after transplantation. Thirty-two weeks later, some animals (primary [1°] recipients) were euthanized; bone marrow (BM) cells were harvested and subsequently transplanted into lethally irradiated (1100 cGy) secondary (2°) recipients.
Analysis of FVIII expression in recipients
After transplantation of 2bF8/MGMT-transduced Sca-1+ cells, animals were analyzed for platelet-FVIII expression. Functional FVIII activity (FVIII:C) levels in recipients’ platelet lysates and plasmas were quantitated as previously described. [20;23] 2bF8 transgene expression was determined by PCR and quantitative real-time PCR (qPCR) analysis as previously reported. [22;23]
In vivo selection of the transduced cells
Beginning 10 weeks after transplantation, animals were treated with O6-benzylguanine (BG) (Sigma, St. Louis, MO, USA) followed by 1,3-bis-2 chloroethyl-1-nitrosourea (BCNU) (Emcure Pharmaceuticals, Pune, India). BG was administered intravenously at a dose of 30 mg kg−1 body weight. BCNU was given intravenously 1 hour after BG administration at a dose of 5 mg kg−1. Animals were treated once every 5 weeks for a total of 3 or 4 treatments. The levels of platelet-FVIII expression and the copy number of the 2bF8 transgene per cell were monitored to examine the efficacy of in vivo selection of 2bF8/MGMT-transduced cells.
Phenotypic correction assessment
The tail clip survival test, the tail bleeding test, and rotational thromboelastometry (ROTEM) analysis of whole blood clotting time (WBCT) were used to assess phenotypic correction of the FVIIInull coagulation defect. These assays were performed at least 6 weeks after the final BG/BCNU treatment. The tail clip survival test was carried out as described in our previous reports. [20–23] For the tail bleeding test, tail tips were clipped using a 1.6 mm diameter template. Fifty microliters of blood was collected before and 6 hours after tail tips were removed, and hemoglobin (Hb) was measured using the Vet blood counter (Scil Animal Care Company, Gurnee, IL, USA). The Hb was normalized by defining the pretest levels of Hb as 100%. For ROTEM analysis, blood samples were drawn from vena cava and WBCT was analyzed as previously described. [31] FVIIInull and wild-type (WT) mice were used as controls.
Immune response studies
At least 8 weeks after the final BG/BCNU treatment, animals were immunized intravenously with recombinant human B-domain deleted FVIII (rhF8, Xyntha, Pfizer Inc, New York, NY, USA) at a dose of 50 U kg−1 weekly for 4 weeks. One week after the last immunization, plasma samples were collected and the titers of anti-FVIII inhibitors were determined by Bethesda assay as previously described. [20] The titers of total anti-FVIII antibodies were determined by ELISA assay as reported. [32] Briefly, a 96-well plate was coated with 100 μL of 2 U mL−1 rhF8 at 4°C overnight. Serial dilutions of mouse plasma were added to the coated wells and incubated at room temperature for 2 hours. Horseradish peroxidase (HRP)-conjugated goat anti–mouse IgG (Pierce, Rockford, IL, USA) was used as the detecting antibody, followed by incubation with ortho-phenylenediamine substrate (Life Technologies, Grand Island, NY, USA). Antibody titers were expressed as the highest dilution of plasma showing a positive result (optical density > 0.3). Samples from FVIIInull mice were used as a control. The anti-FVIII antigen-secreting plasma cells (ASCs) were examined by ELISPOT assay as reported. [33] Briefly, polyvinylidendifluorid-bottom 96-well Multiscreen-IP filtration plates (Millipore Corporation, San Francisco, CA, USA) were coated with rhF8. Serial dilutions of splenocytes or BM cells from rhF8 immunized FVIIInull mice or 2bF8/MGMT-transduced BG/BCNU-treated recipients were seeded in completed RPMI 1640 media and incubated at 37 °C for 5 hours. After incubation, cells were removed and the plate was incubated with HRP conjugated goat anti-mouse IgG at 4 °C overnight. The spots were developed using 3-amino-9-ethylcarbazole (Sigma) and read by ELISPOT reader (Cellular Technology Limited, Heights, OH, USA).
Some animals were subsequently immunized with ovalbumin (OVA) (Sigma) as a non-specific immunogen at a dose of 25 μg per mouse adsorbed onto Imject Alum (Thermo, Rockford, IL, USA) by intraperitoneal injection weekly for 3 weeks. One week after the last immunization, the titers of anti-OVA IgG were determined by ELISA assay as previously described. [34] FVIIInull mice were immunized in parallel as a control.
Statistical analysis
Data are presented as the mean ± SD, and statistical comparisons of experimental groups were evaluated by unpaired Student’s t test. A value of P < 0.05 was considered statistically significant.
Results
Introducing the selectable 2bF8 transgene into HSCs of HA mice
We engineered a new LV, pWPT-2bF8/MGMT, harbouring dual genes, the 2bF8 gene and a drug-resistance gene, the MGMTP140K cassette (Fig. 1A). The 2bF8/MGMT cassette was introduced into FVIIInull HSCs by transduction and syngeneic transplantation. To minimize both the toxicity from preconditioning and the potential for insertional mutagenesis, we first used non-myeloablative conditioning regimens and a low MOI of 1. The 2bF8 expression cassette was detected by semi-quantitative PCR in all 2bF8/MGMT-transduced recipients. The representative PCR analysis results in Fig. 1B show augmentation of the PCR product after BG/BCNU treatment.
To quantitate the average copy number of the 2bF8/MGMT transgene per cell in transduced recipients, qPCR was used to analyze the 2bF8 transgene in peripheral blood cell-derived DNA. 2bF8 proviral DNA was measurable in all recipients. Before BG/BCNU treatment, the copy number of the 2bF8 transgene was barely detectable (0.01 ± 0.02 copy per cell) when HSCs were transduced with 2bF8/MGMT-LV at an MOI of 1. However, the copy number increased significantly after drug-selection treatment (0.42 ± 0.15 copy per cell) (Fig. 1C).
These results demonstrate viable engraftment of 2bF8/MGMT genetically modified HSCs in 2bF8/MGMT-transduced recipients and that 2bF8/MGMT-transduced cells are selectable by BG/BCNU treatment in vivo.
Enhancing platelet-FVIII expression in platelet gene therapy of HA mice via MGMT-mediated in vivo selection
To evaluate the feasibility of the MGMT-mediated drug-selection system in platelet gene therapy of HA, we quantified the FVIII:C expression in FVIIInull mice that received 2bF8/MGMT-transduced HSCs (MOI of 1). As determined by a chromogenic assay on platelet lysates, platelet-FVIII expression in recipients that were pre-conditioned with 660 cGy TBI, which was only 0.22 ± 0.15 mU per 108 platelets before treatment, remarkably increased to 4.33 ± 5.48 mU per 108 platelets after BG/BCNU drug-selective treatments (Fig. 2A). The levels of platelet-FVIII in the untreated transduced control group remained low over the study period. FVIII:C was not detected in the plasma in any of the recipients even with platelet-FVIII as high as 22 mU per 108 platelets. When a low intensity pre-conditioning regimen of 440 cGy TBI was used, the levels of platelet-FVIII increased from 0.07 ± 0.03 to 1.61 ± 0.84 mU per 108 platelets after BG/BCNU treatments (Fig. 2B). These results demonstrate that using the MGMT-mediated in vivo drug-selection system can effectively enrich 2bF8/MGMT-transduced cells and thereby enhance platelet-FVIII expression in platelet gene therapy of HA mice even under non-myeloablative conditioning regimens and with a low MOI of 1.
Fig. 2. Platelet-FVIII expression in 2bF8/MGMT-transduced recipients (MOI of 1).
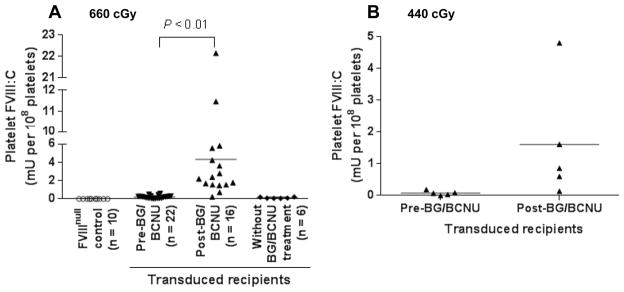
Platelets were isolated from peripheral blood from 2bF8/MGMT-transduced recipients before and after BG/BCNU drug selection treatments. The levels of platelet-FVIII were determined by a chromogenic assay on platelet lysates. The levels of platelet-FVIII expression pre-BCNU treatment were averaged from the time points before BG/BCNU treatments and the levels post-BCNU were averaged from three or more time points after the final BG/BCNU treatment. (A) Platelet-FVIII expression in 2bF8/MGMT-transduced recipients pre-conditioned with 660 cGy TBI. (B) Platelet-FVIII expression in 2bF8/MGMT-transduced recipients pre-conditioned with 440 cGy TBI. These data demonstrate that MGMT-mediated in vivo drug-selection treatments can efficiently enhance platelet-FVIII expression in 2bF8/MGMT-transduced recipients.
Phenotypic correction assessment of 2bF8/MGMT-transduced recipients
To assess phenotypic correction of the FVIIInull coagulation defect in 2bF8/MGMT-transduced recipients, three assays, the tail clip survival test, the tail bleeding test, and ROTEM analysis, were used. Fifteen of 16 transduced BG/BCNU-treated recipients pre-conditioned with 660 cGy TBI survived tail clipping. Five of 6 recipients without BG/BCNU treatment and none of the untransduced FVIIInull controls survived under the same challenge (Fig. 3A). To grade the degree of blood lost after tail clipping, Hb levels were measured before and 6 hours after tail clipping. Five of 12 FVIIInull mice survived beyond 6 hours with only 34.9 ± 10.5% Hb remaining, while all BG/BCNU-treated transduced recipients survived with 68.6 ± 10.5% Hb remaining, which was not significantly different from the WT control group (Fig. 3B). To further confirm that hemostasis was improved in the treated animals, ROTEM analysis was used to analyze the WBCT. As shown in Figure 3C, the WBCT in transduced BG/BCNU-treated animals was significantly shorter than in FVIIInull controls. There was no significant difference between the transduced BG/BCNU-treated group and the WT control group. Taken together, these results demonstrate that the hemophilic phenotype was rescued in 2bF8/MGMT-transduced BG/BCNU-treated animals even using a low MOI of 1.
Fig. 3. Phenotypic correction assessment of 2bF8/MGMT-transduced recipients (MOI of 1).
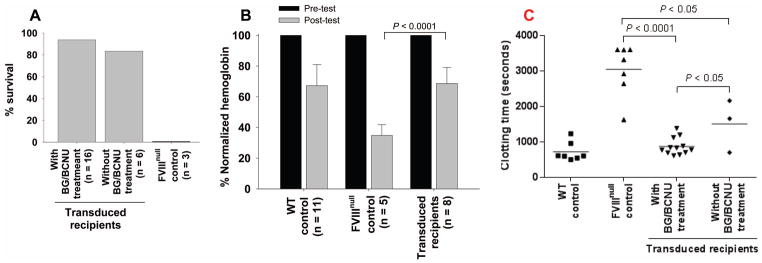
(A) Tail clip survival test. Tail clipping was performed at least 6 weeks after the final BG/BCNU treatment. Fifteen of 16 transduced recipients pre-conditioned with 660 cGy TBI survived tail clipping. (B) Tail bleeding test. Hemoglobin (Hb) levels were measured before and 6 hours after tail clipping. The level of Hb in each animal before the test was defined as 100%. Five of 12 FVIIInull mice survived beyond 6 hours with only 35% Hb remaining, while all BG/BCNU-treated transduced recipients survived with 69% Hb remaining. (C) ROTEM analysis of whole blood clotting time. The clotting time in transduced BG/BCNU-treated animals was significantly shorter than in FVIIInull controls. These results demonstrate that hemostasis was restored in hemophilia A mice after 2bF8/MGMT gene therapy followed by in vivo selection.
Sequential bone marrow transplantation
To ascertain whether platelet-FVIII expression was sustained after 2bF8/MGMT gene therapy followed by drug-selection, secondary transplantation was carried out using BM cells from some of the primary recipients from the 660 cGy group. As expected, proviral DNA was detected in all of the 2° recipients. The average copy number of the 2bF8 transgene was 0.50 ± 0.21 copy per cell, which was not significantly different from that obtained from the 1° recipients post-treatment (Fig. 4A). There was no difference in the levels of platelet-FVIII expression in the 2° recipients and their donors, 1° recipients post-treatment (Fig. 4B). These results demonstrate that long-term engrafting HSCs were genetically modified by 2bF8/MGMT-LV and selected after BG/BCNU treatment.
Fig. 4. Sequential transplantation.
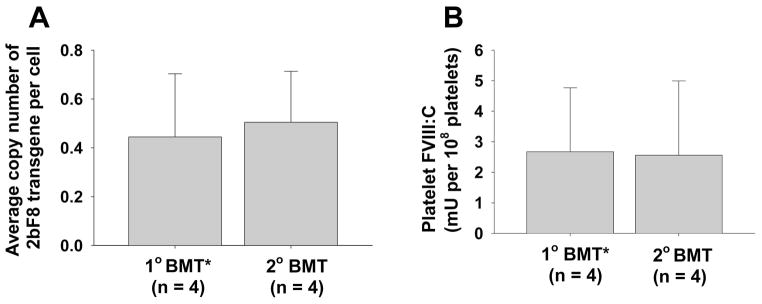
To ascertain whether platelet-FVIII expression was sustained, secondary (2°) transplant was carried out using BM cells from primary (1°) recipients that had been received 2bF8/MGMT-transduced HSCs. (A) Average copy number of 2bF8 proviral DNA per cell in 1° and 2° recipients. (B) Average FVIII:C levels in 1° and 2° recipients. 1° BMT*: data presented in this group were summarized from the primary recipients post-selection from which bone marrow mononuclear cells were collected and transplanted into the secondary recipients. These results demonstrate that the levels of platelet-FVIII expression in the 2° recipients were similar to those obtained from the 1° recipients.
Immune tolerance was established in 2bF8/MGMT-transduced BG/BCNU-treated recipients
To investigate the immune response after 2bF8/MGMT gene therapy in FVIIInull mice, we used the Bethesda assay to determine titers of anti-FVIII inhibitors and the ELISA assay to quantify total anti-FVIII antibodies. As shown in Figures 5A and 5B, no inhibitory antibodies were detected in recipients after 2bF8/MGMT gene therapy. No antibodies were detected in 2bF8/MGMT-transduced BG/BCNU-treated animals even after exogenous rhF8 challenge. One of three 2bF8/MGMT-transduced with no BG/BCNU treatment recipients developed a low titer (6.8 BU mL−1) of inhibitors. In contrast, all FVIIInull control mice developed anti-FVIII antibodies with inhibitor titers of 430 ± 480 BU mL−1 and total anti-FVIII antibody titers of 4203 ± 4109 under the same challenge. We also performed the ELISPOT assay to examine whether there were anti-FVIII antibody secreting cells in the treated animals. As shown in Figure 5C, no ASCs were detected in 2bF8/MGMT-transduced BG/BCNU-treated animals. In contrast, ASCs were detected in both spleen and BM of rhF8-immunized FVIIInull mice.
Fig. 5. The immune response in 2bF8/MGMT-transduced recipients.

To investigate the immune response in 2bF8/MGMT-transduced recipients, plasma samples were collected from 2bF8/MGMT-transduced recipients before and after rhF8 challenge. (A) The titers of anti-FVIII inhibitory antibodies (inhibitors) determined by Bethesda assay. (B) The titers of total anti-FVIII antibodies determined by ELISA assay. (C) The anti-FVIII antibody secreting cells examined by ELISPOT assay. Shown are representative results from 4×106 cells seeded per well. Taken together, figures A, B, and C show that none of the 2bF8/MGMT-transduced BG/BCNU treated recipients pre-conditioned with 660 cGy TBI developed anti-FVIII antibodies even after rhF8 immunization (50 U/kg/week IV × 4). (D) The titers of anti-ovalbumin (OVA) antibodies. To ensure that the immune system was not defective in the 2bF8/MGMT-transduced recipients, we further challenged the animals with OVA and the titers were determined by ELISA. All animals developed high titers of anti-OVA antibodies after OVA challenge. These results demonstrate that immune tolerance developed in the 2bF8/MGMT-transduced recipients and that the immune tolerance is FVIII-specific.
To ensure that the immune systems in the 2bF8/MGMT-transduced recipients were not defective, we further challenged some animals with OVA. All animals developed high titers of anti-OVA antibodies after the OVA challenge, with no significant difference between 2bF8/MGMT-transduced BG/BCNU-treated recipients and FVIIInull controls.
All together, these data demonstrate that immune tolerance developed in the 2bF8/MGMT-transduced recipients and that the immune tolerance is FVIII-specific.
The efficacy of higher MOI in 2bF8/MGMT gene therapy
When a higher MOI of approximately 10 was used to introduce the 2bF8/MGMT expression cassette into Sca-1+ cells, the level of platelet-FVIII expression in recipients was 1.63 ± 0.36 mU per 108 platelets in the 660 cGy group before BG/BCNU treatment. Platelet-FVIII expression levels significantly increased after one treatment (6.52 ± 1.31 mU per 108 platelets) and continued to increase after the second and third treatments, reaching 14.18 ± 12.05 mU per 108 platelets (Fig. 6A). The highest platelet-FVIII expression reached 35.49 mU per 108 platelets, a 21-fold increase over the pre-treatment level of 1.66 mU per 108 platelets. This amount of platelet-FVIII corresponds to 70% of FVIII:C in whole blood in normal mice, assuming a platelet number 109 mL−1 and plasma FVIII:C of 1 U mL−1. The level of platelet-FVIII in the 440 cGy group was 0.53 ± 0.07 mU per 108 platelets before treatment and increased to 2.87 ± 0.20 mU per 108 platelets after three treatments (Fig. 6B). Although platelet-FVIII expression levels continued to increase after the second and third treatments, the enhancements were not as significant as after the first treatment. Albeit the average level of platelet-FVIII in the 660 cGy group appears to be greater than that obtained in the 440 cGy group after BG/BCNU treatment, there was no significant difference between the two groups.
Fig. 6. The efficacy of high MOI (MOI of 10) in 2bF8/MGMT gene therapy.
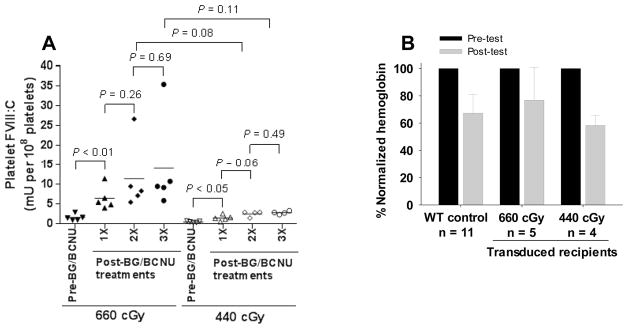
To compare the efficacy of our novel vector 2bF8/MGMT and our non-selectable 2bF8 construct in platelet gene therapy of hemophilia A, we used an MOI of approximately 10, the MOI typically used in our previous 2bF8LV studies. (A) Platelet-FVIII expression in recipients. Platelet-FVIII expression levels in recipients were monitored before and after each BG/BCNU treatment. The levels of platelet-FVIII expression before BG/BCNU treatment were averaged from 2 time points and the levels after the 3rd treatment were averaged from three time points after the final treatment. (B) Tail bleeding test. Hemoglobin (Hb) levels were measured before and 6 hours after tail clipping. The level of Hb in each animal before the test was defined as 100%.
When the tail bleeding test was used to assess the degree of phenotypic correction in these high-MOI-transduced BG/BCNU-treated recipients, the remaining Hb level in the 660 cGy group was 76.8 ± 24.2%, which appears higher than that in the WT control group although there is no statistically significant difference between the two groups. The level of remaining Hb in the 440 cGy group was 58.3 ± 7.5%, which is not significantly different from the 660 cGy group or the WT control (Fig. 6B). These data indicate that we can achieve a similar therapeutic effect post treatment using reduced preconditioning (440 cGy) as we do using higher preconditioning (660 cGy). We also monitored the blood counts on the 2bF8/MGMT-transduced recipients during our study course. Four weeks after transplantation, the number of leukocytes in the 660 cGy group was similar to the 440 cGy group, which was approximately 55% of pre-transplantation levels. Blood counts recovered by 8 weeks after transplantation. Both leukocytes and platelets decreased during BG/BCNU treatment, but gradually recovered. Hemoglobin levels were less affected during BG/BCNU treatment (Fig 7).
Fig. 7. Blood count during study course of 2bF8/MGMT-transduction and BG/BCNU treatment.
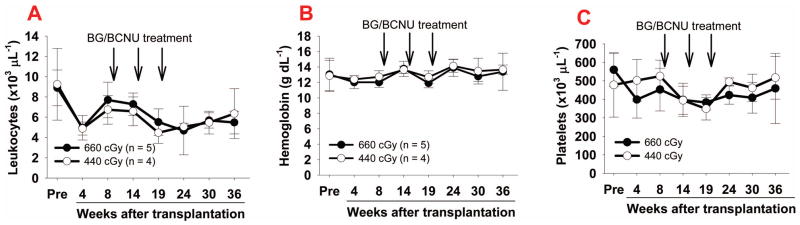
Blood samples were collected from 2F8/MGMT-transduced recipients using sodium citrate as an anticoagulant. Blood cell number and hemoglobin levels were measured using the Vet blood counter. (A) Leukocyte number; (B) Hemoglobin levels; and (C) Platelet number.
Discussion
Any effective gene therapy protocol for HA should provide sustained therapeutic levels of FVIII expression without substantial toxicity. For platelet gene therapy to be successful, utilizing an efficient integrating vector to introduce the transgene into HSCs and employing a sufficient preconditioning to create space for effective engraftment are essential. Thus, the risk of insertional mutagenesis associated with the use of integrating vectors and the toxicities associated with the use of preconditioning must be considered when developing a platelet gene therapy protocol for HA treatment. In the current study, we aimed to enhance platelet-FVIII expression while minimizing the potential toxicities. We developed a novel vector harboring dual genes, the 2bF8 gene and a drug-resistance gene, the MGMTP140K cassette. Our data demonstrate the feasibility of using the MGMT-mediated selection system to enrich 2bF8 genetically modified hematopoietic cells and enhance platelet-FVIII expression in FVIIInull mice even using a relatively low MOI for transduction and under a non-myeloablative conditioning regimen.
Although LVs have great potential to deliver therapeutic genes to HSCs, the size of the transgene affects the titers of vectors and the transduction efficiency on HSCs. [22;34–36] FVIII is an unusually large protein. [37] Even though we use a B-domain deleted FVIII construct, the size of the 2bF8 expression cassette is still relatively large, in contrast to other genes, i.e. 2bF9 or 2bIbα which we are also studying for hemophilia B and Bernard Soulier syndrome gene therapy. [34;36] Indeed, the transduction efficiency is approximately 10% in 2bF8, [22] 20% in 2bF9, [34] and 60% in 2bIbα studies [36] when a similar protocol with a myeloablative conditioning and an MOI of approximately 10 is applied. Our previous studies have demonstrated that platelet gene therapy is a promising therapeutic strategy for HA. [20–25] While therapeutic levels of platelet-FVIII can be achieved using non-selectable 2bF8LV transduction of HSCs at a high MOI followed by syngeneic transplantation in a HA animal models [22–24], severe HA patients may require higher levels of functional platelet-FVIII to be therapeutic, especially for patients with active sport activities. Thus, developing an approach to increase the proportion of transduced platelets and enhance platelet-FVIII expression is desired.
Methods to increase the proportion of transduced HSCs in BM have been under investigation and usually rely on transduction of a drug resistance gene followed by chemotherapeutic treatment to mediate selection. [26;27;38–40] One of the most promising drug-resistance genes is MGMT, which encodes a DNA-repair enzyme that can confer resistance to the cytotoxic effects of nitrosoureas such as BCNU. [41;42] Successful MGMT-based in vivo selection of HSCs using BG and BCNU has been reported in cancer therapy to allow dose intensification and in gene therapy of AIDS, Glanzmann Thrombasthenia, β-thalassemia, and hemophilia B to increase the percentage of gene-modified cells. [29;39;43–47] In the current study, we incorporated the MGMTp140k cassette into our 2bF8LV and used BG/BCNU in vivo chemoselection. To reduce the potential for adverse effects from BCNU chemotherapy, we chose a high dose of BG to sensitize untransduced cells combined with a low dose of BCNU as reported in literature. [39;43–47] The animals did not appear sick after any of chemotherapy treatments. We monitored some recipients for 6 months after the BG/BCNU treatments were completed (12 months after transplantation); these animals were healthy, had normal blood counts, and exhibited no overt evidence of tumor or other illness.
An effective selection system should not only allow for the enhancement of platelet-FVIII expression, but for the reduction in potential for insertional mutagenesis as well because this will make it possible to transplant a lower total number of long-term repopulating HSCs and/or use a low MOI. The question is whether we can achieve sustainable therapeutic results while reducing the MOI. In the current study, the highly selective effectiveness of 2bF8/MGMT-transduced animals in response to BG/BCNU treatment was confirmed by qPCR analysis of the copy number of the proviral DNA and FVIII:C assay analysis of platelet lysates. The average level of platelet-FVIII expression was enhanced 20-fold in the 660 cGy group and 30-fold in the 440 cGy group of 2bF8/MGMT-transduced recipients after BG/BCNU treatments even when a relatively low MOI of 1 was used. The level of platelet-FVIII expression in recipients after in vivo selection (4.33 ± 5.48 mU per 108 platelets) was 2.9-fold higher than that obtained from our non-selectable 2bF8LV-transduced recipients in our previous report using an MOI of 10 and myeloablative conditioning. [23] These data suggest that 2bF8 gene-modified cells can be efficiently enriched and platelet-FVIII expression can be markedly enhanced after BG/BCNU treatment when the the 2bF8/MGMT-mediated gene transfer system is employed.
When HSCs are transduced at a relatively low MOI of 1, the potential for muti-insertions of the transgene into the genome is decreased. In addition, when a lower MOI is employed, fewer numbers of transduced cells may be expanded after selection. Thus, insertion site-mediated genotoxicity and the risk of insertional muta-oncogenesis may be reduced. When an MOI of 10 was employed, which we typically use in our gene therapy protocol with our non-selectable 2bF8LV, the average platelet-FVIII expression level in 2bF8/MGMT-transduced recipients before BG/BCNU treatment is similar to that obtained from 2bF8-transduced recipients in our previous report, [23] demonstrating that the transduction efficiency and expression capacity of 2bF8/MGMT-LV is comparable to 2bF8LV when a similar protocol is applied.
Theoretically, we should be able to reach similar levels of platelet-FVIII post treatment using reduced preconditioning as we do using higher preconditioning as long as the regimen creates enough space for the transduced cells to engraft. When the preconditioning was reduced to 440 cGy, post-selection platelet-FVIII expression appears to be lower than the level reached in the 660 cGy group when either an MOI of 1 or 10 was employed, but there was no significant difference between the two groups in either case. No platelet-FVIII expression was detected in animals preconditioned with 220 cGy TBI which subsequently received 2bF8/MGMT-transduced HSCs using an MOI of 1 even after 4 BG/BCNU treatments (data not shown), indicating insufficient engraftment of transduced cells. Notably, the post-selection level of platelet-FVIII expression in the 440 cGy group with an MOI of 1 is comparable to that obtained from the 2bF8-transduced recipients when a myeloablative regimen and an MOI of 10 was applied in our previous report. [23] Thus, a protocol using 2bF8/MGMT-LV at a low MOI and a sufficient precondition regimen can achieve therapeutic results while reducing toxicities related to platelet gene therapy.
The immune response to the transgene product is a significant concern in gene therapy. Neither inhibitory nor non-inhibitory antibodies developed in transduced recipients, which is consistent with our previous findings. [22;23] To investigate whether immune tolerance was induced in 2bF8/MGMT-transduced recipients after treatment, animals were challenged with exogenous rhF8. No anti-FVIII antibodies were detected by Bethesda, ELISA, or ELISPOT assays. In contrast, when animals were immunized with a non-specific antigen, ovalbumin, all the animals developed anti-ovalbumin antibodies. Our results strongly suggest that platelet gene therapy can not only provide sustained therapeutic platelet-FVIII, but also induce FVIII-specific immune tolerance in HA mice.
In summary, our studies demonstrate that using the MGMT-mediated drug-selection system in 2bF8 gene therapy can efficiently enhance therapeutic platelet-FVIII expression, resulting in sustained phenotypic correction and FVIII-specific immune tolerance induction in HA mice. This effective in vivo selective system allows us to enrich 2bF8-transduced cells and enhance platelet-FVIII expression for HA gene therapy even using a relatively low MOI and a non-myeloablative regimen, which will minimize the toxicities and potential risks related to platelet gene therapy. Further studies to evaluate the efficacy and safety of the 2bF8/MGMT system in a large animal model and in human cells will be warranted.
Acknowledgments
We thank Dr. Haig H. Kazazian at the University of Pennsylvania School of Medicine for the FVIIInull mice. We thank Dr. Christine Baumgartner for advice on the ELISPOT assay and the FVIII antibody ELISA assay. This work was supported by the National Institutes of Health grants HL-102035 (QS) and HL-68138 (DAW), the Children’s Research Institute Funds (QS), the Midwest Athletes against Childhood Cancer Fund (QS), the Rebecca Slye Endowment Fund (QS), the National Hemophilia Foundation JGP Postdoctoral Research Fellowship (YC), and an American Heart Association Award from the Greater Midwest Affiliate 0755827Z (DAW).
Footnotes
Addendum
J. A. Schroeder designed and performed research, analyzed data, and wrote the manuscript; Y. Chen performed research and analyzed data; J. Fang and D. A. Wilcox assisted with the MGMT construct; Q. Shi designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.High KA. Gene transfer as an approach to treating hemophilia. Semin Thromb Hemost. 2003;29:107–20. doi: 10.1055/s-2003-37945. [DOI] [PubMed] [Google Scholar]
- 2.High KA. Gene therapy for haemophilia: a long and winding road. J Thromb Haemost. 2011;9 (Suppl 1):2–11. doi: 10.1111/j.1538-7836.2011.04369.x. [DOI] [PubMed] [Google Scholar]
- 3.Ponder KP. Hemophilia gene therapy: a Holy Grail found. Mol Ther. 2011;19:427–8. doi: 10.1038/mt.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saenko EL, Ananyeva NM, Moayeri M, Ramezani A, Hawley RG. Development of improved factor VIII molecules and new gene transfer approaches for hemophilia A. Curr Gene Ther. 2003;3:27–41. doi: 10.2174/1566523033347417. [DOI] [PubMed] [Google Scholar]
- 5.Balague C, Zhou J, Dai Y, Alemany R, Josephs SF, Andreason G, Hariharan M, Sethi E, Prokopenko E, Jan HY, Lou YC, Hubert-Leslie D, Ruiz L, Zhang WW. Sustained high-level expression of full-length human factor VIII and restoration of clotting activity in hemophilic mice using a minimal adenovirus vector. Blood. 2000;95:820–8. [PubMed] [Google Scholar]
- 6.Guo X, Wang H, Chu H, Wang X, Qu B, Li Z, Qi Z, Wang Z. High level expression of human factor VIII in mammalian cells after retroviral-mediated gene transfer. Chin Med J (Engl) 2001;114:690–3. [PubMed] [Google Scholar]
- 7.Kootstra NA, Matsumura R, Verma IM. Efficient production of human FVIII in hemophilic mice using lentiviral vectors. Mol Ther. 2003;7:623–31. doi: 10.1016/s1525-0016(03)00073-x. [DOI] [PubMed] [Google Scholar]
- 8.Tiede A, Eder M, von DM, Battmer K, Luther S, Kiem HP, Ganser A, Scherr M. Recombinant factor VIII expression in hematopoietic cells following lentiviral transduction. Gene Ther. 2003;10:1917–25. doi: 10.1038/sj.gt.3302093. [DOI] [PubMed] [Google Scholar]
- 9.Moayeri M, Hawley TS, Hawley RG. Correction of murine hemophilia A by hematopoietic stem cell gene therapy. Mol Ther. 2005;12:1034–42. doi: 10.1016/j.ymthe.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye P, Thompson AR, Sarkar R, Shen Z, Lillicrap DP, Kaufman RJ, Ochs HD, Rawlings DJ, Miao CH. Naked DNA transfer of Factor VIII induced transgene-specific, species-independent immune response in hemophilia A mice. Mol Ther. 2004;10:117–26. doi: 10.1016/j.ymthe.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Xu L, Nichols TC, Sarkar R, McCorquodale S, Bellinger DA, Ponder KP. Absence of a desmopressin response after therapeutic expression of factor VIII in hemophilia A dogs with liver-directed neonatal gene therapy. Proc Natl Acad Sci U S A. 2005;102:6080–5. doi: 10.1073/pnas.0409249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hauck B, Xu RR, Xie J, Wu W, Ding Q, Sipler M, Wang H, Chen L, Wright JF, Xiao W. Efficient AAV1-AAV2 hybrid vector for gene therapy of hemophilia. Hum Gene Ther. 2006;17:46–54. doi: 10.1089/hum.2006.17.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu L, Mei M, Ma X, Ponder KP. High expression reduces an antibody response after neonatal gene therapy with B domain-deleted human factor VIII in mice. J Thromb Haemost. 2007;5:1805–12. doi: 10.1111/j.1538-7836.2007.02629.x. [DOI] [PubMed] [Google Scholar]
- 14.Matsui H, Shibata M, Brown B, Labelle A, Hegadorn C, Andrews C, Hebbel RP, Galipeau J, Hough C, Lillicrap D. Ex vivo gene therapy for hemophilia A that enhances safe delivery and sustained in vivo factor VIII expression from lentivirally engineered endothelial progenitors. Stem Cells. 2007;25:2660–9. doi: 10.1634/stemcells.2006-0699. [DOI] [PubMed] [Google Scholar]
- 15.Ide LM, Gangadharan B, Chiang KY, Doering CB, Spencer HT. Hematopoietic stem-cell gene therapy of hemophilia A incorporating a porcine factor VIII transgene and nonmyeloablative conditioning regimens. Blood. 2007;110:2855–63. doi: 10.1182/blood-2007-04-082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsui H, Hegadorn C, Ozelo M, Burnett E, Tuttle A, Labelle A, McCray PB, Jr, Naldini L, Brown B, Hough C, Lillicrap D. A microRNA-regulated and GP64-pseudotyped lentiviral vector mediates stable expression of FVIII in a murine model of Hemophilia A. Mol Ther. 2011;19:723–30. doi: 10.1038/mt.2010.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roth DA, Tawa NE, Jr, O’Brien JM, Treco DA, Selden RF. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med. 2001;344:1735–42. doi: 10.1056/NEJM200106073442301. [DOI] [PubMed] [Google Scholar]
- 18.Powell JS, Ragni MV, White GC, Lusher JM, Hillman-Wiseman C, Moon TE, Cole V, Ramanathan-Girish S, Roehl H, Sajjadi N, Jolly DJ, Hurst D. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102:2038–45. doi: 10.1182/blood-2003-01-0167. [DOI] [PubMed] [Google Scholar]
- 19.Chuah MK, Collen D, Vandendriessche T. Clinical gene transfer studies for hemophilia A. Semin Thromb Hemost. 2004;30:249–56. doi: 10.1055/s-2004-825638. [DOI] [PubMed] [Google Scholar]
- 20.Shi Q, Wilcox DA, Fahs SA, Weiler H, Wells CW, Cooley BC, Desai D, Morateck PA, Gorski J, Montgomery RR. Factor VIII ectopically targeted to platelets is therapeutic in hemophilia A with high-titer inhibitory antibodies. J Clin Invest. 2006;116:1974–82. doi: 10.1172/JCI28416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Q, Fahs SA, Wilcox DA, Kuether EL, Morateck PA, Mareno N, Weiler H, Montgomery RR. Syngeneic transplantation of hematopoietic stem cells that are genetically modified to express factor VIII in platelets restores hemostasis to hemophilia A mice with preexisting FVIII immunity. Blood. 2008;112:2713–21. doi: 10.1182/blood-2008-02-138214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi Q, Wilcox DA, Fahs SA, Fang J, Johnson BD, DU LM, Desai D, Montgomery RR. Lentivirus-mediated platelet-derived factor VIII gene therapy in murine haemophilia A. J Thromb Haemost. 2007;5:352–61. doi: 10.1111/j.1538-7836.2007.02346.x. [DOI] [PubMed] [Google Scholar]
- 23.Kuether EL, Schroeder JA, Fahs SA, Cooley BC, Chen Y, Montgomery RR, Wilcox DA, Shi Q. Lentivirus-mediated platelet gene therapy of murine hemophilia A with pre-existing anti-factor VIII immunity. J Thromb Haemost. 2012;10:1570–80. doi: 10.1111/j.1538-7836.2012.04791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DU LM, Nurden P, Nurden AT, Nichols TC, Bellinger DA, Jensen ES, Haberichter SL, Merricks E, Raymer RA, Fang J, Koukouritaki SB, Jacobi PM, Hawkins TB, Cornetta K, Shi Q, Wilcox DA. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. doi: 10.1038/ncomms3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Q, Kuether EL, Chen Y, Schroeder JA, Fahs SA, Montgomery RR. Platelet gene therapy corrects the hemophilic phenotype in immunocompromised hemophilia A mice transplanted with genetically manipulated human cord blood stem cells. Blood. 2014;123:395–403. doi: 10.1182/blood-2013-08-520478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abonour R, Williams DA, Einhorn L, Hall KM, Chen J, Coffman J, Traycoff CM, Bank A, Kato I, Ward M, Williams SD, Hromas R, Robertson MJ, Smith FO, Woo D, Mills B, Srour EF, Cornetta K. Efficient retrovirus-mediated transfer of the multidrug resistance 1 gene into autologous human long-term repopulating hematopoietic stem cells. Nat Med. 2000;6:652–8. doi: 10.1038/76225. [DOI] [PubMed] [Google Scholar]
- 27.Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, Morecki S, Andolfi G, Tabucchi A, Carlucci F, Marinello E, Cattaneo F, Vai S, Servida P, Miniero R, Roncarolo MG, Bordignon C. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–3. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 28.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10:119–21. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 29.Fang J, Jensen ES, Boudreaux MK, DU LM, Hawkins TB, Koukouritaki SB, Cornetta K, Wilcox DA. Platelet gene therapy improves hemostatic function for integrin alphaIIbbeta3-deficient dogs. Proc Natl Acad Sci U S A. 2011;108:9583–8. doi: 10.1073/pnas.1016394108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Q, Wilcox DA, Morateck PA, Fahs SA, Kenny D, Montgomery RR. Targeting platelet GPIbalpha transgene expression to human megakaryocytes and forming a complete complex with endogenous GPIbbeta and GPIX. J Thromb Haemost. 2004;2:1989–97. doi: 10.1111/j.1538-7836.2004.00961.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang G, Shi Q, Fahs SA, Kuether EL, Walsh CE, Montgomery RR. Factor IX ectopically expressed in platelets can be stored in alpha-granules and corrects the phenotype of hemophilia B mice. Blood. 2010;116:1235–43. doi: 10.1182/blood-2009-11-255612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reipert BM, Ahmad RU, Turecek PL, Schwarz HP. Characterization of antibodies induced by human factor VIII in a murine knockout model of hemophilia A. Thromb Haemost. 2000;84:826–32. [PubMed] [Google Scholar]
- 33.Hausl C, Maier E, Schwarz HP, Ahmad RU, Turecek PL, Dorner F, Reipert BM. Long-term persistence of anti-factor VIII antibody-secreting cells in hemophilic mice after treatment with human factor VIII. Thromb Haemost. 2002;87:840–5. [PubMed] [Google Scholar]
- 34.Chen Y, Schroeder JA, Kuether EL, Zhang G, Shi Q. Platelet gene therapy by lentiviral gene delivery to hematopoietic stem cells restores hemostasis and induces humoral immune tolerance in FIX(null) mice. Mol Ther. 2014;22:169–77. doi: 10.1038/mt.2013.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar M, Keller B, Makalou N, Sutton RE. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther. 2001;12:1893–905. doi: 10.1089/104303401753153947. [DOI] [PubMed] [Google Scholar]
- 36.Kanaji S, Kuether EL, Fahs SA, Schroeder JA, Ware J, Montgomery RR, Shi Q. Correction of Murine Bernard-Soulier Syndrome by Lentivirus-mediated Gene Therapy. Mol Ther. 2012;20:625–32. doi: 10.1038/mt.2011.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufman RJ, Wasley LC, Dorner AJ. Synthesis, processing, and secretion of recombinant human factor VIII expressed in mammalian cells. J Biol Chem. 1988;263:6352–62. [PubMed] [Google Scholar]
- 38.Sorrentino BP, Brandt SJ, Bodine D, Gottesman M, Pastan I, Cline A, Nienhuis AW. Selection of drug-resistant bone marrow cells in vivo after retroviral transfer of human MDR1. Science. 1992;257:99–103. doi: 10.1126/science.1352414. [DOI] [PubMed] [Google Scholar]
- 39.Cai S, Ernstberger A, Wang H, Bailey BJ, Hartwell JR, Sinn AL, Eckermann O, Linka Y, Goebel WS, Hanenberg H, Pollok KE. In vivo selection of hematopoietic stem cells transduced at a low multiplicity-of-infection with a foamy viral MGMT(P140K) vector. Exp Hematol. 2008;36:283–92. doi: 10.1016/j.exphem.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Persons DA, Allay JA, Bonifacino A, Lu T, Agricola B, Metzger ME, Donahue RE, Dunbar CE, Sorrentino BP. Transient in vivo selection of transduced peripheral blood cells using antifolate drug selection in rhesus macaques that received transplants with hematopoietic stem cells expressing dihydrofolate reductase vectors. Blood. 2004;103:796–803. doi: 10.1182/blood-2003-05-1572. [DOI] [PubMed] [Google Scholar]
- 41.Gerson SL. Clinical relevance of MGMT in the treatment of cancer. J Clin Oncol. 2002;20:2388–99. doi: 10.1200/JCO.2002.06.110. [DOI] [PubMed] [Google Scholar]
- 42.Zielske SP, Gerson SL. Lentiviral transduction of P140K MGMT into human CD34(+) hematopoietic progenitors at low multiplicity of infection confers significant resistance to BG/BCNU and allows selection in vitro. Mol Ther. 2002;5:381–7. doi: 10.1006/mthe.2002.0571. [DOI] [PubMed] [Google Scholar]
- 43.Persons DA, Allay ER, Sawai N, Hargrove PW, Brent TP, Hanawa H, Nienhuis AW, Sorrentino BP. Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood. 2003;102:506–13. doi: 10.1182/blood-2003-03-0677. [DOI] [PubMed] [Google Scholar]
- 44.Larochelle A, Choi U, Shou Y, Naumann N, Loktionova NA, Clevenger JR, Krouse A, Metzger M, Donahue RE, Kang E, Stewart C, Persons D, Malech HL, Dunbar CE, Sorrentino BP. In vivo selection of hematopoietic progenitor cells and temozolomide dose intensification in rhesus macaques through lentiviral transduction with a drug resistance gene. J Clin Invest. 2009;119:1952–63. doi: 10.1172/JCI37506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai S, Wang H, Bailey B, Hartwell JR, Silver JM, Juliar BE, Sinn AL, Baluyut AR, Pollok KE. Differential Secondary Reconstitution of In Vivo-Selected Human SCID-Repopulating Cells in NOD/SCID versus NOD/SCID/gamma chain Mice. Bone Marrow Res. 2011;2011:252953. doi: 10.1155/2011/252953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adair JE, Beard BC, Trobridge GD, Neff T, Rockhill JK, Silbergeld DL, Mrugala MM, Kiem HP. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci Transl Med. 2012;4:133ra57. doi: 10.1126/scitranslmed.3003425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao H, Pestina TI, Nasimuzzaman M, Mehta P, Hargrove PW, Persons DA. Amelioration of murine beta-thalassemia through drug selection of hematopoietic stem cells transduced with a lentiviral vector encoding both gamma-globin and the MGMT drug-resistance gene. Blood. 2009;113:5747–56. doi: 10.1182/blood-2008-10-186684. [DOI] [PMC free article] [PubMed] [Google Scholar]