

Abstract
Inflammation is a physiological response of the immune system to injury or infection but may become chronic. In general, inflammation is self-limiting and resolves by activating a termination program named resolution of inflammation. It has been argued that unresolved inflammation may be the basis of a variety of chronic inflammatory diseases. Resolution of inflammation is an active process that is fine-tuned by the production of proresolving mediators and the shutdown of intracellular signaling molecules associated with cytokine production and leukocyte survival. Apoptosis of leukocytes (especially granulocytes) is a key element in the resolution of inflammation and several signaling molecules are thought to be involved in this process. Here, we explore key signaling molecules and some mediators that are crucial regulators of leukocyte survival in vivo and that may be targeted for therapeutic purposes in the context of chronic inflammatory diseases.
1. Introduction
Inflammation is a reaction of an organism to cell and tissue damage caused by various types of agents (sterile or not, including autoimmune events). Inflammation may also be physiological and is thought to be crucial for the maintenance of tissue homeostasis [1–5]. Important microcirculatory events occur during the inflammatory process, including vascular permeability, changes in the movement, recruitment and accumulation of leukocytes, and the release of inflammatory mediators [6]. After elimination of the harmful agent, the inflammatory process is usually resolved, as seen by the reduction in the number of leukocytes in the inflammatory site and the reversal of vascular changes. Resolution is necessary to restore the original architecture and function of a given tissue. Failure to resolve can cause persistent inflammation with consequent maintenance or increase of tissue destruction [3]. It has been argued that unresolved inflammation or excessive initial inflammation may be the basis of a variety of chronic inflammatory diseases [7].
The resolution of inflammation is an active process that is coordinated and controlled by a variety of extracellular and intracellular molecules [4]. With the termination of the inflammatory stimulus, the reduction of proinflammatory mediators occurs at the site through the decreased synthesis and increased catabolism of these molecules [4, 8]. The release of proresolving mediators also occurs which prevents further migration and increases apoptotic events of leukocytes (primarily granulocytes) [9]. In parallel, some proresolving molecules are able to induce the incoming of nonphlogistic macrophages to further perpetuate efferocytosis of apoptotic granulocytes. In doing so, proresolving molecules reprogram macrophages to perform more restorative and resolutive roles, thus amplifying the production of proresolving molecules and promoting resolution [10, 11]. These events mark the beginning of the resolution process, which is essential to reestablish tissue homeostasis [4, 8, 10, 12].
Some leucocytes, such as granulocytes (mainly neutrophils and eosinophils) and macrophages, are profoundly involved in the inflammatory response. Granulocytes release toxic compounds and also act as phagocytes together with macrophages to remove the agent causing inflammation. However, for the inflammatory process to be successful and self-limiting (with the goal of restoring tissue homeostasis), the actions of neutrophils and eosinophils must be finely regulated [3, 4, 8]. Thus, apoptosis of granulocytes followed by the efferocytosis (phagocytosis of apoptotic cells) by macrophages and an active resolution process are obvious avenues to achieve this goal [6, 10, 12].
Several signaling molecules, including PI3K/Akt, NF-κB, MAPKs, and CDKs, have been shown to be involved in enhancing granulocyte survival in vivo and in vitro [13–16]. The rationale behind enhanced granulocyte survival involves delaying death of these cells to enable efficient effector function, such as bacterial killing. However, if not finely controlled, prolonged activation of survival pathways and prevention of apoptosis in granulocytes may eventually delay inflammation resolution. In contrast with the molecules described above, proresolving mediators, including Annexin A1 (AnxA1), hydrogen peroxide (H2O2), cyclic adenosine monophosphate (cAMP), TNF-related apoptosis-inducing ligand (TRAIL) elevating agents (see Figure 1), and other specialized lipid mediators (lipoxin A4, resolvins, maresins, and protectins), perform the opposite action; that is, they induce granulocyte apoptosis. Recent studies have shown that strategies that modulate apoptosis-controlling proteins may promote the resolution of the inflammatory process [17–22]. Therefore, potential therapeutic strategies that modulate the resolution pathways may further represent a useful pharmacological arsenal for the treatment and prevention of various acute and chronic inflammatory diseases. Here, we discuss some aspects of the complex signaling network and some interventions that interfere with key signaling molecules associated with leukocyte survival and consequently contribute to inflammation resolution and return to homeostasis.
Figure 1.
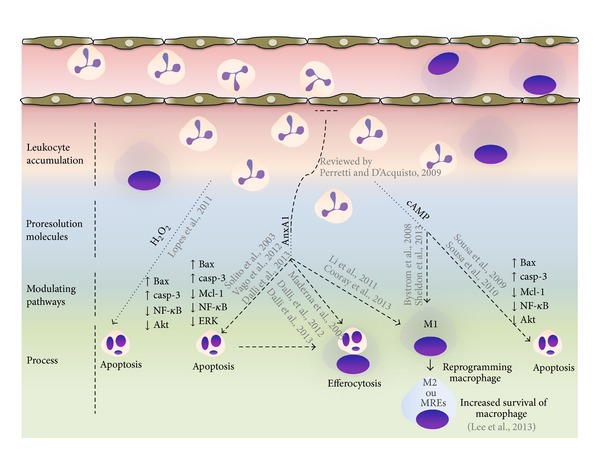
Targets to promote granulocyte apoptosis and inflammation resolution. During early phase of inflammation production of proinflammatory mediators and activation of signal survival pathways (PI3K/Akt, NF-κB, MAPKs, and CDKs) promotes leukocyte accumulation and survival in the inflammatory site. While inflammatory response evolves, local activation/release of proresolution mediators occurs and pathways (H2O2, AnxA1, and cAMP) that control further granulocyte ingress and turn on a resolution program. These resolution molecules, in addition to proresolving lipid mediators which are not highlighted in this cartoon, downregulate survival pathways and activate an apoptosis-associated program in granulocytes. Resolution molecules are also able to promote efferocytosis and coordinate reprogramming of macrophages. These events will reestablish tissue homeostasis.
2. Signaling Molecules as Crucial Regulators of the Resolution of Inflammation
2.1. Cyclin-Dependent Kinases
Cyclin-dependent kinases (CDK) are serine/threonine protein kinases that bind to cyclin to mediate the phosphorylation reactions that are associated with the progression and regulation of the cell cycle [23]. Surprisingly, terminally differentiated cells, including neutrophils, also express CDKs [24]. Previous studies have demonstrated that human neutrophils express CDK isoforms and their activity is associated with neutrophil lifespan [24–28]. More recent data have shown that CDK inhibitors (CDKi) drive granulocyte apoptosis and resolve inflammation by downregulating Mcl-1 and upregulating proapoptotic proteins such as Bim [24, 26, 27, 29–31]. In several models of inflammation, including passively induced arthritis, bleomycin-induced lung injury, and carrageenan-elicited acute pleurisy, R-roscovitine, a selective inhibitor of cyclin-dependent kinases CDK2, CDK5, CDK7, and CDK9, enhanced the resolution of established inflammation. This resolution was associated with an increase in neutrophil apoptosis in a caspase-dependent manner [24]. In murine models, it has also been reported that the induction of neutrophil apoptosis with R-roscovitine in conjunction with antibiotic therapy reduces markers of neuronal damage of pneumococcal meningitis [30]. Moreover, neutrophil clearance mediated through CDK inhibition reduced the lung inflammation induced by lipoteichoic acid, Streptococcus pneumonia, and bleomycin-induced lung injury models [26, 32]. R-roscovitine inhibits the CDK7- and CDK9-dependent phosphorylation of RNA polymerase II to block the transcriptional capacity of neutrophils, which can be a key mechanism associated with neutrophil apoptosis after CDK inhibition [26]. The studies discussed above clearly demonstrate that CDK inhibitors induce apoptosis and in vivo clearance of nonproliferating cells, such as granulocytes. AT7519 is a more recently investigated CDK inhibitor that has been evaluated in clinical trials for anticancer therapy [33]. A study by Alessandri et al. demonstrated that AT7519 induced eosinophil apoptosis and enhanced the resolution of allergic pleurisy [31]. AT7519 was also capable of inducing neutrophil apoptosis and accelerating the resolution of inflammation induced by LPS or Escherichia coli without the impairment of bacterial clearance [34]. Importantly, AT7519 has already been tested to treat patients with refractory solid tumors [33]. Altogether, these findings suggest that CDK may be a useful therapeutic strategy for the treatment of inflammatory diseases.
2.2. Mitogen-Activated Protein Kinases (MAPK) and Extracellular-Signal-Regulated Kinase (ERK)
The mitogen-activated protein kinases (MAPK) represent a family of serine threonine kinases that phosphorylate and activate transcription factors present in the cytoplasm or nucleus to drive the expression of genes and consequently biological responses. There are three main MAPK-activated signaling cascades that lead to differential gene expression: extracellular-signal-regulated kinase ERK1/2, p38 MAP kinase, and c-Jun N-terminal kinases (JNKs); these cascades are activated by several stimuli to regulate proliferation, differentiation, cell survival, mitosis, apoptosis, and other cell functions [35, 36]. The MEK/ERK signaling pathway has been targeted in an attempt to promote resolution of acute inflammation. Thus, it has been demonstrated that treatment with U0126, a potent and selective MEK1/2 inhibitor (a kinase upstream ERK1/2), was able to reduce inflammatory parameters in a murine model of allergic asthma [37] and LPS-induced lung injury [38]. Additionally, the use of a specific ERK1/2 inhibitor (PD98059) augmented the resolution of inflammation in a pleurisy model; this was associated with the inhibition of the production of neutrophil survival factors at the site of inflammation and the increased neutrophil apoptosis [39]. A recent study in vitro demonstrated that MSK1/2 (mitogen- and stress-activated protein kinase 1 and 2)—two kinases phosphorylated by both ERK1/2 and p38 MAPK—are associated with control on the induction of cyclooxygenase- (COX-) 2 mRNA and the IL-10 production through CREB (cAMP responsive element-binding protein) in macrophages stimulated or not with LPS [40]. However, there is no evidence that MSK1/2 are relevant for resolution of inflammation in vivo. Therefore, although the anti-inflammatory effects of MAPK inhibitors have been clearly defined, the potential of these inhibitors to promote inflammation resolution needs to be better clarified using other models of in vivo inflammation.
2.3. Cyclic Adenosine Monophosphate
Cyclic adenosine monophosphate (cAMP) is a ubiquitous second messenger produced after adenylate cyclase activation that has been shown to play an important role in modulating the activity of cells involved in the inflammatory process, primarily through PKA activity [41]. Intracellular levels of cAMP are physiologically modulated by agonist ligands (such as PGE2, adenosine, and β-adrenergic) and are fine-tuned and controlled by phosphodiesterases (PDEs), which are intracellular enzymes that hydrolyze cAMP [41, 42]. In addition to known anti-inflammatory proprieties of cAMP-elevating agents [41–44], emerging data support a role for cAMP in some steps of the resolution process [20, 45–47]. Our research group demonstrated an important role for cAMP in inducing the resolution of acute inflammation by modulating the apoptosis of granulocytes in vivo [20, 45]. cAMP elevation, which is mimicked by the administration of cAMP mimetic compounds or promoted by rolipram (a PDE4 inhibitor), induced the resolution of both eosinophilic [45] and neutrophilic inflammation [20], which was mediated by PKA and dependent on granulocyte apoptosis. The resolution induced by increasing cAMP levels has been associated with the modulation of several molecular pathways, which are important for leukocyte survival. For example, it has been shown that high concentrations of cAMP decrease prosurvival intracellular molecules, including MCL-1, PI3K, and NF-κB and increase levels of proapoptotic molecules: BAX and cleaved caspase-3 [20]. Lower levels of cAMP may account for the lack of resolution of inflammation in a murine model of chronic granulomatous disease [48].
Emerging concepts about the role of cAMP in inflammation resolution came first from Bystrom and cols [46], who demonstrated the participation of cAMP in reprogramming inflammatory macrophages to resolution-phase macrophages. In agreement with these findings, a recent paper showed that cAMP contributes to resolution by polarizing M1 to M2 macrophages [49]. In addition to inducing macrophage reprogramming, enhanced levels of cAMP induced by binding to lysophosphatidylserine (lyso-PS) expressed on apoptotic neutrophils are also involved in efferocytosis [50, 51]. Lyso-PS acts on the macrophage G2A receptor and enhances the clearance of these neutrophils by signaling through the PKA-dependent increase of Rac1 activity via an increased production of PGE2 and cAMP [52]. However, whether the above-described mechanisms of cAMP may be applied in in vivo situations of inflammation remains to be determined.
cAMP may function as an intermediate of the effects of certain proresolutive molecules. For example, one study suggests that RvD1 is able to increase intracellular levels of cAMP and rescue macrophage apoptosis in a cAMP-dependent manner [47]. A recent study by our group demonstrated that treatment with a PDE4 inhibitor, which enhances cAMP, induced resolution of inflammation that was associated with increased levels of AnxA1 [21]. Altogether these data suggest that cAMP is a crucial control molecule in the resolution of inflammation. Not only is cAMP induced by proresolving molecules but it may also induce the release of proresolving molecules, thus acting at multiple regulatory levels to induce resolution. Therefore, cAMP-elevating drugs may represent a useful therapeutic strategy to induce the resolution of inflammation.
2.4. Phosphoinositide 3-Kinases
Phosphoinositide 3-kinases (PI3Ks) are a family of intracellular lipid kinases that phosphorylate the 3-OH group of inositol membrane lipids, thus regulating many aspects of cell function, including cell metabolism, survival, and polarity. This family can be divided into three main classes (I, II, and III) based on structural and biochemical characteristics [53, 54]. In mammals, isoforms of PI3K are related to signal transduction, and each isoform plays a different role [53]. Class I is subdivided into two subclasses, IA and IB. PI3Kγ is the only member of class IB and is the most highly expressed in cells of the immune system. This isoform is composed of the p110γ catalytic subunit and the p101 regulatory subunit and is activated by the Gβ γ subunits of G proteins [55, 56]. PI3Ks are known to be important in many cell processes related to the immune system, including cell activation, migration [55, 57, 58], and cell survival, via the phosphorylation of Akt/protein kinase B (PBK) [20, 45]; PI3Ks are also activated by antigen, cytokine, and chemokine receptors [59].
Our group has evaluated the role of PI3Kγ in inducing and maintaining the inflammatory process in experimental models. In a model of allergic pleurisy in mice, the inhibition of PI3K cleared accumulated eosinophils and increased the number of apoptotic events. Experiments using adoptive transfer of bone marrow cells showed that PI3Kγ on leukocytes was required for the maintenance of eosinophil influx at the later stages of eosinophilic inflammation [17]. These studies suggest that PI3Kγ on leukocytes is relevant for the maintenance of inflammation and that inhibitors of these enzymes could potentially impart on resolution of inflammation.
However, there are no published studies demonstrating that selective blockade of PI3Kγ is really proresolutive in vivo. Blockers of PI3Kγ may have anti-inflammatory effects in vivo, as we have shown in a model of bleomycin-induced pulmonary inflammation [19]. However, a definite demonstration that these enzymes are relevant in vivo is lacking as most published studies have only studied the effects of pan PI3K inhibitors, especially wortmannin and LY294002, in preclinical models of inflammation resolution. There are now many new selective PI3K inhibitors in development [60]. Whether such drugs with greater selectivity and safety profile will resolve inflammation in vivo needs to be studied.
2.5. Nuclear Factor Kappa B
Nuclear factor kappa B (NF-κB) is a transcription factor that regulates immune response to both injury and infection [61, 62]. NF-κB is a convergence point of several signal transduction pathways by conveying the signals of these molecules to the nucleus and promoting transcriptional activation of genes associated with inflammation and cell survival [62]. The activity of NF-κB is primarily regulated through interactions with inhibitory IκB proteins. The phosphorylation of IκB results in its proteasome degradation and the release of NF-κB (usually composed of p50/p65 heterodimers) for nuclear translocation and activation of gene transcription [61, 62]. Over 750 inhibitors of the NF-κB pathway have been identified, including a variety of natural and synthetic molecules. These molecules act by inhibiting NF-κB nuclear translocation/or transactivation or through IκB super repression [63–67].
Recently, numerous investigations have supported the role of miRNAs in the regulation of NF-κB. miRNAs have been found to be involved in NF-κB signaling by targeting NF-κB regulators and effectors [68, 69]. Recent studies have shown that NF-κB inhibitors may attenuate inflammatory parameters in different experimental models of inflammation [70]. For example, NF-κB inhibitors possess anti-inflammatory effects in models of lipopolysaccharide-induced lung injury [71], traumatic brain injury [72], colitis [73], and pulmonary arterial hypertension [74]. Fewer studies have evaluated the effects of NF-κB inhibitors in the resolution of inflammation. Our research group showed that inhibition of NF-κB promotes resolution in established murine models of neutrophilic and eosinophilic inflammation [22, 45]. The resolution of inflammation induced by NF-κB inhibitors in the models of arthritis [22] and allergic pleurisy [45] was associated with enhanced apoptosis of inflammatory cells.
NF-κB activation usually results in the upregulation of antiapoptotic genes that may lead to cell survival [64]. However, NF-κB may also control genes associated with survival and anti-inflammation [70, 75]. In this sense, a few studies have shown that NF-κB inhibitors failed to promote the resolution of inflammation [20] and actually prolonged the inflammatory response by preventing leukocyte apoptosis [70, 76]. Greten and cols also described that NF-κB could also function as a negative posttranslational regulator of inflammasome activation. Therefore, it is clear that NF-κB may have dual role, both pro- and antiresolution, in models of inflammation. This duality of function of NF-κB is likely the result of the central role of this molecule in the convergence of several inflammatory signals [70]. Whether manipulating NF-κB in inflammation will ultimately result in beneficial functions clearly deserves further investigation.
The discussion above suggests that several signaling pathways have been implicated in leukocyte survival during inflammatory response. It is important to note that each specific molecule associated with a signaling pathway cascade may not work in a disconnected manner. Crosstalk between signaling pathways is likely to be essential for leukocyte survival and much more work is needed to understand the interaction between potential resolution inducing pathways, especially in the context of the complex in vivo situation of an inflammatory response. However, as demonstrated above, there are molecules which are crucial for resolution of inflammation and whose effects may be indeed exploited therapeutically [77, 78].
3. Molecules Involved in Apoptosis of Granulocytes and Resolution of the Inflammatory Response
3.1. Annexin A1
Annexin A1 (AnxA1) is a 37 kDa glucocorticoid-induced protein firstly identified by its action on phospholipase- (PL-) A2 inhibition and prevention of eicosanoid synthesis [79]. AnxA1 is a protein member of the annexin superfamily, which exerts its anti-inflammatory activity by binding to receptor ALX (named FPR2, formyl peptide receptor-2, murine), which is also shared with lipoxins [80]. During the initial steps of acute inflammation, AnxA1 limits the recruitment of leukocytes and the production of proinflammatory mediators [80]. During the resolution phase, AnxA1 acts by inducing the apoptosis of neutrophils and this effect is associated with increased expression of cleaved caspase-3 and BAX and decreased expression of pERK1/2, NF-κB, and MCL-1 [21, 81–83] and increasing efferocytosis by macrophages [83–85]. Interestingly, activation of FPR2 by AnxA1 and LXA4 skewed M1 macrophages to M2-like cells [86]. In this context of macrophage modulation, it was demonstrated that AnxA1 released from apoptotic cells contributed to the immunomodulatory effect of these cells on inflammation cells by damping inflammatory monocyte activation [87]. Additionally, AnxA1 may induce indirectly the chemoattraction of monocytes [88]. These effects—migration of monocytes and skewing towards a M2-like phenotype—may be relevant in the context of inflammation but remain to be determined in vivo.
The N-terminal region of AnxA1 is the major effector portion responsible for the anti-inflammatory action of the protein [80]. However, once in the extravascular space, the major part of the active AnxA1 (37 kDa) contained in neutrophils is cleaved by proteases, particularly elastase and proteinase-3, yielding the inactive AnxA1 (of 33 kDa form) [89, 90]. As a strategy to deliver anxA1 in vivo, we and others have explored the therapeutic potential of an AnxA1 peptidomimetic Ac2-26, which retains the biological activity of the entire protein. In a model of acute inflammation triggered by LPS, the administration of Ac2-26 at the peak of inflammation resolved inflammation by inducing caspase-dependent apoptosis of inflammatory cells [21]; this mechanism was also demonstrated using a longer peptide, AnxA12–50 [83]. An AnxA1 cleavage-resistant protein and an AnxA12–50 peptide with a mutation on the cleavage site were demonstrated to be more effective in improving several parameters of inflammation compared with a full length protein and a peptide that was not mutated at the sites of proteases action [83, 90].
One intriguing characteristic of the FPR receptor is that it recognizes both proinflammatory and proresolving signals, thus integrating contrasting cues to determine the course of inflammation [91, 92]. Cooray and cols revealed this intriguing receptor characteristic and showed that the anti-inflammatory signal triggered by AnxA1 (Ac2-26 peptide) and LXA4 promotes FPR2 homodimerization and resolution activities by inducing p38-induced IL-10 production; this stands in contrast with proinflammatory signals, such as SAA, that bind to the receptor alone. Interestingly, Ac2-26 peptide, which is a nonselective FPR ligand (binding to both FPR2 and FPR1), is able to promote the dimerization of FPR1 and FPR2 and change the proinflammatory nature of FPR1 by transducing JNK/caspase-3 proapoptotic signal and promoting resolution of inflammation [93]. These latter findings would help explain the restorative role of Ac2-26 peptide by acting through FPR1 and orchestrating epithelial repair in a model of mucosal inflammation [94]. Thus, AnxA1, its peptidomimetics, or AnxA1-inducer drugs may have great therapeutic potential as resolution inducing drugs in vivo.
3.2. Hydrogen Peroxide
The nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) expressed in phagocytes is a multisubunit enzyme complex that generates hydrogen peroxide (H2O2) and other reactive oxygen species (ROS) [95]. Accumulating data has suggested that ROS are not merely injurious but can also downregulate inflammation and contribute to the maintenance of tissue homeostasis [96, 97]. Consistent with this observation, various lines of evidence have indicated a critical role of H2O2 for the natural resolution of inflammation and regeneration/repair of tissue by inducing apoptosis in different cell types such as neutrophil [22], hepatocyte [98], myocytes [97], and endothelial [99] and lymphoma cells [100].
Our group has investigated the effects of H2O2 in the context of the resolution of inflammation. Lopes et al. demonstrated that H2O2 resolves neutrophilic inflammation by activating BAX and caspase-3 and the shutting down NF-κB and PI3K pathways. Consistently with the latter observation, deficiency of the gp91phox component of NADPH oxidase was associated with increased inflammation in a model of antigen-induced arthritis. In vitro, H2O2 has been shown to induce programmed cell death in various cell types, including leukocytes. In these cells, H2O2 appears to decrease survival signaling pathways, including PI3K/Akt, the transcription factor NF-κB, and mitochondrial pathways [22, 101–104]. The situation in vivo is much less well known and studies will be needed to determine the precise molecular pathways that control H2O2 production and the extracellular and intracellular signaling mechanisms through which H2O2 promotes resolution of inflammation. In this sense, a recent study showing that H2O2 may induce the expression of AnxA1 raises the hypothesis that AnxA1 could be involved in the proresolving abilities of this molecule [105]. Therefore, although the proresolving role of H2O2 is of great interest, further studies on its source and mechanisms of action are clearly needed.
3.3. TNF-Related Apoptosis-Inducing Ligand
The TNF-related apoptosis-inducing ligand (TRAIL) is a cytokine that belongs to the TNF superfamily that was discovered in 1995 [106–108]. TRAIL is able to interact with two proapoptotic death receptors, TRAIL-R1/DR4 and TRAIL-R2/DR5, as well as three decoy receptors without functional death domains [109, 110]. The role of TRAIL in biological systems is complex. Several studies have demonstrated a key role of TRAIL in controlling a number of different types of cancer [109–115]. However, some studies have shown that TRAIL has important functions in the immune system, including an immunoregulatory function [116–120]. TRAIL is also involved in the control of some autoimmune diseases [121, 122]. For example, the neutralization of endogenous TRAIL may prevent the resolution of granulomatous experimental autoimmune thyroiditis [123].
A few studies have demonstrated that TRAIL is able to promote apoptosis of human and murine neutrophils [108, 124] and may, hence, promote inflammation resolution. In this regard, it has been shown that the duration of neutrophilic inflammation is enhanced in TRAIL-deficient mice [108]. In addition, TRAIL-deficient mice showed an aberrant inflammatory response associated with reduced apoptosis of inflammatory cells and increased collagen deposition in a model of chronic pulmonary inflammation induced by bleomycin [125]. TRAIL was also found to modulate allergic inflammation. The treatment with antiTRAIL antibody blocked apoptosis of T helper type 2 (Th2) cells and eosinophils and enhanced the inflammatory response [126]. Although conclusive evidence is lacking for a role of TRAIL in the resolution of inflammation [127], the effects of TRAIL are associated with the apoptosis of leukocytes, suggesting TRAIL's potential therapeutic use for the treatment of established inflammatory diseases. Agonistic antibodies have been produced to treat many cancers; the monoclonal antiTRAIL-R1 antibody (mapatumumab) [128] is currently in clinical development, and five antiTRAIL-R2 antibodies are also being tested (lexatumumab, Apomab, TRA-8, AMG 655, and LBY135) [129–133]. Thus, these antibodies could potentially be explored in the context of inflammation resolution.
4. Concluding Remarks
Given the importance of inflammation and its resolution, many studies have sought to better understand the molecular scenario involved in these processes. Some of the actors involved in the resolution of inflammation were mentioned in the present review and were demonstrated to be potential targets of therapeutic approaches. The resolution of inflammation is a vital process that enables the return to homeostasis of the immune system and the organ affected by inflammation, avoiding the development of chronic and autoimmune diseases [8, 134]. In vitro studies provide essential information about molecular machinery that helps to elucidate how different intracellular molecules control leukocyte survival in inflammatory sites. However, they do not cover the entire complexity of in vivo settings, which include intracellular pathways and molecules that are interrelated or codependent [78]. Moreover, resolution of inflammation, an in vivo phenomenon, is much more complex than simple apoptosis of leukocytes and includes switching off proinflammatory pathways, efferocytosis, and the function of cells other than leukocytes. For example, some proresolving mediators act in macrophages by rescuing from apoptosis and by activating them to induce phagocytosis of apoptotic leukocytes (efferocytosis) [53]. There is also evidence to suggest that nonhematopoietic cells may be involved in the context of the resolution of inflammation [135]. However, studies evaluating the role of epithelial cells and other nonprofessional phagocytic cells in the resolution of inflammation are lacking.
Future studies should try to integrate current findings with single signaling molecules with more complex signaling pathways and how they interact with each other, all of these in the complex in vivo situation. Consideration should be given not only to pathways associated with apoptosis of leukocytes but also to molecules and pathways associated with triggering efferocytosis. One must also keep in mind that cells other than leukocytes may respond to resolution inducing molecules in vivo by releasing secondary mediators which themselves could be more relevant as final mediators of resolution. New animal models in which natural resolution of inflammation does not occur are clearly needed, especially in models accompanied by a degree of chronic fibrosis. Most reported studies have been performed in systems in which resolution eventually occurs. Whether strategies which speed the resolution of inflammation will also resolve inflammation and prevent or reverse fibrosis in a nonresolving chronic model needs to be determined. Finally, one will also need to take the difficult task of translating experimental findings into human diseases. Whether proresolving strategies will be accompanied by significant degree of immunosuppression is currently not known and will need to be addressed in the future.
Acknowledgments
The authors would like to acknowledge funding from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Comissão de Aperfeiçoamento de Pessoal do Ensino Superior (CAPES, Brazil), Fundação do Amparo a Pesquisa de Minas Gerais (FAPEMIG, Brazil), The Instituto Nacional de Ciência e Tecnologia (INCT in Dengue), and the European Community's Seventh Framework Programme (FP7-2007-2013, Timer consortium) under Grant agreement HEALTH-F4-2011-281608.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Lirlândia Pires Sousa and Vanessa Pinho contributed equally to the coordination of this work.
References
- 1.Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140(6):771–776. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Katzenstein AA, Myers JL. Idiopathic pulmonary fibrosis: Clinical relevance of pathologic classification. The American Journal of Respiratory and Critical Care Medicine. 1998;157(4):1301–1315. doi: 10.1164/ajrccm.157.4.9707039. [DOI] [PubMed] [Google Scholar]
- 3.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nature Reviews Drug Discovery. 2004;3(5):401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nature Immunology. 2005;6(12):1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 5.Isobe Y, Kato T, Arita M. Emerging roles of eosinophils and eosinophil-derived lipid mediators in the resolution of inflammation. Frontiers in Immunology. 2012;3 doi: 10.3389/fimmu.2012.00270.Article 270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chertov O, Yang D, Zack Howard OM, Oppenheim JJ. Leukocyte granule proteins mobilize innate host defenses and adaptive immune responses. Immunological Reviews. 2000;177:68–78. doi: 10.1034/j.1600-065x.2000.17702.x. [DOI] [PubMed] [Google Scholar]
- 7.Nathan C, Ding A. Nonresolving Inflammation. Cell. 2010;140(6):871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 8.Serhan CN, Brain SD, Buckley CD, et al. Resolution of inflammation: state of the art, definitions and terms. The FASEB Journal. 2007;21(2):325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40(3):315–327. doi: 10.1016/j.immuni.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gautier EL, Ivanov S, Lesnik P, et al. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood. 2013;122(15):2714–2722. doi: 10.1182/blood-2013-01-478206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. Journal of Clinical Investigation. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ariel A, Serhan CN. New lives given by cell death: Macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Frontiers in Immunology. 2012;3:p. 4. doi: 10.3389/fimmu.2012.00004.Article 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham SM, Clark AR. Dual-specificity phosphatase 1: a critical regulator of innate immune responses. Biochemical Society Transactions. 2006;34, part 6:1018–1023. doi: 10.1042/BST0341018. [DOI] [PubMed] [Google Scholar]
- 14.Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. Journal of Biological Chemistry. 1997;272(27):16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- 15.Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. Journal of Immunology. 2002;169(11):6408–6416. doi: 10.4049/jimmunol.169.11.6408. [DOI] [PubMed] [Google Scholar]
- 16.Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. Journal of Immunology. 2006;176(3):1899–1907. doi: 10.4049/jimmunol.176.3.1899. [DOI] [PubMed] [Google Scholar]
- 17.Pinho V, Souza DG, Barsante MM, et al. Phosphoinositide-3 kinases critically regulate the recruitment and survival of eosinophils in vivo: importance for the resolution of allergic inflammation. Journal of Leukocyte Biology. 2005;77(5):800–810. doi: 10.1189/jlb.0704386. [DOI] [PubMed] [Google Scholar]
- 18.Pinho V, Russo RDC, Amaral FA, et al. Tissue- and stimulus-dependent role of phosphatidylinositol 3-kinase isoforms for neutrophil recruitment induced by chemoattractants in vivo. Journal of Immunology. 2007;179(11):7891–7898. doi: 10.4049/jimmunol.179.11.7891. [DOI] [PubMed] [Google Scholar]
- 19.Russo RC, Garcia CC, Barcelos LS, et al. Phosphoinositide 3-kinase γ plays a critical role in bleomycin-induced pulmonary inflammation and fibrosis in mice. Journal of Leukocyte Biology. 2011;89(2):269–282. doi: 10.1189/jlb.0610346. [DOI] [PubMed] [Google Scholar]
- 20.Sousa LP, Lopes F, Silva DM, et al. PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-κB-independent manner. Journal of Leukocyte Biology. 2010;87(5):895–904. doi: 10.1189/jlb.0809540. [DOI] [PubMed] [Google Scholar]
- 21.Sousa LP, Vago JP, Nogueira CRC, et al. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. Journal of Leukocyte Biology. 2012;92(2):249–258. doi: 10.1189/jlb.0112008. [DOI] [PubMed] [Google Scholar]
- 22.Lopes F, Coelho FM, Costa VV, et al. Resolution of neutrophilic inflammation by H2O2 in antigen-induced arthritis. Arthritis and Rheumatism. 2011;63(9):2651–2660. doi: 10.1002/art.30448. [DOI] [PubMed] [Google Scholar]
- 23.Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends in Pharmacological Sciences. 2002;23(9):417–425. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- 24.Rossi AG, Sawatzky DA, Walker A, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nature Medicine. 2006;12(9):1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- 25.Leitch AE, Haslett C, Rossi AG. Cyclin-dependent kinase inhibitor drugs as potential novel anti-inflammatory and pro-resolution agents. British Journal of Pharmacology. 2009;158(4):1004–1016. doi: 10.1111/j.1476-5381.2009.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leitch AE, Lucas CD, Marwick JA, Duffin R, Haslett C, Rossi AG. Cyclin-dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death and Differentiation. 2012;19(12):1950–1961. doi: 10.1038/cdd.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leitch AE, Riley NA, Sheldrake TA, et al. The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signalling and drive neutrophil apoptosis. European Journal of Immunology. 2010;40(4):1127–1138. doi: 10.1002/eji.200939664. [DOI] [PubMed] [Google Scholar]
- 28.Rossi M, Duan S, Jeong Y, et al. Regulation of the CRL4(Cdt2) ubiquitin ligase and cell-cycle exit by the SCFFbxo11 ubiquitin ligase. Molecular Cell. 2013;49(6):1159–1166. doi: 10.1016/j.molcel.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duffin R, Leitch AE, Sheldrake TA, et al. The CDK inhibitor, R-roscovitine, promotes eosinophil apoptosis by down-regulation of Mcl-1. The FEBS Letters. 2009;583(15):2540–2546. doi: 10.1016/j.febslet.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 30.Koedel U, Frankenberg T, Kirschnek S, et al. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathogens. 2009;5(5) doi: 10.1371/journal.ppat.1000461.e1000461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alessandri AL, Duffin R, Leitch AE, et al. Induction of eosinophil apoptosis by the cyclin-dependent kinase inhibitor AT7519 promotes the resolution of eosinophil-dominant allergic inflammation. PLoS ONE. 2011;6(9) doi: 10.1371/journal.pone.0025683.e25683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoogendijk AJ, Roelofs JJTH, Duitman J, et al. R-roscovitine reduces lung inflammation induced by Lipoteichoic acid and Streptococcus pneumoniae. Molecular Medicine. 2012;18(7):1086–1095. doi: 10.2119/molmed.2012.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahadevan D, Plummer R, Squires MS, et al. A phase I pharmacokinetic and pharmacodynamic study of AT7519, a cyclin-dependent kinase inhibitor in patients with refractory solid tumors. Annals of Oncology. 2011;22(9):2137–2143. doi: 10.1093/annonc/mdq734. [DOI] [PubMed] [Google Scholar]
- 34.Lucas CD, Dorward DA, Tait MA, et al. Downregulation of Mcl-1 has anti-inflammatory pro-resolution effects and enhances bacterial clearance from the lung. Mucosal Immunology. 2014;7:857–868. doi: 10.1038/mi.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pearson G, Robinson F, Gibson TB, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrine Reviews. 2001;22(2):153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 36.Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochimica et Biophysica Acta—Proteins and Proteomics. 2005;1754(1-2):253–262. doi: 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Duan W, Chan JHP, Wong CH, Leung BP, Wong WSF. Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. Journal of Immunology. 2004;172(11):7053–7059. doi: 10.4049/jimmunol.172.11.7053. [DOI] [PubMed] [Google Scholar]
- 38.Schuh K, Pahl A. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochemical Pharmacology. 2009;77(12):1827–1834. doi: 10.1016/j.bcp.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 39.Sawatzky DA, Willoughby DA, Colville-Nash PR, Rossi AG. The involvement of the apoptosis-modulating proteins ERK 1/2, Bcl-x L and Bax in the resolution of acute inflammation in vivo. The American Journal of Pathology. 2006;168(1):33–41. doi: 10.2353/ajpath.2006.050058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacKenzie KF, van den Bosch MWM, Naqvi S, et al. MSK1 and MSK2 inhibit lipopolysaccharide-induced prostaglandin production via an interleukin-10 feedback loop. Molecular and Cellular Biology. 2013;33(7):1456–1467. doi: 10.1128/MCB.01690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soderling SH, Beavo JA. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Current Opinion in Cell Biology. 2000;12(2):174–179. doi: 10.1016/s0955-0674(99)00073-3. [DOI] [PubMed] [Google Scholar]
- 42.Schudt C, Hatzelmann A, Beume R, Tenor H. Phosphodiesterases as Drug Targets. Vol. 204. Berlin, Germany: Springer; 2011. Phosphodiesterase inhibitors: history of pharmacology; pp. 1–46. (Handbook of Experimental Pharmacology). [DOI] [PubMed] [Google Scholar]
- 43.Teixeira MM, Gristwood RW, Cooper N, Hellewell PG. Phosphodiesterase (PDE)4 inhibitors: anti-inflammatory drugs of the future? Trends in Pharmacological Sciences. 1997;18(5):164–170. doi: 10.1016/s0165-6147(97)01049-3. [DOI] [PubMed] [Google Scholar]
- 44.Souza DG, Cassali GD, Poole S, Teixeira MM. Effects of inhibition of PDE4 and TNF-α on local and remote injuries following ischaemia and reperfusion injury. British Journal of Pharmacology. 2001;134(5):985–994. doi: 10.1038/sj.bjp.0704336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sousa LP, Carmo AF, Rezende BM, et al. Cyclic AMP enhances resolution of allergic pleurisy by promoting inflammatory cell apoptosis via inhibition of PI3K/Akt and NF-κB. Biochemical Pharmacology. 2009;78(4):396–405. doi: 10.1016/j.bcp.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 46.Bystrom J, Evans I, Newson J, et al. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood. 2008;112(10):4117–4127. doi: 10.1182/blood-2007-12-129767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HN, Surh YJ. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochemical Pharmacology. 2013;86(6):759–769. doi: 10.1016/j.bcp.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 48.Rajakariar R, Newson J, Jackson EK, et al. Nonresolving inflammation in gp91phox-/- mice, a model of human chronic granulomatous disease, has lower adenosine and cyclic adenosine 5′-monophosphate. Journal of Immunology. 2009;182(5):3262–3269. doi: 10.4049/jimmunol.0801739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheldon KE, Shandilya H, Kepka-Lenhart D, Poljakovic M, Ghosh A, Morris SM., Jr. Shaping the murine macrophage phenotype: IL-4 and cyclic AMP synergistically activate the arginase I promoter. The Journal of Immunology. 2013;191(5):2290–2298. doi: 10.4049/jimmunol.1202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frasch SC, Fernandez-Boyanapalli RF, Berry KZ, et al. Signaling via macrophage G2A enhances efferocytosis of dying neutrophils by augmentation of rac activity. Journal of Biological Chemistry. 2011;286(14):12108–12122. doi: 10.1074/jbc.M110.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frasch SC, Berry KZ, Fernandez-Boyanapalli R, et al. NADPH oxidase-dependent generation of lysophosphatidylserine enhances clearance of activated and dying neutrophils via G2A. The Journal of Biological Chemistry. 2008;283(48):33736–33749. doi: 10.1074/jbc.M807047200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frasch SC, Bratton DL. Emerging roles for lysophosphatidylserine in resolution of inflammation. Progress in Lipid Research. 2012;51(3):199–207. doi: 10.1016/j.plipres.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature Reviews Genetics. 2006;7(8):606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 54.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nature Reviews Molecular Cell Biology. 2012;13(3):195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 55.Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annual Review of Immunology. 2004;22:563–598. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- 56.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Experimental Cell Research. 1999;253(1):239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 57.Sasaki T, Irie-Sasaki J, Jones RG, et al. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287(5455):1040–4046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 58.Hirsch E, Katanaev VL, Garlanda C, et al. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science. 2000;287(5455):1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 59.Fruman DA, Cantley LC. Phosphoinositide 3-kinase in immunological systems. Seminars in Immunology. 2002;14(1):7–18. doi: 10.1006/smim.2001.0337. [DOI] [PubMed] [Google Scholar]
- 60.Shao T, Wang J, Chen JG, et al. Discovery of 2-methoxy-3-phenylsulfonamino-5-(quinazolin-6-yl or quinolin-6-yl)benzamides as novel PI3K inhibitors and anticancer agents by bioisostere. European Journal of Medicinal Chemistry. 2014;75:96–105. doi: 10.1016/j.ejmech.2014.01.053. [DOI] [PubMed] [Google Scholar]
- 61.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes & Development. 2012;26(3):203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends in Immunology. 2004;25(6):280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 63.Gilmore TD, Herscovitch M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene. 2006;25(51):6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 64.Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Molecular Cancer. 2013;12(1, article 86) doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu Q, Wu H, Chim SM, et al. SC-514, a selective inhibitor of IKKβ attenuates RANKL-induced osteoclastogenesis and NF-κB activation. Biochemical Pharmacology. 2013;86(12):1775–1783. doi: 10.1016/j.bcp.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 66.Watanabe R, Azuma RW, Suzuki J, et al. Inhibition of NF-kappaB activation by a novel IKK inhibitor reduces the severity of experimental autoimmune myocarditis via suppression of T-cell activation. The American Journal of Physiology—Heart and Circulatory Physiology. 2013;305(12):H1761–H1771. doi: 10.1152/ajpheart.00159.2013. [DOI] [PubMed] [Google Scholar]
- 67.Lee CS, Jeong EB, Kim YJ, et al. Quercetin-3-O-(2′′-galloyl)-α-l-rhamnopyranoside inhibits TNF-α-activated NF-κB-induced inflammatory mediator production by suppressing ERK activation. International Immunopharmacology. 2013;16(4):481–487. doi: 10.1016/j.intimp.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 68.Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-κB signaling. Journal of Molecular Cell Biology. 2011;3(3):159–166. doi: 10.1093/jmcb/mjr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boldin MP, Baltimore D. MicroRNAs, new effectors and regulators of NF-κB. Immunological Reviews. 2012;246(1):205–220. doi: 10.1111/j.1600-065X.2011.01089.x. [DOI] [PubMed] [Google Scholar]
- 70.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-κB in the resolution of inflammation. Nature Medicine. 2001;7(12):1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 71.Chian CF, Chiang CH, Chuang CH, et al. Inhibitor of nuclear factor-kappaB, SN50, attenuates lipopolysaccharide-induced lung injury in an isolated and perfused rat lung model. Translational Research. 2014;163(3):211–220. doi: 10.1016/j.trsl.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 72.Sun YX, Dai DK, Liu R, et al. Therapeutic effect of SN50, an inhibitor of nuclear factor-κB, in treatment of TBI in mice. Neurological Sciences. 2013;34(3):345–355. doi: 10.1007/s10072-012-1007-z. [DOI] [PubMed] [Google Scholar]
- 73.El-Salhy M, Umezawa K, Gilja OH, Hatlebakk JG, Gundersen D, Hausken T. Amelioration of severe TNBS induced colitis by novel AP-1 and NF-κB inhibitors in rats. The Scientific World Journal. 2014;2014:8 pages. doi: 10.1155/2014/813804.813804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hosokawa S, Haraguchi G, Sasaki A, et al. Pathophysiological roles of nuclear factor kappaB (NF-kB) in pulmonary arterial hypertension: effects of synthetic selective NF-kB inhibitor IMD-0354. Cardiovascular Research. 2013;99(1):35–43. doi: 10.1093/cvr/cvt105. [DOI] [PubMed] [Google Scholar]
- 75.Lawrence T, Fong C. The resolution of inflammation: anti-inflammatory roles for NF-κB. International Journal of Biochemistry and Cell Biology. 2010;42(4):519–523. doi: 10.1016/j.biocel.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 76.Greten FR, Arkan MC, Bollrath J, et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ . Cell. 2007;130(5):918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sousa LP, Alessandri AL, Pinho V, et al. Pharmacological strategies to resolve acute inflammation. Current Opinion in Pharmacology. 2013;13(4):625–631. doi: 10.1016/j.coph.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 78.Alessandri AL, Sousa LP, Lucas CD, Rossi AG, Pinho V, Teixeira MM. Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacology and Therapeutics. 2013;139(2):189–212. doi: 10.1016/j.pharmthera.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 79.Flower RJ, Blackwell GJ. Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nature. 1979;278(5703):456–459. doi: 10.1038/278456a0. [DOI] [PubMed] [Google Scholar]
- 80.Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nature Reviews Immunology. 2009;9(1):62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 81.Parente L, Solito E. Annexin 1: more than an anti-phospholipase protein. Inflammation Research. 2004;53(4):125–132. doi: 10.1007/s00011-003-1235-z. [DOI] [PubMed] [Google Scholar]
- 82.Solito E, Kamal A, Russo-Marie F, Buckingham JC, Marullo S, Perretti M. A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. The FASEB Journal. 2003;17(11):1544–1546. doi: 10.1096/fj.02-0941fje. [DOI] [PubMed] [Google Scholar]
- 83.Dalli J, Consalvo AP, Ray V, et al. Proresolving and tissue-protective actions of annexin A1-based cleavage-resistant peptides are mediated by formyl peptide receptor 2/lipoxin A4 receptor. Journal of Immunology. 2013;190(12):6478–6487. doi: 10.4049/jimmunol.1203000. [DOI] [PubMed] [Google Scholar]
- 84.Maderna P, Yona S, Perretti M, Godson C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac2-26. Journal of Immunology. 2005;174(6):3727–3733. doi: 10.4049/jimmunol.174.6.3727. [DOI] [PubMed] [Google Scholar]
- 85.Dalli J, Jones CP, Cavalcanti DM, Farsky SH, Perretti M, Rankin SM. Annexin A1 regulates neutrophil clearance by macrophages in the mouse bone marrow. The FASEB Journal. 2012;26(1):387–396. doi: 10.1096/fj.11-182089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y, Cai L, Wang H, et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene. 2011;30(36):3887–3899. doi: 10.1038/onc.2011.112. [DOI] [PubMed] [Google Scholar]
- 87.Pupjalis D, Goetsch J, Kottas DJ, Gerke V, Rescher U. Annexin A1 released from apoptotic cells acts through formyl peptide receptors to dampen inflammatory monocyte activation via JAK/STAT/SOCS signalling. EMBO Molecular Medicine. 2011;3(2):102–114. doi: 10.1002/emmm.201000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blume KE, Soeroes S, Keppeler H, et al. Cleavage of annexin A1 by ADAM10 during secondary necrosis generates a monocytic “find-me” signal. Journal of Immunology. 2012;188(1):135–145. doi: 10.4049/jimmunol.1004073. [DOI] [PubMed] [Google Scholar]
- 89.Oliani SM, Perretti M. Cell localization of the anti-inflammatory protein annexin 1 during experimental inflammatory response. Italian Journal of Anatomy and Embryology. 2001;106(2, supplement 1):69–77. [PubMed] [Google Scholar]
- 90.Pederzoli-Ribeil M, Maione F, Cooper D, et al. Design and characterization of a cleavage-resistant Annexin A1 mutant to control inflammation in the microvasculature. Blood. 2010;116(20):4288–4296. doi: 10.1182/blood-2010-02-270520. [DOI] [PubMed] [Google Scholar]
- 91.Filep JG. Resolution of inflammation: leukocytes and molecular pathways as potential therapeutic targets. Frontiers in Immunology. 2013;4:p. 256. doi: 10.3389/fimmu.2013.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Filep JG. Glucocorticoid protection against myocardial ischemia-reperfusion injury: central role for the PGD2-Nrf2 pathway. Hypertension. 2014;63(1):22–23. doi: 10.1161/HYPERTENSIONAHA.113.01832. [DOI] [PubMed] [Google Scholar]
- 93.Cooray SN, Gobbetti T, Montero-Melendez T, et al. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(45):18232–18237. doi: 10.1073/pnas.1308253110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leoni G, Alam A, Alexander Neumann P, et al. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. The Journal of Clinical Investigation. 2013;123(1):443–454. doi: 10.1172/JCI65831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gardiner GJ, Deffit SN, McLetchie S, et al. A role for NADPH oxidase in antigen presentation. Frontiers in Immunology. 2013;4, article 295 doi: 10.3389/fimmu.2013.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van der Vliet A, Janssen-Heininger YM. Hydrogen peroxide as a damage signal in tissue injury and inflammation: murderer, mediator, or messenger? Journal of Cellular Biochemistry. 2014;115(3):427–435. doi: 10.1002/jcb.24683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang K, Jiang L, Deng G, et al. Heat shock protein 70 interacts with nucleolin and inhibits its cleavage, down-regulation and apoptosis induced by hydrogen peroxide in myocytes. The Journal of Biological Chemistry. 2010 doi: 10.1074/jbc.A109.009480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim S, Jung H, Hyun D, Park E, Kim Y, Lim C. Glutathione reductase plays an anti-apoptotic role against oxidative stress in human hepatoma cells. Biochimie. 2010;92(8):927–932. doi: 10.1016/j.biochi.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 99.Fang W, Li H, Zhou L. Protective effects of prostaglandin e 1 on human umbilical vein endothelial cell injury induced by hydrogen peroxide. Acta Pharmacologica Sinica. 2010;31(4):485–492. doi: 10.1038/aps.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Klamt F, Zdanov S, Levine RL, et al. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nature Cell Biology. 2009;11(10):1241–1246. doi: 10.1038/ncb1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li J, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40(4):477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 102.Chen C, Chow M, Huang W, Lin Y, Chang Y. Flavonoids inhibit tumor necrosis factor-α-induced up-regulation of intercellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells through activator protein-1 and nuclear factorκB: Structure-activity relationships. Molecular Pharmacology. 2004;66(3):683–693. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 103.Carreras MC, Poderoso JJ. Mitochondrial nitric oxide in the signaling of cell integrated responses. The American Journal of Physiology—Cell Physiology. 2007;292(5):C1569–C1580. doi: 10.1152/ajpcell.00248.2006. [DOI] [PubMed] [Google Scholar]
- 104.Groeger G, Quiney C, Cotter TG. Hydrogen peroxide as a cell-survival signaling molecule. Antioxidants and Redox Signaling. 2009;11(11):2655–2671. doi: 10.1089/ars.2009.2728. [DOI] [PubMed] [Google Scholar]
- 105.Yu CJ, Ko CJ, Hsieh CH, et al. Proteomic analysis of osteoarthritic chondrocyte reveals the hyaluronic acid-regulated proteins involved in chondroprotective effect under oxidative stress. Journal of Proteomics. 2014;99:40–53. doi: 10.1016/j.jprot.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 106.Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3(6):673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 107.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. The Journal of Biological Chemistry. 1996;271(22):12687–12690. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 108.McGrath EE, Marriott HM, Lawrie A, et al. TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. Journal of Leukocyte Biology. 2011;90(5):855–865. doi: 10.1189/jlb.0211062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pennarun B, Kleibeuker JH, Oenema T, Stegehuis JH, de Vries EGE, de Jong S. Inhibition of IGF-1R-dependent PI3K activation sensitizes colon cancer cells specifically to DR5-mediated apoptosis but not to rhTRAIL. Analytical Cellular Pathology. 2010;33(5-6):229–244. doi: 10.3233/ACP-CLO-2010-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pennarun B, Meijer A, de Vries EGE, Kleibeuker JH, Kruyt F, de Jong S. Playing the DISC: turning on TRAIL death receptor-mediated apoptosis in cancer. Biochimica et Biophysica Acta. 2010;1805(2):123–140. doi: 10.1016/j.bbcan.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 111.Zhou K, Yan Y, Zhao S. Esophageal cancer-selective expression of TRAIL mediated by MREs of miR-143 and miR-122. Tumor Biology. 2014;35(6):5787–5795. doi: 10.1007/s13277-014-1768-5. [DOI] [PubMed] [Google Scholar]
- 112.Liu Y, Yang Z, Gong C, et al. Quercetin enhances apoptotic effect of TRAIL in ovarian cancer cells through ROS mediated CHOP-death receptor 5 pathway. Cancer Science. 2014;105(5):520–527. doi: 10.1111/cas.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lin WC, Tsai HF, Liao HJ, et al. Helicobacter pylori sensitizes TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human gastric epithelial cells through regulation of FLIP. Cell Death and Disease. 2014;5 doi: 10.1038/cddis.2014.81.e1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Holland PM. Death receptor agonist therapies for cancer, which is the right TRAIL? Cytokine & Growth Factor Reviews. 2014;25(2):185–193. doi: 10.1016/j.cytogfr.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 115.Jennewein C, Karl S, Baumann B, Micheau O, Debatin K-M, Fulda S. Identification of a novel pro-apoptotic role of NF-κB in the regulation of TRAIL- and CD95-mediated apoptosis of glioblastoma cells. Oncogene. 2012;31(11):1468–1474. doi: 10.1038/onc.2011.333. [DOI] [PubMed] [Google Scholar]
- 116.Collison A, Foster PS, Mattes J. Emerging role of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) as a key regulator of inflammatory responses. Clinical and Experimental Pharmacology and Physiology. 2009;36(11):1049–1053. doi: 10.1111/j.1440-1681.2009.05258.x. [DOI] [PubMed] [Google Scholar]
- 117.Aktas O, Schulze-Topphoff U, Zipp F. The role of TRAIL/TRAIL receptors in central nervous system pathology. Frontiers in Bioscience. 2007;12(8):2912–2921. doi: 10.2741/2281. [DOI] [PubMed] [Google Scholar]
- 118.Steinwede K, Henken S, Bohling J, et al. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. Journal of Experimental Medicine. 2012;209(11):1937–1952. doi: 10.1084/jem.20120983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zheng SJ, Jiang J, Shen H, Chen YH. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. Journal of Immunology. 2004;173(9):5652–5658. doi: 10.4049/jimmunol.173.9.5652. [DOI] [PubMed] [Google Scholar]
- 120.Benedict CA, Ware CF. Trail: not just for tumors anymore? The Journal of Experimental Medicine. 2012;209(11):1903–1906. doi: 10.1084/jem.20122235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Anel A, Martinez-Lostao L. Apo2l/trail: New insights in the treatment of autoimmune disorders. Recent Patents on Inflammation and Allergy Drug Discovery. 2011;5(3):184–199. doi: 10.2174/187221311797264883. [DOI] [PubMed] [Google Scholar]
- 122.Salehi E, Vodjgani M, Massoud A, et al. Increased expression of TRAIL and its receptors on peripheral T-cells in type 1 diabetic patients. Iranian Journal of Immunology. 2007;4(4):197–205. [PubMed] [Google Scholar]
- 123.Fang Y, Sharp GC, Yagita H, Braley-Mullen H. A critical role for TRAIL in resolution of granulomatous experimental autoimmune thyroiditis. Journal of Pathology. 2008;216(4):505–513. doi: 10.1002/path.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Renshaw SA, Parmar JS, Singleton V, et al. Acceleration of human neutrophil apoptosis by TRAIL. The Journal of Immunology. 2003;170(2):1027–1033. doi: 10.4049/jimmunol.170.2.1027. [DOI] [PubMed] [Google Scholar]
- 125.McGrath EE, Lawrie A, Marriott HM, et al. Deficiency of tumour necrosis factor-related apoptosis-inducing ligand exacerbates lung injury and fibrosis. Thorax. 2012;67(9):796–803. doi: 10.1136/thoraxjnl-2011-200863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Faustino L, Fonseca DM, Florsheim EB, et al. Tumor necrosis factor-related apoptosis-inducing ligand mediates the resolution of allergic airway inflammation induced by chronic allergen inhalation. Mucosal Immunology. 2014 doi: 10.1038/mi.2014.9. [DOI] [PubMed] [Google Scholar]
- 127.Leitch AE, Lucas CD, Rossi AG. Editorial: neutrophil apoptosis: hot on the TRAIL of inflammatory resolution. Journal of Leukocyte Biology. 2011;90(5):841–843. doi: 10.1189/jlb.0511222. [DOI] [PubMed] [Google Scholar]
- 128.von Pawel J, Harvey JH, Spigel DR, et al. Phase II trial of mapatumumab, a fully human agonist monoclonal antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1), in combination with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Clinical Lung Cancer. 2013;15(3):188.e2–196.e2. doi: 10.1016/j.cllc.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 129.Engesæter B, Engebraaten O, Flørenes VA, Mælandsmo GM. Dacarbazine and the agonistic TRAIL receptor-2 antibody lexatumumab induce synergistic anticancer effects in melanoma. PLoS ONE. 2012;7(9) doi: 10.1371/journal.pone.0045492.e45492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jin H, Yang R, Ross J, et al. Cooperation of the agonistic DR5 antibody apomab with chemotherapy to inhibit orthotopic lung tumor growth and improve survival. Clinical Cancer Research. 2008;14(23):7733–7740. doi: 10.1158/1078-0432.CCR-08-0670. [DOI] [PubMed] [Google Scholar]
- 131.Amm HM, Zhou T, Steg AD, Kuo H, Li Y, Buchsbaum DJ. Mechanisms of drug sensitization to TRA-8, an agonistic death receptor 5 antibody, involve modulation of the intrinsic apoptotic pathway in human breast cancer cells. Molecular Cancer Research. 2011;9(4):403–417. doi: 10.1158/1541-7786.MCR-10-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fuchs CS, Fakih M, Schwartzberg L, et al. TRAIL receptor agonist conatumumab with modified FOLFOX6 plus bevacizumab for first-line treatment of metastatic colorectal cancer: a randomized phase 1b/2 trial. Cancer. 2013;119(24):4290–4298. doi: 10.1002/cncr.28353. [DOI] [PubMed] [Google Scholar]
- 133.Natoni A, MacFarlane M, Inoue S, et al. TRAIL signals to apoptosis in chronic lymphocytic leukaemia cells primarily through TRAIL-R1 whereas cross-linked agonistic TRAIL-R2 antibodies facilitate signalling via TRAIL-R2. British Journal of Haematology. 2007;139(4):568–577. doi: 10.1111/j.1365-2141.2007.06852.x. [DOI] [PubMed] [Google Scholar]
- 134.Ortega-Gómez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Molecular Medicine. 2013;5(5):661–674. doi: 10.1002/emmm.201202382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gall A, Treuting P, Elkon KB, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36(1):120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]