

Abstract
During colitis, activation of two inflammatory T cell subsets, Th17 and Th1 cells, promotes ongoing intestinal inflammatory responses. n-6 polyunsaturated fatty acid- (PUFA-) derived eicosanoids, such as prostaglandin E2 (PGE2), promote Th17 cell-mediated inflammation, while n-3 PUFA antagonize both Th17 and Th1 cells and suppress PGE2 levels. We utilized two genetic mouse models, which differentially antagonize PGE2 levels, to examine the effect on Th17 cells and disease outcomes in trinitrobenzene sulfonic acid- (TNBS-) induced colitis. Fat-1 mice contain the ω3 desaturase gene from C. elegans and synthesize n-3 PUFA de novo, thereby reducing the biosynthesis of n-6 PUFA-derived eicosanoids. In contrast, Fads1 Null mice contain a disrupted Δ5 desaturase gene and produce lower levels of n-6 PUFA-derived eicosanoids. Compared to Wt littermates, Fat-1 and Fads1 Null mice exhibited a similar colitic phenotype characterized by reduced colonic mucosal inflammatory eicosanoid levels and mRNA expression of Th17 cell markers (IL-17A, RORγτ, and IL-23), decreased percentages of Th17 cells and, improved colon injury scores (P ≤ 0.05). Thus, during colitis, similar outcomes were obtained in two genetically distinct models, both of which antagonize PGE2 levels via different mechanisms. Our data highlight the critical impact of n-6 PUFA-derived eicosanoids in the promotion of Th17 cell-mediated colonic inflammation.
1. Introduction
Inflammatory bowel disease (IBD) manifests as two clinical conditions, ulcerative colitis (UC) and Crohn's disease (CD). The induction and persistence of chronic inflammation during IBD is attributed to the activation of two inflammatory T cell subsets (Th17 and Th1 cells) and production of their signature cytokines, IL-17 and IFNγ, respectively [1–4]. The role of Th17 cells in the pathogenesis of IBD has been documented in humans with active disease [5–7] and in mouse models in which disease severity was reduced by blockade or deficiency of IL-23 and/or IL-17 [8, 9].
A recent case-control study found that high intakes of n-6 polyunsaturated fatty acids (PUFA) increased the risk of developing UC [10] which was attributed, in part, to the immunomodulatory effects of arachidonic acid- (AA-) derived eicosanoids [11]. In IBD patients (CD and UC) increased levels of AA and its eicosanoid metabolites, such as PGE2, leukotriene B4 (LTB4), and thromboxane B2 (TXB2), were found in the mucosa of the gastrointestinal tract [12–17]. In UC patients, the concentration of PGE2 increased in proportion to the degree of mucosal injury or disease severity [17]. Moreover, in IBD patients, the combination of PGE2, IL-23, and IL-1β works synergistically to enhance IL-17A secretion from CD161+ CD4+ T cells [18] which infiltrate the gastrointestinal tract [19–21]. In the trinitrobenzene sulfonic acid- (TNBS-) induced mouse colitis model, which induces T cell-mediated immune responses within the colonic mucosa [22] and is driven by inflammatory Th17 cells [23], both serum and colonic mucosal PGE2 levels were elevated [24]. PGE2 was shown to exacerbate colonic inflammatory processes and colitis severity in this model through the activation of the IL-23/IL17 axis and by increasing local Th17 cell numbers [25]. Through alterations in the cytokine microenvironment, PGE2 can influence inflammatory T cell development directly by skewing naïve T cell differentiation and effector function toward the production of proinflammatory Th17 and Th1 cell subsets [18, 26–29] and indirectly by inducing antigen presenting cells to favor IL-23 production [25, 30, 31], thereby promoting the differentiation and maintenance of Th17 cells. Other n-6 PUFA-derived eicosanoids have also been shown to promote Th17 cell development [32], thereby demonstrating partial functional redundancy in the immunomodulatory effects of the AA-derived eicosanoid profile. Collectively, these data indicate that AA-derived eicosanoids may drive the activation of Th17 cells during IBD and any treatment strategy designed to antagonize their mucosal levels could reduce Th17 cell activation and the severity of the disease phenotype.
Fish oil (FO) derived long chain n-3 PUFA exert anti-inflammatory effects [33–35] and have been shown to enhance remission of chronic intestinal inflammation [36]. Moreover, an estimated 50% of IBD patients utilize self-prescribed oral complementary alternative medicines/diets, such as FO [37]. Dietary n-3 PUFA accumulate in cell membranes, partly at the expense of AA, thereby reducing the available substrate for the synthesis of AA-derived eicosanoids [38–41] while concomitantly serving as substrates for the production of n-3 PUFA-derived anti-inflammatory resolvins, docosatrienes, and neuroprotectins [42]. Further, n-3 PUFA have been demonstrated to reduce splenic CD4+ T cell ex vivo polarization into Th1 [43, 44] and Th17 cells [45]. Therefore, n-3 PUFA may suppress colitis-associated Th17 cell activation, in part, by reducing mucosal AA-derived eicosanoid levels. To test this hypothesis, we utilized two genetic mouse models which antagonize AA-derived eicosanoid production: (i) the Fat-1 transgenic mouse which produces long chain n-3 PUFA de novo [46] and exhibits reduced colonic AA-derived eicosanoid levels [47] and (ii) the Fads1 Null mouse, which exhibits systemic disruption of the Fads1 (Δ5 desaturase) gene, reciprocally altering the tissue level of dihomo-γ-linolenic (DGLA) and AA, resulting in decreased AA-derived eicosanoid levels [48]. In this study, we determined the effect of antagonizing AA-derived eicosanoids on local [colon and mesenteric lymph node (MLN)] and systemic (splenic) levels of the relevant CD4+ T cell effector subsets (Th17, Th1, Th2, and Tregs) and Th17 cell-mediated colonic cytokine expression in response to TNBS-induced colitis.
2. Materials and Methods
2.1. Animals and Diets
Fads1 and Fat-1 transgenic mice, both on a C57BL/6 background, were generated in collaboration with the Texas Institute for Genomic Medicine (Texas A&M University) and Dr. Jing Kang (Harvard University), respectively. Fads1 knockout mice [genotypes: wild-type (Wt), heterozygous (Het), and null (Null)] represent a Δ5 desaturase knockout strain that produces AA deficiency without the underlying complication of essential fatty acid deficiency [i.e., linoleic acid (LA) or DGLA] [48]. Fat-1 transgenic mice (genotypes: Wt and Fat-1) synthesize long chain n-3 PUFA de novo [46]. Littermate specific pathogen-free male and female mice from both strains were genotyped, phenotyped, and housed as previously described [46–48]. All mice were fed a commercial 10% safflower oil diet (D03092902R; Research Diets, New Brunswick, NJ, USA), wherein GC fatty acid analysis of the diet confirmed that it is free of AA and contained trace levels of n-3 PUFA (0.17% α-linolenic acid) but was adequate in all other nutrients. All procedures adhered to U.S. Public Health Service Policy and were approved by the Institutional Animal Care and Use Committee at Texas A&M University.
2.2. Colitis Induction and Histological Scoring
Colonic inflammation was induced by exposure to 2,4,6-trinitrobenzene sulfonic acid (TNBS; Sigma Aldrich, St. Louis, MO, USA) as previously described [49]. In brief, mice were individually housed and 100 μL of a 1% (w/v) presensitizing dosage of TNBS dissolved in a 4 : 1 volume ratio of acetone and olive oil (Azienda, Florence, Italy) was applied topically onto a shaved 1.5 × 1.5 cm field between the shoulders. The site was selected to prevent the animals from ingesting the TNBS which could induce oral tolerance [49]. After 7 d, mice were anesthetized with isoflurane to effect and were under anesthesia for ≤5 min during which time a 100 μL enema containing 2.5% (w/v) TNBS in a 1 : 1 volume ratio of water and absolute ethanol was administered. Saline control mice were exposed to the presensitization vehicle topically (4 : 1 volume ratio of acetone and olive oil) followed 7 d later by a 100 μL saline enema. All mice were sacrificed 3 d after TNBS enema by CO2 asphyxiation. Colons were excised (distal to the cecum and proximal to the anus) and flushed with sterile PBS and the mucosa was scraped from one longitudinal colon half for gene expression analysis. The other longitudinal colon half was fixed in 4% paraformaldehyde, Swiss-rolled, paraffin embedded, and stained with hematoxylin and eosin. The degree of colon injury (score 0–3 per colon region, i.e, proximal, middle, and distal) was graded in a blinded manner by a board-certified pathologist (B. Weeks) in accordance with the criteria outlined previously [47].
2.3. Flow Cytometry Analysis of T Cell Subsets
Splenic and MLN mononuclear cells were isolated by lympholyte-M enrichment (Cedarlane, Burlington, NC, USA) as described [50]. Colonic tissues were digested using Type IV collagenase (Sigma Aldrich) as described elsewhere [51] and lymphocytes were enriched over 70/45% Percoll gradient (Sigma Aldrich). Surface and intracellular staining were performed as reported previously [45, 52]. Cells were surface-stained with APC-anti-CD4 (clone L3T4, eBioscience, San Diego, CA, USA) followed by intracellular detection of PE-anti-FOXP3 (clone FJK-16s), PE-anti-IL-17A (clone eBio17B7), PE-anti-IFNγ (clone XMG1.2), or PE-anti-IL-4 (clone 11B11) (eBioscience). Isotype controls utilized were PE-IgG2a κ, PE-IgG1κ, and APC-IgG2b κ (eBioscience). Flow cytometric analysis was conducted using a BD Accuri C6 flow cytometer (BD Bioscience, San Jose, CA, USA).
2.4. RNA Isolation and Measurement of mRNA Expression
RNA was isolated from colon mucosal scrapings using the RNA 4-PCR kit (Ambion/Life Technologies, Grand Island, NY, USA). Real-time RT-PCR was used to quantify mRNA expression and amplification was performed using the Taqman Universal PCR master mix and Taqman gene expression kits (Applied Biosystems/Life Technologies, Grand Island, NY, USA) were used for amplification of IL-1β (Mm00434228_m1), IL-6 (Mm00446190_m1), IL-17A (Mm00439618_m1), IL-17F (Mm00521423_m1), IL-21 (Mm00517640_m1), IL-22 (Mm01226722_g1), IL-23 (Mm00518984_m1), IL23R (Mm_00519943_m1), IFNγ (Mm01168134_m1), IL-27 (Mm00461162_m1), TNFα (Mm00443260_g1), CCL2 (MCP-1, Mm00441242_m1), IL-4 (Mm00445259_m1), IL-10 (Mm00439614_m1), TGFβ1 (Mm01178820_m1), Rorc (RORγ τ, Mm01261022_m1), Tbx21 (T-bet, Mm00450960_m1), FOXP3 (Mm00475162_m1), CCL20 (Mm01268754_m1), and CCR6 (Mm99999114_s1). Amplification of mRNA (fluorescence) was recorded over 40 cycles and the corresponding cycle numbers (Ct) were used to calculate mRNA expression according to the calculation: 2(40−Ct). Target gene expression was normalized to ribosomal 18S expression (Mm03928990_g1).
2.5. Colonic Mucosal Eicosanoid Profiling
Eicosanoids were extracted from colonic scraped mucosa from TNBS-treated Fat-1 and Fads1 mice as previously described [47, 48, 53]. Liquid chromatography/tandem mass spectroscopic analyses were performed using a QuattroUltima mass spectrometer (Waters, Milford, MA, USA) equipped with an Agilent 1100 binary pump high-performance liquid chromatography system (Agilent Technologies, Santa Clara, CA, USA) according to a modified version of the method of Yang et al. [53]. Eicosanoids of interest were chromatographically separated using a Luna 3 μm phenyl-hexyl 4.6 × 100 mm analytic column (Phenomenex, Torrance, CA, USA) [48]. Eicosanoids were detected and quantified by multiple reaction mode monitoring of the transitions m/z as described elsewhere [48, 54].
2.6. Statistics
The predetermined upper limit of probability for statistical significance throughout this investigation was P ≤ 0.05, and analyses were conducted using the SAS system for Windows Version 9.0 (SAS Institute, Cary, NC, USA). Data were analyzed by either one-way ANOVA or two-way ANOVA (main effects: genotype and treatment) followed, if justified, by testing using Least Squares Means. Data sets not exhibiting a normal distribution were analyzed using the Kruskal-Wallis test (χ 2 approximation) followed, if justified, by the statistical probability outcome (P ≤ 0.05) using Wilcoxon two-sample testing.
3. Results
3.1. Colonic Mucosal Inflammatory AA-Derived Eicosanoid Profile Is Antagonized in Fat-1 and Fads1 Null Mice
The colonic mucosal eicosanoid profiles from TNBS-treated Fat-1 (Table 1) and Fads1 (Table 2) mice are shown. In Fat-1 mice, colonic mucosal levels of n-6 PUFA-derived eicosanoids, specifically PGE2, prostaglandins (PG) D2 and F2α, 15-hydroxy-eicosatetraenoic acid (HETE), arachidonoyl ethanolamine (AEA), and arachidonoyl glycerol (AG), were reduced significantly, whereas the n-3 PUFA-derived PGE3 was increased compared to Wt (P < 0.05). In Fads1 Null mice, increased levels of PGE1 and decreased levels PGE2, D2, Fα, thromboxane (TX) B2, 5-HETE, AEA, and 2AG (P < 0.05) were observed compared to Wt littermates. Collectively, these data demonstrate that the local inflammatory (n-6 PUFA-derived) eicosanoid profile in response to TNBS-induced colitis is antagonized in both genetic mouse models via two different mechanisms, that is, by increasing n-3 PUFA content in Fat-1 mice and by inducing AA deficiency in Fads1 Null mice, as seen previously [47, 48].
Table 1.
Colonic mucosal eicosanoid and cannabinoid profiles in Wt and Fat-1 TNBS-treated mice1.
| Eicosanoid (ng/mg protein) |
Wt | Fat-1 |
|---|---|---|
| PGE1 | 0.68 ± 0.16 | 1.00 ± 0.35 |
| PGE2 | 13.79 ± 3.46 | 7.50 ± 1.79∗ |
| PGE3 | 0.00 ± 0.00 | 0.24 ± 0.03∗ |
| PGD2 | 44.37 ± 2.09 | 23.18 ± 5.69∗ |
| PGF2α | 4.16 ± 0.64 | 2.40 ± 0.59∗ |
| 6-keto-PGF1α | 3.65 ± 0.73 | 2.05 ± 0.35 |
| TXB2 | 1.02 ± 0.16 | 0.85 ± 0.28 |
| 13-PGE1 | 0.02 ± 0.01 | 0.00 ± 0.00 |
| 13-PGE2 | 11.33 ± 2.36 | 7.68 ± 1.27 |
| 13-HODE | 12.29 ± 2.68 | 8.33 ± 0.87 |
| 5-HETE | 1.01 ± 0.18 | 0.93 ± 0.35 |
| 12-HETE | 7.74 ± 2.22 | 8.40 ± 3.69 |
| 15-HETE | 0.97 ± 0.13 | 0.50 ± 0.15∗ |
| AEA | 44.10 ± 3.89 | 19.97 ± 6.35∗ |
| 2-AG | 0.50 ± 0.05 | 0.24 ± 0.03∗ |
1Mean values ± SEM of TNBS-treated Fat-1 mice (n = 4-5/genotype) and values marked with an asterisk differ from Wt (P ≤ 0.05). PG, prostaglandin; TX, thromboxane; 13-PGE1, 13,14-dihydro 15-keto-PGE1; 13-PGE2, 13,14-dihydro 15-keto-PGE2; HODE, hydroxyl-octadecadienoic acid; HETE, hydroxyl-eicosatetraenoic acid; AEA, arachidonoyl ethanolamine; and AG, arachidonoyl glycerol.
Table 2.
Colonic mucosal eicosanoid and cannabinoid profiles in Fads1 TNBS-treated mice1.
| Eicosanoid (ng/mg protein) |
Wt | Het | Null |
|---|---|---|---|
| PGE1 | 1.91 ± 1.09∗ | 1.87 ± 0.50∗ | 16.36 ± 6.88‡ |
| PGE2 | 15.70 ± 4.92∗ | 14.19 ± 1.66∗ | 1.92 ± 0.77‡ |
| PGE3 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| PGD2 | 43.12 ± 4.51∗ | 39.14 ± 2.51∗ | 1.38 ± 0.06‡ |
| PGF2α | 5.94 ± 0.98∗ | 4.15 ± 0.57∗ | 0.11 ± 0.03‡ |
| 6-keto-PGF1α | 3.78 ± 0.98∗ | 3.00 ± 0.74∗ | 0.41 ± 0.11‡ |
| TXB2 | 1.08 ± 0.23∗ | 1.26 ± 0.21∗ | 0.17 ± 0.02‡ |
| 13-PGE1 | 0.11 ± 0.05∗ | 0.07 ± 0.03∗ | 0.78 ± 0.14‡ |
| 13-PGE2 | 14.34 ± 1.62∗ | 7.71 ± 0.94‡ | 0.69 ± 0.09§ |
| 13-HODE | 14.05 ± 2.63 | 23.18 ± 7.63 | 9.25 ± 1.67 |
| 5-HETE | 1.04 ± 0.16∗ | 2.65 ± 1.35∗ | 0.12 ± 0.01‡ |
| 12-HETE | 6.47 ± 1.88∗ | 47.66 ± 12.84‡ | 2.57 ± 1.40∗ |
| 15-HETE | 1.06 ± 0.10 | 3.04 ± 1.50 | 0.11 ± 0.02 |
| AEA | 0.59 ± 0.04∗ | 0.52 ± 0.07∗ | 0.04 ± 0.00‡ |
| 2-AG | 34.88 ± 6.74∗ | 56.17 ± 12.53∗ | 2.32 ± 0.57‡ |
1Mean values ± SEM of TNBS-treated Wt, Het, and Null Fads1 mice (n = 5/genotype). Values not sharing a superscript symbol differ (P ≤ 0.05). PG, prostaglandin; TX, thromboxane; 13-PGE1, 13,14-dihydro 15-keto-PGE1; 13-PGE2, 13,14-dihydro 15-keto-PGE2; HODE, hydroxyl-octadecadienoic acid; HETE, hydroxyl-eicosatetraenoic acid; AEA, arachidonoyl ethanolamine; and AG, arachidonoyl glycerol.
3.2. Fat-1 Mice Are More Resistant to TNBS-Induced Colon Injury
Body weight and colon length at the time of sacrifice are shown in Supplemental Table 1 (see Supplementary Material available online at http://dx.doi.org/10.1155/2014/917149). Compared to saline controls, TNBS-treated mice exhibited lower body weights and colon shortening, but they did not differ between genotypes (P > 0.05). The degree of colon injury following exposure to TNBS was assessed based on colonic histological changes in a blinded manner by a board-certified pathologist (B. Weeks). Representative distal colon images from Wt and Fat-1 saline control and TNBS-treated mice are shown (Figures 1(a)–1(c)). The saline control treatment did not induce colonic damage and both Wt and Fat-1 mice exhibited an injury score of 0 in all regions of the colon. In TNBS-treated mice, injury scores were lower in the proximal and middle regions of the colon compared to the distal colon; however, they did not differ between Wt and Fat-1 mice (proximal colon, P = 0.45, and middle colon, P = 0.54, results not shown). However, within the distal colon (site of TNBS administration via rectal enema), the degree of colonic injury was significantly reduced in Fat-1 mice compared to Wt (P = 0.05, Figure 1(d)). This outcome is consistent with the TNBS model exerting the majority of histopathological damage within the distal colon [55] and with the ability of n-3 PUFA to enhance the resolution of inflammatory processes and reduce colonic injury by promoting mucosal repair [47, 56].
Figure 1.
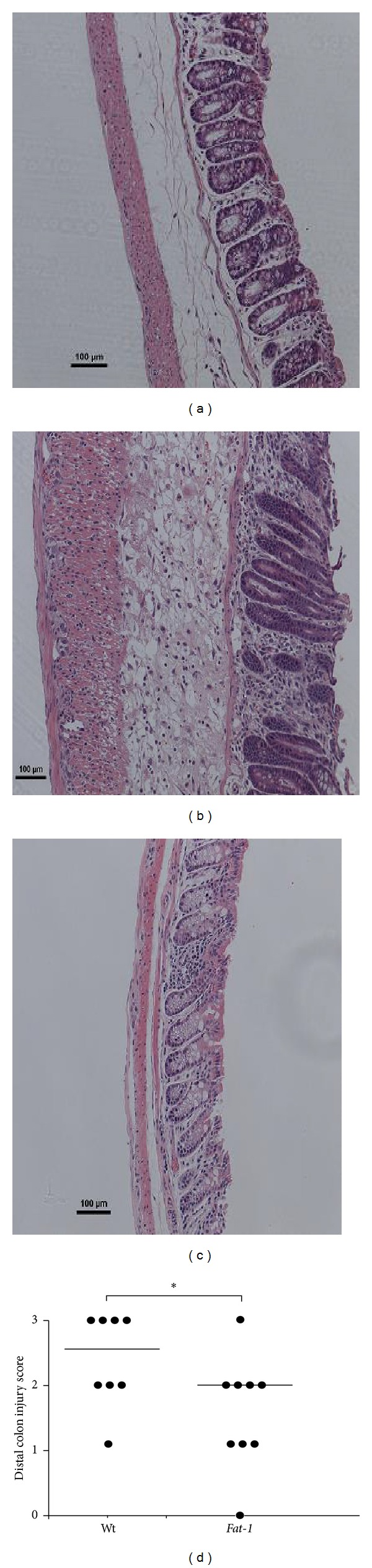
The degree of distal colon histological injury/damage in Fat-1 and Wt TNBS-treated mice (n = 8-9/genotype). ((a) and (b)) Representative images (100x magnification, scale bar = 100 μm) of TNBS-treated Fat-1 and Wt mice, (c) Wt saline control treated (injury score = 0) distal colons, and (d) injury scores (0–3) in the distal colon from Wt and Fat-1 TNBS-treated mice, respectively. Data were analyzed using the Kruskal-Wallis test (P ≤ 0.05). Median values are shown and significant differences between genotypes are marked with an asterisk (P ≤ 0.05).
3.3. Colitis-Associated Changes in CD4+ T Cell Subsets in Local and Systemic Anatomical Sites in Wt and Fat-1 Mice
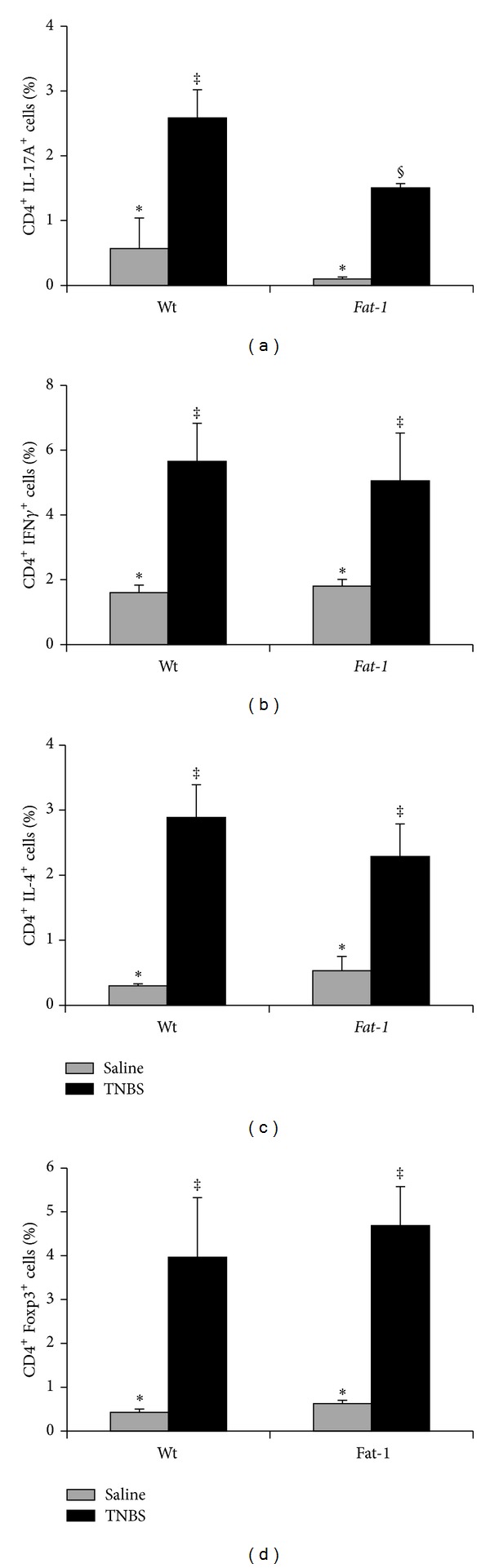
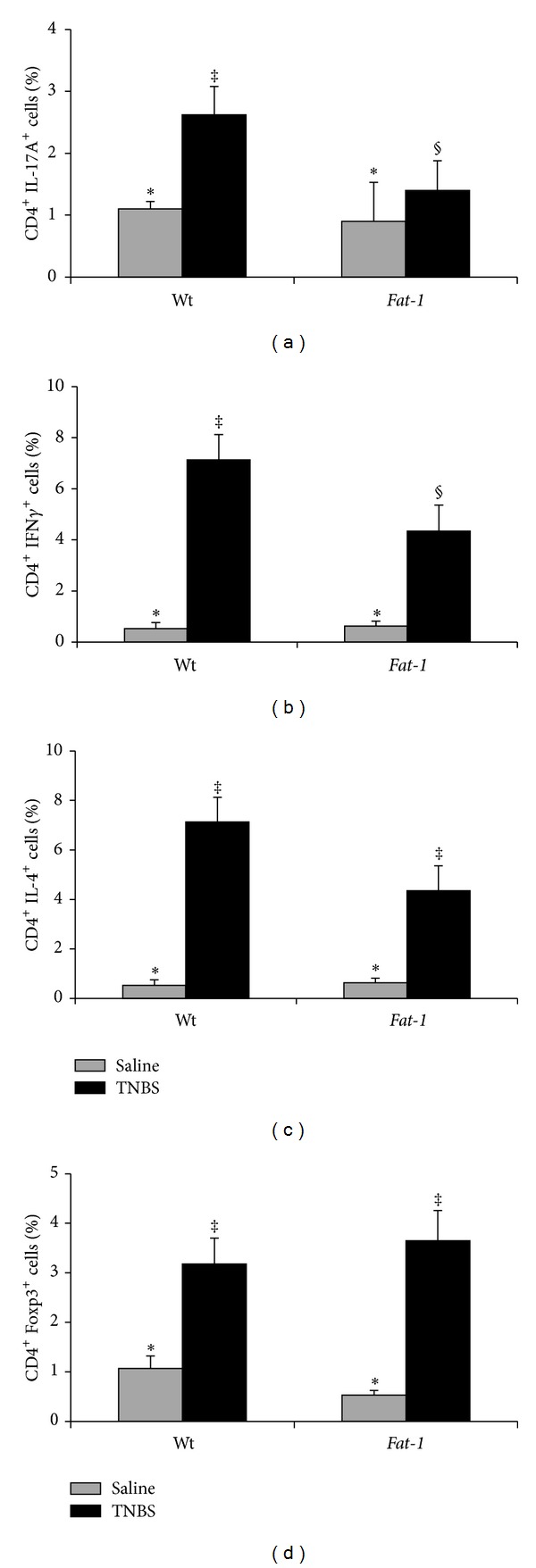
Following the induction of colitis, the effect of n-3 PUFA on the resident CD4+ T cell effector subset populations (i.e., Th1, Th2, Th17, and Treg) was documented both locally (colon lamina propria and MLN) and systemically (spleen). Representative dot plots for T cell subsets isolated from Wt TNBS-treated colon, MLN, and spleen are shown in Supplemental Figure 1. In all tissue sites, TNBS exposure increased the percentage of all CD4+ T cell subsets compared to the saline control group (treatment: P < 0.05, Figures 2–4). Within the colon, the percentages of proinflammatory Th17 and Th1 cells were reduced in Fat-1 mice compared to Wt (P < 0.05, Figures 2(a) and 2(b)), whereas the percentages of Th2 and Treg cells did not differ between groups (P > 0.05, Figures 2(c) and 2(d)). In the MLN, which drains and is anatomically proximal to the inflamed colon, the percentages of both Th17 and Th1 cells were reduced in Fat-1 mice compared to Wt (P < 0.05) (Figures 3(a) and 3(b)), whereas Treg and Th2 cells were unaffected (P > 0.05, Figures 3(c) and 3(d)). In the spleen, only the percentage of Th17 cells was reduced in the Fat-1 mouse (P < 0.05) compared to Wt (Figure 4(a)). Splenic Th1, Th2, and Treg cell populations did not differ between groups (P > 0.05, Figures 4(b)–4(d)). Collectively, these data demonstrate that proinflammatory Th17 and Th1 cells are selectively antagonized by n-3 PUFA in both local and systemic sites during colitis.
Figure 2.
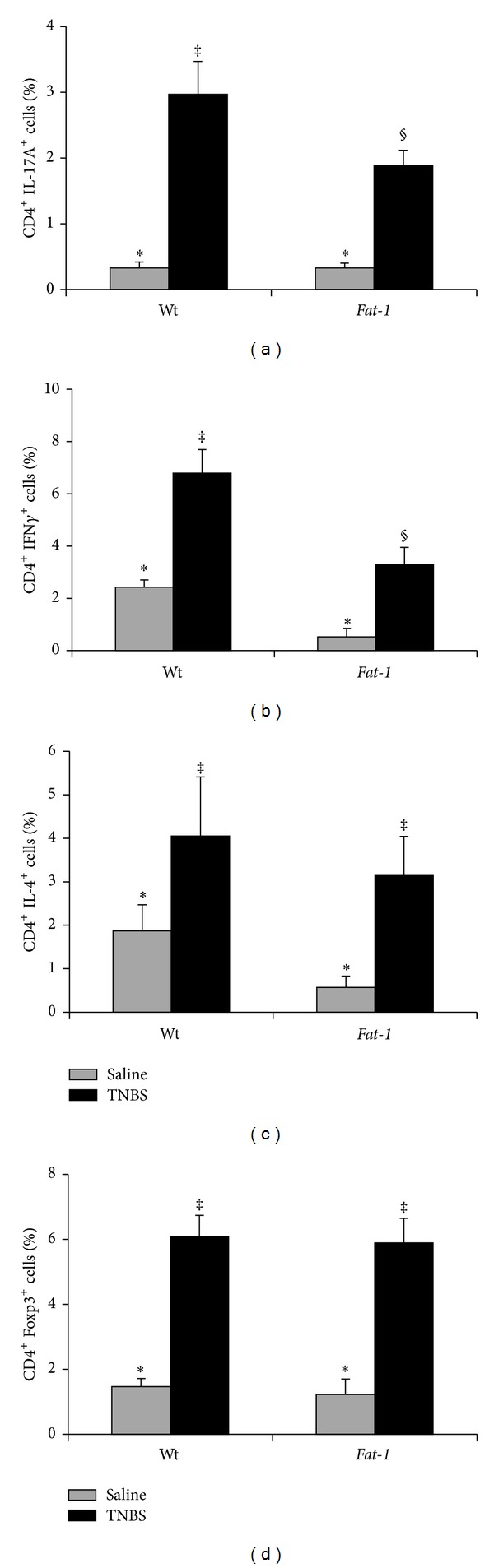
Percentage of colon lamina propria CD4+ T cell subsets in Wt and Fat-1 TNBS-treated mice (black bars, n = 8–16 pooled samples/genotype comprised of 2-3 colons) and saline controls (grey bars, n = 4 pooled samples/genotype). (a) Th17 cells (CD4+ IL-17A+), (b) Th1 cells (CD4+ IFNγ +), (c) Th2 cells (CD4+ IL-4+), and (d) Tregs (CD4+ Foxp3+). Data were analyzed by two-way ANOVA (main effects: genotype and treatment); bars represent means ± SEM. Bars not sharing a symbol differ (P ≤ 0.05).
Figure 4.
Percentage of splenic CD4+T cell subsets in Wt and Fat-1 TNBS-treated mice (black bars, n = 10–12 spleens/genotype) and saline controls (grey bars, n = 4/genotype). (a) Th17 cells (CD4+ IL-17A+), (b) Th1 cells (CD4+ IFNγ +), (c) Th2 cells (CD4+ IL-4+), and (d) Tregs (CD4+ Foxp3+). Data were analyzed by two-way ANOVA (main effects: genotype and treatment). Bars represent means ± SEM. Bars not sharing a symbol differ (P ≤ 0.05).
Figure 3.
Percentage of mesenteric lymph node (MLN) CD4+ T cell subsets in Wt and Fat-1 TNBS-treated mice (black bars, n = 9–12 pooled samples/genotype comprised of 2–4 MLNs) and saline controls (grey bars, n = 4 pooled samples/genotype). (a) Th17 cells (CD4+ IL-17A+), (b) Th1 cells (CD4+ IFNγ +), (c) Th2 cells (CD4+ IL-4+), and (d) Tregs (CD4+ Foxp3+). Data were analyzed by two-way ANOVA (main effects: genotype and treatment); bars represent means ± SEM. Bars not sharing a symbol differ (P ≤ 0.05).
3.4. The Colonic Mucosal Cytokine Microenvironment Is Modified in a Manner Consistent with Reduced Th17 Cell Activation in Fat-1 Mice
To gain insight into how effector T cell populations are changing in response to TNBS-induced colitis, gene expression of critical transcription factors and cytokines that make up the inflammatory colonic milieu were assessed in both Wt and Fat-1 TNBS-treated mice (Table 3). Colonic mRNA expression of RORγ τ, the master transcription factor that directs both the differentiation of Th17 cells and the expression of hallmark Th17 cytokines [57], was reduced significantly in Fat-1 mice (P = 0.03). Conversely, n-3 PUFA had no significant effect on the mRNA expression of other key transcription factors associated with other T cell subsets, namely, Foxp3 (Tregs) and T-bet (Th1 cells) (P > 0.05). Gene expression of the Th17 cell signature cytokine, IL-17A, was decreased significantly in the Fat-1 mouse versus Wt (P = 0.04), whereas IL-17F and IL-22 did not differ between groups (P > 0.05). Additionally, mRNA expression of IL-21, which promotes Th17 cell differentiation and proliferation [58, 59] and controls both Th1 and Th17 cell responses [2, 60–62], was reduced in the Fat-1 mouse (P = 0.01), consistent with the reduced percentage of Th1 and Th17 cells in the colon lamina propria (Figures 2(b) and 2(c)). Colonic mRNA expression of IL-23 showed a trend towards a significant reduction in the Fat-1 mouse (P = 0.06), whereas the expression of IL-23R did not differ between groups (P = 0.95). Interestingly, n-3 PUFA significantly upregulated mRNA expression of IL-27 (P = 0.04), a key cytokine that has been shown to antagonize Th17 cell development [60, 62]. Consistent with the anti-inflammatory effects of n-3 PUFA [33–35], colonic mRNA expression of IL-10 was upregulated in the Fat-1 mouse (P = 0.04), whereas TGFβ1 expression was unaffected (P = 0.44). Fat-1 mRNA levels of classic inflammatory cytokines (IL-1β and TNFα) and chemokines (MCP-1) were reduced compared to Wt levels (P < 0.05), whereas colonic mRNA levels of IL-6, IFNγ, and IL-4 did not differ between genotypes (P > 0.05).
Table 3.
Colonic mucosal mRNA expression in Wt and Fat-1 TNBS-treated mice1.
| Gene | Wt | Fat-1 |
|---|---|---|
| IL-17A | 6.54 ± 2.95 | 2.04 ± 0.73∗ |
| IL-17F | 6.14 ± 1.33 | 5.96 ± 1.03 |
| IL-21 | 0.63 ± 0.09 | 0.15 ± 0.07∗ |
| IL-22 | 0.48 ± 0.17 | 0.95 ± 0.32 |
| IL-23 | 1.63 ± 0.42 | 1.01 ± 0.41 |
| IL-23R | 1.72 ± 0.44 | 1.76 ± 0.28 |
| IL-27 | 3.68 ± 0.84 | 7.50 ± 1.49∗ |
| CCL20 | 4.93 ± 1.59 | 4.25 ± 0.61 |
| CCR6 | 0.45 ± 0.97 | 0.7 ± 0.48 |
| RORγτ | 15.79 ± 1.29 | 12.25 ± 0.82∗ |
| Tbet | 2.30 ± 0.79 | 1.42 ± 0.46 |
| Foxp3 | 1.74 ± 0.97 | 3.24 ± 0.63 |
| IL-10 | 1.08 ± 0.68 | 2.64 ± 0.52∗ |
| TGFβ1 | 5.56 ± 0.66 | 6.91 ± 2.26 |
| MCP-1 | 8.34 ± 3.75 | 3.03 ± 0.58∗ |
| IL-1β | 1.28 ± 0.87 | 0.39 ± 0.09∗ |
| TNFα | 1.90 ± 1.3 | 0.73 ± 0.18∗ |
| IL-6 | 2.88 ± 0.86 | 2.66 ± 0.61 |
| IFNγ | 0.63 ± 0.22 | 0.34 ± 0.15 |
| IL-4 | 0.20 ± 0.11 | 0.28 ± 0.25 |
1Mean values ± SEM of TNBS-treated Wt and Fat-1 mice (n = 8–10/genotype). All genes were measured within the same samples and expression was normalized to ribosomal 18S (in arbitrary units). For each gene, values marked with an asterisk differ from Wt (P ≤ 0.05).
3.5. TNBS Colitis Induced Colon Injury Is Reduced in Fads1 Null Mice
The degree of TNBS-induced colon injury was assessed histologically in Fads1 Wt, Het, and Null mice (representative distal colon images, Figures 5(a)–5(c)). Histological injury scores were 0 throughout the colon in saline control treated mice from all three Fads1 genotypes. Within TNBS-treated mice, the degree of colon injury did not differ between genotypes in the proximal (P = 0.10) and middle (P = 0.31) regions of the colon. However, distal colon injury scores were reduced significantly in Fads1 Null mice compared to both Wt and Het mice (P = 0.04) (Figure 5(d)). Colon length and final body weights were lower in TNBS-treated mice compared to saline controls but did not differ between genotypes (P > 0.05) as shown in Supplemental Table 2. These data suggest that antagonizing colon mucosal eicosanoid levels via AA deficiency reduces the severity of colon histological structural damage induced during TNBS colitis.
Figure 5.
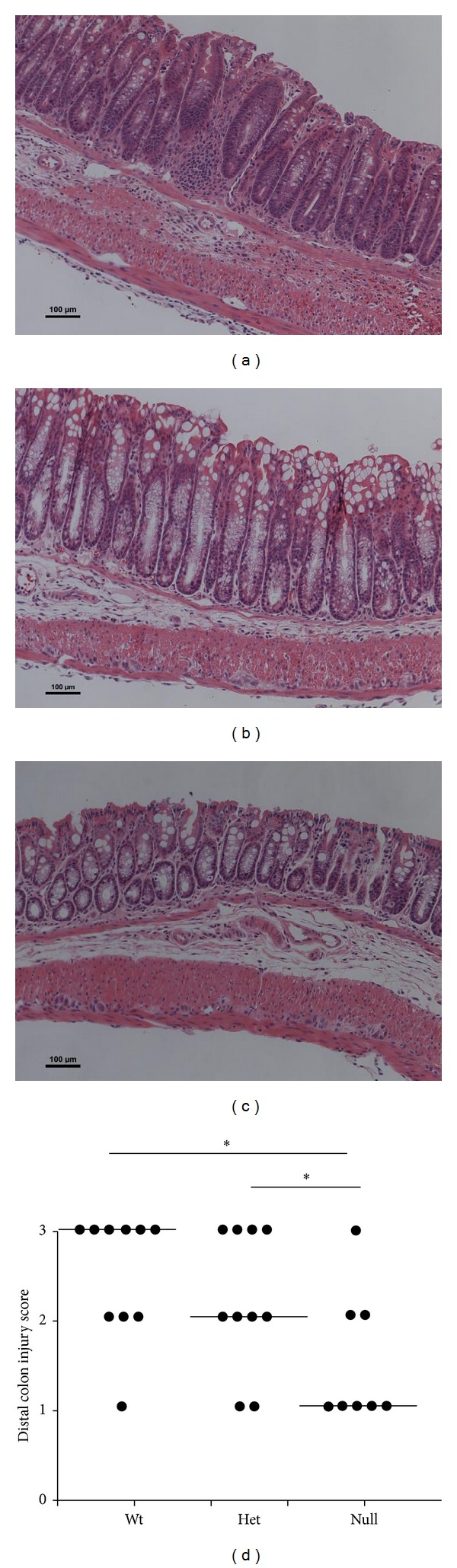
Distal colon histological injury/damage scores assessed in Wt, Het, and Null Fads1 TNBS-treated mice (n = 8–10/genotype) in a blinded manner by a board-certified pathologist (B. Weeks). ((a)–(c)) Representative images (100x magnification, scale bar = 100 μm) of Wt, Het, and Null Fads1 TNBS-treated distal colons, respectively. (d) Distal colon injury scores (0–3). Data were analyzed using the Kruskal-Wallis test (P ≤ 0.05) followed by Wilcoxon two-sample testing. Median values are shown and significant differences between genotypes are marked by an asterisk (P ≤ 0.05).
3.6. Changes in Splenic T Cell Subsets in the Fads1 Mouse
Due to limitations in mouse numbers and the necessity to pool colons to obtain sufficient cells for analysis, quantification of T cell subsets in the colon and MLN was not possible. Therefore, changes in systemic T cell subsets (i.e., Th1, Th2, Th17, and Treg) following exposure to TNBS were determined in the spleen and representative dot plots are shown in Supplemental Figure 2. As expected, TNBS treatment increased all splenic CD4+ T cell subsets compared to saline controls (P < 0.05). The percentages of splenic Th17 and Th1 cells were reduced in Fads1 Nullmice compared to both Wt and Het mice (P < 0.05, Figures 6(a) and 6(b)). Interestingly, the percentage of splenic Th2 cells was also reduced in the Fads1 Null mouse compared to both Wt and Het TNBS-treated mice (P = 0.04, Figure 6(c)), whereas splenic Tregs were unaffected (Wt versus Null, P > 0.05, Figure 6(d)). There was no difference between Wt and Het mice in the percentage of splenic Th17, Th1, and Th2 cells (Figures 6(a)–6(c)), whereas the percentage of splenic Treg cells was increased in Het mice compared to both Wt and Fads1 Null (P = 0.008, Figure 6(d)).
Figure 6.
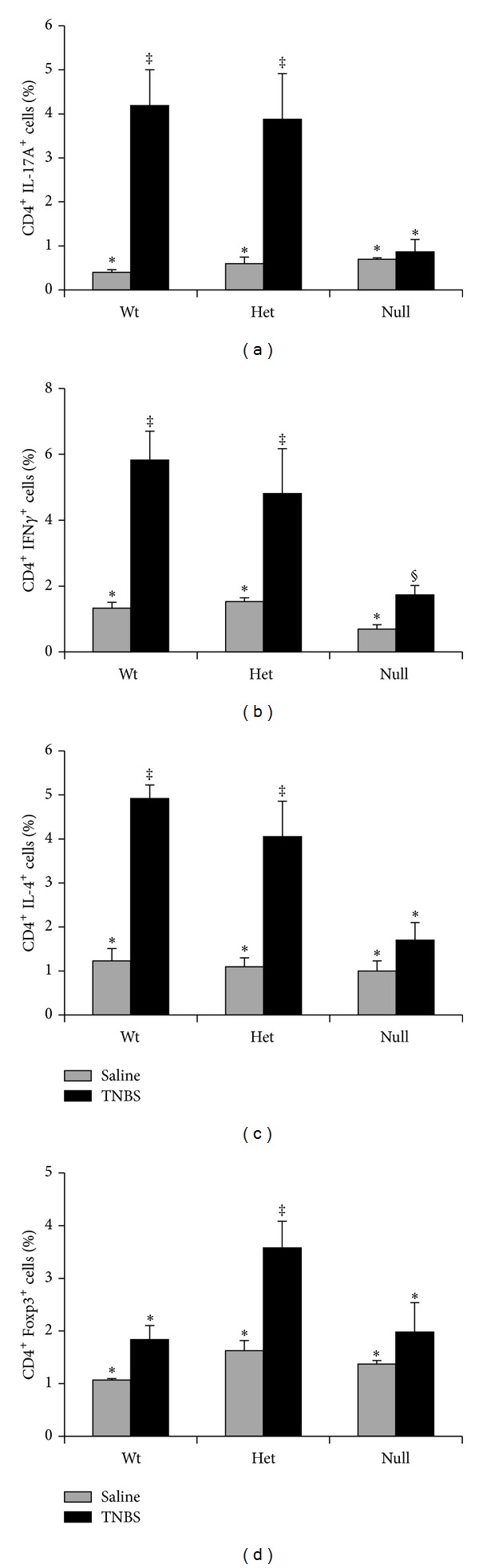
Percentage of splenic CD4+ T cell subsets in Wt, Het, and Null Fads1 TNBS-treated mice (black bars, n = 6–9 mice/genotype) and saline controls (grey bars, n = 3-4 mice/genotype). (a) Th17 cells (CD4+ IL-17A+), (b) Th1 cells (CD4+ IFNγ +), (c) Th2 cells (CD4+ IL-4+), and (d) Tregs (CD4+ Foxp3+). Data were analyzed by two-way ANOVA (main effects: genotype and treatment). Bars represent means ± SEM. Bars not sharing a symbol differ (P ≤ 0.05).
3.7. The Colonic Mucosal Cytokine Microenvironment Is Modified in a Manner Consistent with Reduced Th17 Cell Activation in Fads1Null Mice
The mRNA expression levels of key cytokines and transcription factors related to specific T cell subsets and inflammatory status in the colons of TNBS-treated Fads1 mice are shown in Table 4. mRNA levels did not differ between Wt and Het mice for any genes except the Th1 cell master transcription factor Tbet, which was reduced in both Het and Null mice compared to Wt (P = 0.004). Additionally, mRNA expression of the Th17 cell master transcription factor RORγ τ was reduced in Fads1 Null mice compared to Wt (P = 0.03). Further, in Fads1 Null mice, mRNA levels of the Th17 cell signature cytokines, IL-17A and IL-17F, and IL-23, which maintains an established Th17 cell phenotype, were all reduced compared to Wt (P < 0.05). mRNA levels of the inflammatory mediator IL-6 were also reduced in Fads1 Null mice compared to Wt (P < 0.05). Collectively, these data indicate that, in mice devoid of AA-derived eicosanoids, colonic mRNA levels of Th17 cell related cytokines and transcription factors are suppressed following TNBS treatment, coinciding with an improved clinical outcome in Fads1 Null mice.
Table 4.
Colonic mucosal mRNA expression in Wt, Het, and Null Fads1 TNBS-treated mice1.
| Gene | Wt | Het | Null |
|---|---|---|---|
| IL-17A | 7.62 ± 4.70∗ | 1.10 ± 0.32‡ | 0.31 ± 0.01‡ |
| IL-17F | 12.95 ± 3.71∗ | 5.46 ± 1.07‡ | 4.19 ± 0.98‡ |
| IL-21 | 1.53 ± 0.80 | 0.70 ± 0.18 | 0.47 ± 0.14 |
| IL-22 | 1.48 ± 1.05 | 0.44 ± 0.33 | 0.10 ± 0.10 |
| IL-23 | 3.88 ± 0.43∗ | 3.58 ± 0.57∗‡ | 2.19 ± 0.39‡ |
| IL-23R | 0.73 ± 0.16 | 0.40 ± 0.05 | 0.65 ± 0.21 |
| IL-27 | 2.75 ± 0.67 | 1.86 ± 0.42 | 1.80 ± 0.46 |
| CCL20 | 1.42 ± 0.49 | 1.36 ± 0.57 | 1.13 ± 0.30 |
| CCR6 | 1.22 ± 0.38 | 0.82 ± 0.24 | 0.91 ± 0.44 |
| RORγτ | 11.07 ± 2.04∗ | 8.75 ± 1.34∗ | 5.28 ± 0.17‡ |
| Tbet | 8.35 ± 1.32∗ | 3.30 ± 0.66‡ | 4.12 ± 0.69‡ |
| Foxp3 | 2.95 ± 0.65 | 2.52 ± 0.23 | 1.81 ± 0.27 |
| IL-10 | 13.57 ± 3.91∗ | 9.08 ± 2.77∗ | 3.95 ± 1.38‡ |
| TGFβ1 | 1.80 ± 0.27 | 1.68 ± 0.26 | 1.31 ± 0.13 |
| MCP-1 | 25.57 ± 8.37∗ | 32.41 ± 14.00∗ | 8.86 ± 3.49‡ |
| IL-1β | 44.13 ± 12.57 | 31.30 ± 13.19 | 24.51 ± 12.95 |
| TNFα | 9.85 ± 1.81 | 9.89 ± 3.82 | 5.65 ± 1.72 |
| IL-6 | 11.54 ± 4.07∗ | 7.11 ± 3.34∗‡ | 2.10 ± 0.73‡ |
| IFNγ | 1.89 ± 1.01 | 0.52 ± 0.16 | 0.62 ± 0.21 |
| IL-4 | 0.64 ± 0.36 | nd | 0.38 ± 0.73 |
1Mean values ± SEM of TNBS-treated Wt, Het, and Null Fads1 mice (n = 6–9/genotype). All genes were measured within the same samples and expression was normalized to ribosomal 18S (in arbitrary units). For each gene, values not sharing a superscript symbol differ between genotypes (P ≤ 0.05). nd, not detected.
4. Discussion
In the TNBS colitis model, PGE2 has been shown to exacerbate colonic inflammatory processes and colitis severity through activation of the IL-23/IL17 axis and by increasing local Th17 cell numbers [18, 25, 30, 31]. Moreover, PGE2 coordinates locally with cytokines present within the tissue microenvironment to directly promote Th17 cell differentiation and effector function [18, 26–29]. Further, other n-6 PUFA-derived eicosanoids have also been shown to promote Th17 cell development [32]. In Fat-1 mice (Table 1), levels of n-6-derived eicosanoids were reduced and n-3 PUFA-derived PGE3 levels were increased compared to Wt littermates. Additionally, in Fads1 Null mice, the biosynthesis of AA-derived eicosanoids was reduced (Table 2) and colonic mucosal PGE2 levels were negligible compared to Wt. Interestingly, despite the different mechanisms through which colonic tissue AA-derived eicosanoid levels, including PGE2, were antagonized in the two genetic mouse models utilized in this study, the TNBS-induced inflammatory phenotype observed in both models was similar. This highlights the critical role which PGE2 and other AA-derived eicosanoids may be playing in activating inflammatory T cell subsets and perpetuating colitis-associated inflammation and disease severity. In both mouse models (Fat-1 transgenic and Fads1 Null mice), colonic mucosal AA-derived eicosanoid levels were reduced (Tables 1 and 2), colon injury scores improved (Figures 1(d) and 5(d)), the percentage of splenic inflammatory Th17 cells was decreased (Figures 4(a) and 6(a)), and colonic mucosal mRNA expression of Th17 cell markers and inflammatory mediators (IL-17A, RORγ τ, IL-23, and MCP-1) were reduced (Tables 3 and 4). Further, in both models, loss of AA-derived eicosanoids was accompanied by reduced Th17 cell numbers and a remodeled colonic mucosal gene expression profile that was consistent with suppressed inflammatory potential and Th17 cell polarization, activation, and maintenance [2, 9, 57, 58, 61–65]. These changes coincided with reduced TNBS-induced mucosal injury, thereby demonstrating the critical roles which AA and its metabolites, for example, PGE2, and Th17 cells play in perpetuating TNBS colitis severity. The similarities between the two models support the conclusion that the combination of inflammatory cytokines and noncytokine immunomodulators (i.e., AA-derived eicosanoids) present in the local inflammatory microenvironment during T cell differentiation and activation determines the ultimate phenotype of Th17 cells, as previously demonstrated in the TNBS model [26].
Enriched n-3 PUFA tissue levels in Fat-1 mice [46] corresponded to reduced colitis severity and the decreased percentages of proinflammatory Th17 and Th1 cells within the colon lamina propria (Figures 2(a) and 2(b)), the MLN (Figures 3(a) and 3(b)), and the spleen (Figures 4(a) and 4(b)). These results demonstrate that the activation and/or polarization of T cell subsets known to drive IBD [1–4] are directly antagonized by n-3 PUFA both locally and systemically. Conversely, the percentages of Tregs and Th2 cells were unaffected (Figures 2–4), indicating that the beneficial effect of n-3 PUFA was likely not due to enhanced Treg function or a shift in polarization from Th1 to Th2 cells. Interestingly, in Fat-1 mice, changes in the colonic gene expression profile were consistent with reduced Th17 cell activation, proliferation, maintenance, and inflammatory capacity [57, 63, 65] evidenced by reduced expression of IL-17A, IL-21, IL-23, MCP-1, IL-1β, and TNFα (Table 3) and increased expression of the Th17 cell-antagonizing cytokine, IL-27, and the anti-inflammatory cytokine, IL-10.
In Fads1 Null mice, due to the limitations in cell numbers, only splenic T cell subsets, representative of the systemic response, were assessed and the percentages of Th17, Th1, and Th2 cells were reduced compared to Wt littermates (Figures 6(a)–6(d)). Interestingly, the percentage of splenic Tregs was not affected in Fads1 Null mice compared to Wt and was actually elevated in Het mice (Figure 6(d)), indicating that not all CD4+ T cell effector subsets are reduced in Fads1 Null mice, as regulatory capabilities appear to be sustained. Moreover, the percentage of all T cell subsets detected in Fads1 Null saline control treated mice did not differ from either Wt or Het, indicating that the subsequent TNBS-induced reductions in cell numbers observed in Fads1 Null mice were not a byproduct of reduced splenic T cell numbers or activation. The reduced percentage of splenic Th17 and Th1 cells in Fads1 Null mice coincided with reduced colonic AA-derived eicosanoid levels including PGE2 (Table 1) and reduced colonic injury scores (Figure 5(d)). Interestingly, both the Fat-1 and Fads1 Null TNBS-treated mice exhibited suppressed IL-17A, IL-23, and RORγ τ colonic mRNA levels compared to Wt littermate controls, consistent with reduced inflammation and Th17 cell polarization, function, and maintenance (Tables 3 and 4) [57, 63, 65]. Collectively, the mucosal gene expression data indicate that the local percentage of activated Th17 cells in the colon in response to TNBS-induced colitis was likely also reduced in the Fads1 Null mouse, similar to the outcome in the Fat-1 mouse, although future studies confirming this interpretation are required.
AA is enriched in the membrane phospholipids of cells involved in the inflammatory response, and therefore, this fatty acid serves as the major precursor for eicosanoid mediators [66], which modulate both the intensity and duration of inflammatory responses [67]. The AA-derived eicosanoids have been shown to exert largely proinflammatory effects, although some anti-inflammatory effects have been reported [66, 67]. Ultimately, the eicosanoid profile and resulting functional outcome are contextually dependent upon the cell types present within the tissue microenvironment and the nature of the inflammatory stimulus. In this study, we showed that antagonism of AA-derived eicosanoids (specifically PGE2) by two distinct mechanisms results in fewer proinflammatory Th1 and Th17 cells both in local colitis-associated tissue sites and, systemically, a remodeled colonic phenotype consistent with reduced Th17 cell activation and maintenance, and reduced TNBS-induced mucosal damage and disease severity. PGE2-mediated reduction of mucosal damage has been reported in the dextran sodium sulfate- (DSS-) induced acute colitis model [68, 69]. This beneficial effect may be mediated through the EP4 receptor [70, 71] and is likely attributable to the ability of PGE2 to stimulate epithelial cell proliferation in the presence of DSS [72]. However, Fads1 Null mice, which are devoid of PGE2, exhibit reduced viability in response to DSS and are unable to stimulate mucosal repair mechanisms [48]. Since DSS is toxic to the gut epithelial cells and affects the integrity of the mucosal barrier [49], it is more appropriately utilized as a mucosal wounding model, which is further supported by the lack of adaptive immune system involvement in the acute phase [73], thereby questioning the applicability of the DSS model to human IBD.
Conversely, the TNBS colitis model recapitulates many of the macroscopic and histological characteristics of human IBD [74, 75]. CD4+ T cell activation/tissue accumulation is a key feature of the immunopathology [49, 74, 75], and specifically, the Th17 cell subset has been shown to drive the inflammatory pathology [23]. Moreover, inhibition of CD4+ T cells via CD4 mimetics or anti-CD4 monoclonal antibody prevents disease development [76]. Administration of 16,16-dimethyl prostaglandin E2 or Enprostil (PGE2 analogue) to TNBS-treated rats has been shown to acutely reduce histological damage scores and myeloperoxidase activity [77, 78], although these effects are not sustained for longer durations after exposure [78]. However, Th17 cell activation and Th17 cell driven inflammation mediated by PGE2 during TNBS-induced colitis have been documented previously [18, 26–29]. Other AA-derived eicosanoids may play a contributing role to the activation of Th17 cells as documented elsewhere [32]. Therefore, future studies investigating the specific contributions of other n-6-derived eicosanoids are warranted. The effects of PGE2 are mediated through receptors (EP1-EP4), which are expressed on multiple cell types [79] including T cells, dendritic cells [25, 26, 28], and the colonic mucosa [80]. Previous data indicate that the PGE2-dependent effect on Th17 cells is meditated through the EP2 and EP4 receptors [25, 26, 29], suggesting that antagonism of these receptors could be a potential therapeutic target for IBD treatment, although future studies are required.
In conclusion, our observations demonstrate the utility of antagonizing colonic AA-derived eicosanoids as a mechanism to reduce inflammatory T cell activation and improve colitis-associated immunopathology. Moreover, these results suggest that dietary n-3 PUFA could be used alone, or as an adjunctive therapy, in improving the clinical outcome of colonic mucosal Th17 cell-mediated pathologies.
Supplementary Material
Final body weights and colon lengths of saline control and TNBS-treated Fat-1 and Fads1 mice are shown in Supplemental Tables 1 and 2, respectively. Representative dot plots depicting the detection of CD4+ T cell subsets in TNBS-treated Fat-1 and Fads1 mice are shown in Supplemental Figures 1 and 2, respectively.
Acknowledgments
The authors thank Dr. Jing Kang, Harvard School of Medicine, for providing Fat-1 breeder mice. This research was supported in part by a Postdoctoral Fellowship PDF-388466-2010 Award to Jennifer M. Monk by the Natural Sciences and Engineering Research Council (NSERC) of Canada and Grants from the Cancer Prevention and Research Institute of Texas (CPRIT), RP120028, the National Institutes of Health (NIH), CA59034, CA129444, and the U.S. Department of Agriculture CSREES Special Grant, “Designing Foods for Health,” 2010-34402-20875.
Abbreviations
- IBD:
Inflammatory bowel disease
- CD:
Crohn's disease
- FO:
Fish oil
- PUFA:
Polyunsaturated fatty acids
- EPA:
Eicosapentaenoic acid
- DHA:
Docosahexaenoic acid
- Tregs:
Regulatory T cells
- TNBS:
2,4,6-Trinitrobenzene sulfonic acid
- PGE2:
Prostaglandin E2
- PG:
Prostaglandin
- TX:
Thromboxane
- HODE:
Hydroxyl-octadecadienoic acid
- HETE:
Hydroxyl-eicosatetraenoic acid
- AEA:
Arachidonoyl ethanolamine
- AG:
Arachidonoyl glycerol
- DSS:
Dextran sodium sulfate.
Conflict of Interests
The authors declare that there is no conflict of interests.
References
- 1.Liu ZJ, Yadav PK, Su JL, Wang JS, Fei K. Potential role of Th17 cells in the pathogenesis of in flammatory bowel disease. World Journal of Gastroenterology. 2009;15(46):5784–5788. doi: 10.3748/wjg.15.5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarra M, Pallone F, MacDonald TT, Monteleone G. IL-23/IL-17 axis in IBD. Inflammatory Bowel Diseases. 2010;16(10):1808–1813. doi: 10.1002/ibd.21248. [DOI] [PubMed] [Google Scholar]
- 3.Shih DQ, Targan SR. Immunopathogenesis of inflammatory bowel disease. World Journal of Gastroenterology. 2008;14(3):390–400. doi: 10.3748/wjg.14.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zenewicz LA, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends in Molecular Medicine. 2009;15(5):199–207. doi: 10.1016/j.molmed.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Eastaff-Leung N, Mabarrack N, Barbour A, Cummins A, Barry S. Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in inflammatory bowel disease. Journal of Clinical Immunology. 2010;30(1):80–89. doi: 10.1007/s10875-009-9345-1. [DOI] [PubMed] [Google Scholar]
- 6.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52(1):65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hölttä V, Klemetti P, Sipponen T, et al. IL-23/IL-17 immunity as a hallmark of Crohn's disease. Inflammatory Bowel Diseases. 2008;14(9):1175–1184. doi: 10.1002/ibd.20475. [DOI] [PubMed] [Google Scholar]
- 8.Elson CO, Cong Y, Weaver CT, et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132(7):2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- 9.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. Journal of Clinical Investigation. 2006;116(5):1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tjonneland A, Overvad K, Bergmann MM, et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: a nested case-control study within a European prospective cohort study. Gut. 2009;58(12):1606–1611. doi: 10.1136/gut.2008.169078. [DOI] [PubMed] [Google Scholar]
- 11.Sakata D, Yao C, Narumiya S. Prostaglandin E2, an immunoactivator. Journal of Pharmacological Sciences. 2010;112(1):1–5. doi: 10.1254/jphs.09r03cp. [DOI] [PubMed] [Google Scholar]
- 12.Ahrenstedt O, Hallgren R, Knutson L. Jejunal release of prostaglandin E2 in Crohn's disease: relation to disease activity and first-degree relatives. Journal of Gastroenterology and Hepatology. 1994;9(6):539–543. doi: 10.1111/j.1440-1746.1994.tb01557.x. [DOI] [PubMed] [Google Scholar]
- 13.Ligumsky M, Karmeli F, Sharon P, Zor U, Cohen F, Rachmilewitz D. Enhanced thromboxane A2 and prostacyclin production by cultured rectal mucosa in ulcerative colitis and its inhibition by steroids and sulfasalazine. Gastroenterology. 1981;81(3):444–449. [PubMed] [Google Scholar]
- 14.Rampton DS, Sladen GE, Youlten LJF. Rectal mucosal prostaglandin E2 release and its relation to disease activity, electrical potential difference, and treatment in ulcerative colitis. Gut. 1980;21(7):591–596. doi: 10.1136/gut.21.7.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharon P, Ligumsky M, Rachmilewitz D, Zor U. Role of prostaglandins in ulcerative colitis. Enhanced production during active disease and inhibition by sulfasalazine. Gastroenterology. 1978;75(4):638–640. [PubMed] [Google Scholar]
- 16.Sharon P, Stenson WF. Enhanced synthesis of leukotriene B4 by colonic mucosa in inflammatory bowel disease. Gastroenterology. 1984;86(3):453–460. [PubMed] [Google Scholar]
- 17.Wiercinska-Drapalo A, Flisiak R, Prokopowicz D. Mucosal and plasma prostaglandin E2 in ulcerative colitis. Hepato-Gastroenterology. 1999;46(28):2338–2342. [PubMed] [Google Scholar]
- 18.Barrie A, Khare A, Henkel M, et al. Prostaglandin E2 and IL-23 plus IL-1β differentially regulate the Th1/Th17 immune response of human CD161+CD4+ memory T cells. Clinical and Translational Science. 2011;4(4):268–273. doi: 10.1111/j.1752-8062.2011.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cosmi L, de Palma R, Santarlasci V, et al. Human interleukin 17-producing cells originate from a CD161 +CD4+ T cell precursor. The Journal of Experimental Medicine. 2008;205(8):1903–1916. doi: 10.1084/jem.20080397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinschek MA, Boniface K, Sadekova S, et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. Journal of Experimental Medicine. 2009;206(3):525–534. doi: 10.1084/jem.20081712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maggi L, Santarlasci V, Capone M, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. European Journal of Immunology. 2010;40(8):2174–2181. doi: 10.1002/eji.200940257. [DOI] [PubMed] [Google Scholar]
- 22.Neurath M, Fuss I, Strober W. TNBS-colitis. International Reviews of Immunology. 2000;19(1):51–62. doi: 10.3109/08830180009048389. [DOI] [PubMed] [Google Scholar]
- 23.Winer S, Paltser G, Chan Y, et al. Obesity predisposes to Th17 bias. European Journal of Immunology. 2009;39(9):2629–2635. doi: 10.1002/eji.200838893. [DOI] [PubMed] [Google Scholar]
- 24.Khan I, Al-Awadi FM, Thomas N, Haridas S, Anim JT. Cyclooxygenase-2 inhibition and experimental colitis: beneficial effects of phosphorothioated antisense oligonucleotide and meloxicam. Scandinavian Journal of Gastroenterology. 2002;37(12):1428–1436. doi: 10.1080/003655202762671314. [DOI] [PubMed] [Google Scholar]
- 25.Sheibanie AF, Yen J, Khayrullina T, et al. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23→IL-17 axis. Journal of Immunology. 2007;178(12):8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 26.Boniface K, Bak-Jensen KS, Li Y, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. The Journal of Experimental Medicine. 2009;206(3):535–548. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chizzolini C, Chicheportiche R, Alvarez M, et al. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood. 2008;112(9):3696–3703. doi: 10.1182/blood-2008-05-155408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Napolitani G, Acosta-Rodriguez EV, Lanzavecchia A, Sallusto F. Prostaglandin E2 enhances Th17 responses via modulation of IL-17 and IFN-γ production by memory CD4+ T cells. European Journal of Immunology. 2009;39(5):1301–1312. doi: 10.1002/eji.200838969. [DOI] [PubMed] [Google Scholar]
- 29.Yao C, Sakata D, Esaki Y, et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through TH1 cell differentiation and TH17 cell expansion. Nature Medicine. 2009;15(6):633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 30.Khayrullina T, Yen JH, Jing H, Ganea D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. The Journal of Immunology. 2008;181:721–735. doi: 10.4049/jimmunol.181.1.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheibanie AF, Tadmori I, Jing H, Vassiliou E, Ganea D. Prostaglandin E2 induces IL-23 production in bone marrow-derived dendritic cells. The FASEB Journal. 2004;18:1318–1320. doi: 10.1096/fj.03-1367fje. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, Qin J, Wei P, et al. Effects of leukotriene B4 and prostaglandin E2 on the differentiation of murine Foxp3+ T regulatory cells and Th17 cells. Prostaglandins Leukotrienes and Essential Fatty Acids. 2009;80(4):195–200. doi: 10.1016/j.plefa.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Calder PC. Mechanisms of action of (n-3) fatty acids. The Journal of Nutrition. 2012;142:592S–599S. doi: 10.3945/jn.111.155259. [DOI] [PubMed] [Google Scholar]
- 34.Chapkin RS, Davidson LA, Ly L, Weeks BR, Lupton JR, McMurray DN. Immunomodulatory effects of (n-3) fatty acids: putative link to inflammation and colon cancer. The Journal of Nutrition. 2007;137(1):200S–204S. doi: 10.1093/jn/137.1.200S. [DOI] [PubMed] [Google Scholar]
- 35.Sijben JWC, Calder PC. Differential immunomodulation with long-chain n-3 PUFA in health and chronic disease. The Proceedings of the Nutrition Society. 2007;66(2):237–259. doi: 10.1017/S0029665107005472. [DOI] [PubMed] [Google Scholar]
- 36.Turner D, Shah PS, Steinhart AH, Zlotkin S, Griffiths AM. Maintenance of remission in inflammatory bowel disease using omega-3 fatty acids (fish oil): a systematic review and meta-analyses. Inflammatory Bowel Diseases. 2011;17(1):336–345. doi: 10.1002/ibd.21374. [DOI] [PubMed] [Google Scholar]
- 37.Kong SC, Hurlstone DP, Pocock CY, et al. The incidence of self-prescribed oral complementary and alternative medicine use by patients with gastrointestinal diseases. Journal of Clinical Gastroenterology. 2005;39(2):138–141. [PubMed] [Google Scholar]
- 38.Caughey GE, Mantzioris E, Gibson RA, Cleland LG, James MJ. The effect on human tumor necrosis factor alpha and interleukin 1 beta production of diets enriched in n-3 fatty acids from vegetable oil or fish oil. The American Journal of Clinical Nutrition. 1996;63:116–122. doi: 10.1093/ajcn/63.1.116. [DOI] [PubMed] [Google Scholar]
- 39.Endres S, Ghorbani R, Kelley VE, et al. The effect of dietary supplementation with n-3 polyunsaturated fatty acids on the synthesis of interleukin-1 and tumor necrosis factor by mononuclear cells. The New England Journal of Medicine. 1989;320(5):265–271. doi: 10.1056/NEJM198902023200501. [DOI] [PubMed] [Google Scholar]
- 40.Lee TH, Hoover RL, Williams JD, et al. Effect of dietary enrichment with eicosapentaenoic and docosahexaenoic acids on in vitro neutrophil and monocyte leukotriene generation and neutrophil function. The New England Journal of Medicine. 1985;312(19):1217–1224. doi: 10.1056/NEJM198505093121903. [DOI] [PubMed] [Google Scholar]
- 41.Sperling RI, Benincaso AI, Knoell CT, Larkin JK, Austen KF, Robinson DR. Dietary ω-3 polyunsaturated fatty acids inhibit phosphoinositide formation and chemotaxis in neutrophils. The Journal of Clinical Investigation. 1993;91(2):651–660. doi: 10.1172/JCI116245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serhan CN, Arita M, Hong S, Gotlinger K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids. 2004;39(11):1125–1132. doi: 10.1007/s11745-004-1339-7. [DOI] [PubMed] [Google Scholar]
- 43.Zhang P, Kim W, Zhou L, et al. Dietary fish oil inhibits antigen-specific murine Th1 cell development by suppression of clonal expansion. The Journal of Nutrition. 2006;136(9):2391–2398. doi: 10.1093/jn/136.9.2391. [DOI] [PubMed] [Google Scholar]
- 44.Zhang P, Smith R, Chapkin RS, McMurray DN. Dietary (n-3) polyunsaturated fatty acids modulate murine Th1/Th2 balance toward the Th2 pole by suppression of Th1 development. The Journal of Nutrition. 2005;135:1745–1751. doi: 10.1093/jn/135.7.1745. [DOI] [PubMed] [Google Scholar]
- 45.Monk JM, Hou TY, Turk HF, et al. Dietary n-3 polyunsaturated fatty acids (PUFA) decrease obesity-associated Th17 cell-mediated inflammation during colitis. PLoS ONE. 2012;7(11) doi: 10.1371/journal.pone.0049739.e49739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427(6974):p. 504. doi: 10.1038/427504a. [DOI] [PubMed] [Google Scholar]
- 47.Jia Q, Lupton JR, Smith R, et al. Reduced colitis-associated colon cancer in fat-1 (n-3 fatty acid desaturase) transgenic mice. Cancer Research. 2008;68(10):3985–3991. doi: 10.1158/0008-5472.CAN-07-6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan YY, Monk JM, Hou TY, et al. Characterization of an arachidonic acid-deficient (Fads1 knockout) mouse model. Journal of Lipid Research. 2012;53(7):1287–1295. doi: 10.1194/jlr.M024216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nature Protocols. 2007;2(3):541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 50.Monk JM, Kim W, Callaway E, et al. Immunomodulatory action of dietary fish oil and targeted deletion of intestinal epithelial cell PPAR δ in inflammation-induced colon carcinogenesis. The American Journal of Physiology Gastrointestinal and Liver Physiology. 2012;302(1):G153–G167. doi: 10.1152/ajpgi.00315.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denning TL, Norris BA, Medina-Contreras O, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. Journal of Immunology. 2011;187(2):733–747. doi: 10.4049/jimmunol.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monk JM, Jia Q, Callaway E, et al. Th17 cell accumulation is decreased during chronic experimental colitis by (n-3) PUFA in fat-1 mice. The Journal of Nutrition. 2012;142(1):117–124. doi: 10.3945/jn.111.147058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang P, Chan D, Felix E, et al. Determination of endogenous tissue inflammation profiles by LC/MS/MS: COX- and LOX-derived bioactive lipids. Prostaglandins Leukotrienes and Essential Fatty Acids. 2006;75(6):385–395. doi: 10.1016/j.plefa.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 54.Hughes D, Otani T, Yang P, et al. NAD+-dependent 15-hydroxyprostaglandin dehydrogenase regulates levels of bioactive lipids in non-small cell lung cancer. Cancer Prevention Research. 2008;1(4):241–249. doi: 10.1158/1940-6207.CAPR-08-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kruschewski M, Foitzik T, Perez-Cantó A, Hũbotter A, Buhr HJ. Changes of colonic mucosal microcirculation and histology in two colitis models: An experimental study using intravital microscopy and a new histological scoring system. Digestive Diseases and Sciences. 2001;46(11):2336–2343. doi: 10.1023/a:1012334727509. [DOI] [PubMed] [Google Scholar]
- 56.Nieto N, Torres MI, Ŕos A, Gil A. Dietary polyunsaturated fatty acids improve histological and biochemical alterations in rats with experimental ulcerative colitis. The Journal of Nutrition. 2002;132(1):11–19. doi: 10.1093/jn/132.1.11. [DOI] [PubMed] [Google Scholar]
- 57.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Seminars in Immunology. 2007;19(6):409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce proinflammatory T H17 cells. Nature. 2007;448(7152):484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448(7152):480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 60.Batten M, Li J, Yi S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nature Immunology. 2006;7(9):929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 61.Monteleone G, Monteleone I, Fina D, et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-γ production in Crohn's disease. Gastroenterology. 2005;128(3):687–694. doi: 10.1053/j.gastro.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 62.Stumhofer JS, Laurence A, Wilson EH, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nature Immunology. 2006;7(9):937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 63.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annual Review of Immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 64.Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORγt and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nature Immunology. 2008;9(11):1297–1306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nature Immunology. 2007;8(9):967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 66.Calder PC. Omega-3 fatty acids and inflammatory processes. Nutrients. 2010;2(3):355–374. doi: 10.3390/nu2030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. Journal of Clinical Investigation. 2001;108(1):15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hirata I, Murano M, Nitta M, Sasaki S, Toshina K, et al. Estimation of mucosal inflammatory mediators in rat DSS-induced colitis. Possible role of PGE(2) in protection against mucosal damage. Digestion. 2001;63(supplement 1):73–80. doi: 10.1159/000051915. [DOI] [PubMed] [Google Scholar]
- 69.Sasaki S, Hirata I, Maemura K, et al. Prostaglandin E2 inhibits lesion formation in dextran sodium sulphate- induced colitis in rats and reduces the levels of mucosal inflammatory cytokines. Scandinavian Journal of Immunology. 2000;51(1):23–28. doi: 10.1046/j.1365-3083.2000.00623.x. [DOI] [PubMed] [Google Scholar]
- 70.Kabashima K, Saji T, Murata T, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. The Journal of Clinical Investigation. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nitta M, Hirata I, Toshina K, Murano M, Maemura K, et al. Expression of the EP4 prostaglandin E2 receptor subtype with rat dextran sodium sulphate colitis: colitis suppression by a selective agonist, ONO-AE1-329. Scandinavian Journal of Immunology. 2002;56:66–75. doi: 10.1046/j.1365-3083.2002.01096.x. [DOI] [PubMed] [Google Scholar]
- 72.Tessner TG, Cohn SM, Schloemann S, Stenson WF. Prostaglandins prevent decreased epithelial cell proliferation associated with dextran sodium sulfate injury in mice. Gastroenterology. 1998;115:874–882. doi: 10.1016/s0016-5085(98)70259-8. [DOI] [PubMed] [Google Scholar]
- 73.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107(6):1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 74.Blumberg RS, Saubermann LJ, Strober W. Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Current Opinion in Immunology. 1999;11(6):648–656. doi: 10.1016/s0952-7915(99)00032-1. [DOI] [PubMed] [Google Scholar]
- 75.Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Advanced Drug Delivery Reviews. 2007;59:1073–1083. doi: 10.1016/j.addr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 76.Okamoto S, Watanabe M, Yamazaki M, et al. A synthetic mimetic of CD4 is able to suppress disease in a rodent model of immune colitis. European Journal of Immunology. 1999;29:355–366. doi: 10.1002/(SICI)1521-4141(199901)29:01<355::AID-IMMU355>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 77.Allgayer H, Deschryver K, Stenson WF. Treatment with 16,16'-dimethyl prostaglandin E2 before and after induction of colitis with trinitrobenzenesulfonic acid in rats decreases inflammation. Gastroenterology. 1989;96:1290–1300. doi: 10.1016/s0016-5085(89)80016-2. [DOI] [PubMed] [Google Scholar]
- 78.Onizuka Y, Murase K, Furusu H, Isomoto H, Mizuta Y. Effect of intrarectal prostaglandin E2 analogue (enprostil) on trinitrobenzenesulphonic acid-induced colitis in rats. Journal of International Medical Research. 2000;28:28–35. doi: 10.1177/147323000002800105. [DOI] [PubMed] [Google Scholar]
- 79.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacology & therapeutics. 2004;103(2):147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 80.Dey I, Lejeune M, Chadee K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. British Journal of Pharmacology. 2006;149:611–623. doi: 10.1038/sj.bjp.0706923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Final body weights and colon lengths of saline control and TNBS-treated Fat-1 and Fads1 mice are shown in Supplemental Tables 1 and 2, respectively. Representative dot plots depicting the detection of CD4+ T cell subsets in TNBS-treated Fat-1 and Fads1 mice are shown in Supplemental Figures 1 and 2, respectively.