

Abstract
Background. Endogenous cannabinoid anandamide (AEA) protects neurons from oxidative injury in rodent models; however the mechanism of AEA-induced neuroprotection remains to be determined. Activation of neuronal NADPH oxidase 2 (Nox2) contributes to oxidative damage of the brain, and inhibition of Nox2 can attenuate cerebral oxidative stress. We aimed to determine whether the neuronal Nox2 was involved in protection mediated by AEA. Methods. The mouse hippocampal neuron cell line HT22 was exposed to hydrogen peroxide (H2O2) to mimic oxidative injury of neurons. The protective effect of AEA was assessed by measuring cell metabolic activity, apoptosis, lactate dehydrogenase (LDH) release, cellular morphology, intracellular reactive oxygen species (ROS), and antioxidant and oxidant levels and Nox2 expression. Results. HT22 cells exposed to H2O2 demonstrated morphological changes, decreased LDH release, reduced metabolic activity, increased levels of intracellular ROS and oxidized glutathione (GSSG), reduced levels of superoxide dismutase (SOD), and reduced glutathione (GSH) and increased expression of Nox2. AEA prevented these effects, a property abolished by simultaneous administration of CB1 antagonist AM251 or CB1-siRNA. Conclusion. Nox2 inhibition is involved in AEA-induced cytoprotection against oxidative stress through CB1 activation in HT22 cells.
1. Introduction
Oxidative stress is implicated in the pathology of many central nervous system (CNS) disorders, including Alzheimer's disease, Parkinson's disease, and ischemic stroke [1–3]. Hydrogen peroxide (H2O2) is produced at nearly every stage of the oxidative cycle and widely applied to induce oxidative stress in vitro [4]. H2O2-induced oxidative stress can cause lipid peroxidation, mitochondria injury, and DNA damage [5, 6].
NADPH oxidase (Nox) is a membrane-associated enzyme complex consisting of several subunits including NADPH oxidase 2 (Nox2). Activation of neuronal Nox2 contributes to oxidative damage of the CNS [7], and inhibition of Nox2 can attenuate cerebral oxidative stress injury [8]. We have previously demonstrated that inhibition of Nox2 reduced the damage induced by oxygen glucose-deprivation to a mouse hippocampal neuron cell line, HT22 [9].
Endogenous cannabinoid anandamide (N-arachidonoylethanolamine, AEA) mimics the bioactivity of Δ9-tetrahydrocannabinol (THC), the principal psychoactive component of marijuana [10]. There are two main cannabinoid receptors, CB1 and CB2 [11]. In the CNS, CB1 is mainly expressed in neurons, and CB2 in glial cells, such as microglia and astrocytes [11]. It was recently demonstrated in rodent models that AEA conferred neuroprotection by activating cannabinoid receptors. AEA could protect the newborn brain against excitotoxicity by activating CB1 [12] and attenuated cytotoxic edema caused by administration of Na+/K+-ATPase inhibitor [10]. We have previously reported that electroacupuncture pretreatment induces neuroprotection by stimulating release of AEA through a protein kinase C epsilon-mediated pathway [13]. However, the precise mechanism by which AEA mediated protection in the CNS remains undefined. The aim of this study was to determine whether AEA could protect HT22 cells against H2O2-induced injury and whether Nox2 was involved in the AEA-induced protection from oxidative stress via activation of CB1.
2. Materials and Methods
2.1. Materials
The HT22 cell line was a gift from Xuzhou Medical College (Xuzhou, China). The primary anti-CB1 antibody and primary anti-Nox2 antibody were purchased from Abcam Ltd. (Cambridge, UK), the primary anti-cleaved caspase-3 antibody was obtained from Santa Cruz (USA), and bovine serum albumin (BSA) and the cy3-labeled secondary antibody were purchased from Beijing Cowin Bioscience Co., Ltd. (Beijing, China). The AEA, AM251, Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), apocynin, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-tetrazolium bromide (MTT), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The 4′,6-diamidino-2-phenylindole (DAPI) and ROS Reagent kit were obtained from Beyotime (Nantong, China). The lactate dehydrogenase (LDH), superoxide dismutase (SOD), and reduced glutathione (GSH) and oxidized glutathione (GSSG) kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
2.2. Cell Culture
HT22 cells were cultured in DMEM with 10% FBS (v/v), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. The medium was replaced every 2 days. When cell density reached roughly 70–80%, cells were exposed to the indicated drugs for 3 h. Then some evaluations were performed.
2.3. Experimental Protocols
To find a suitable H2O2 concentration, the HT22 cells were assigned into seven groups (Figure 1(a)). Except for control group, the other six groups were exposed to different concentrations of H2O2 for 3 h, ranging from 50 μM to 1000 μM. Then MTT assay was taken to determine the injury degree of the cells. To find a suitable AEA concentration, the cells were divided into six groups. Except for control and H2O2 only groups, the other four groups were exposed to 200 μM H2O2 plus different concentrations of AEA (Figure 1(b)). Then MTT assay was taken to evaluate the injury degree of the cells.
Figure 1.
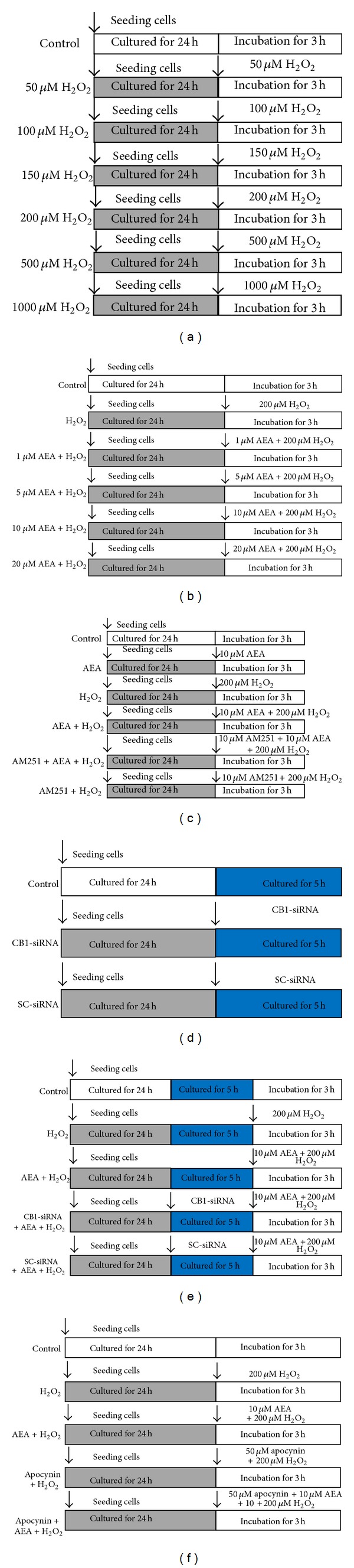
Experimental protocol diagram. (a) The HT22 cells were assigned into seven groups. The control cells were cultured in drug-free medium, and the other six groups were exposed to different concentrations of H2O2 for 3 h, ranging from 50 μM to 1000 μM. MTT assay was taken to determine the injury degree. (b) The cells were divided into six groups; except for control and H2O2 only groups, the other four groups were exposed to 200 μM H2O2 plus different concentrations of AEA for 3 h. MTT assay was taken to evaluate the injury degree. (c) The cells were assigned into six groups, including control, AEA, H2O2, AEA + H2O2, AM251 + AEA + H2O2, and AM251 + H2O2 groups. After an incubation of 3 h, MTT assay, LDH release, and western blotting were taken to determine the roles of CB1 and Nox2 in AEA-induced protection. (d) The cells were divided into three groups, including control, CB1-siRNA, and SC-siRNA groups; after an incubation of 5 h, western blotting was used to evaluate the silencing rate of CB1 protein expression. (e) Then, the cells were divided into five groups, including control, H2O2, AEA + H2O2, CB1-siRNA + AEA + H2O2, and SC-siRNA + AEA + H2O2; the cell injury was evaluated by MTT and LDH release at 3 h after incubation, and ROS generation was evaluated by measuring fluorescence intensity. (f) The cells were divided into five groups, including control, H2O2, AEA + H2O2, apocynin + AEA + H2O2, and apocynin + AEA + H2O2; western blotting, MTT assay, and LDH release were taken to measure Nox2 expression and cell injury.
Then the cells were assigned into six groups, including control, AEA, H2O2, AEA + H2O2, AM251 + AEA + H2O2, and AM251 + H2O2 groups. After an incubation of 3 h, MTT assay, LDH release, and western blotting were taken to determine the roles of CB1 and Nox2 in AEA-induced protection (Figure 1(c)). To further investigate the role of CB1 in AEA-induced protection against H2O2 in HT22 cells, the HT22 cells were divided into three groups, including control, CB1-siRNA, and SC-siRNA groups; after an incubation of 5 h, western blotting was used to evaluate the silencing rate of CB1 protein expression (Figure 1(d)). Then, the cells were divided into five groups, including control, H2O2, AEA + H2O2, CB1-siRNA + AEA + H2O2, and SC-siRNA + AEA + H2O2. Then cells injury was evaluated by MTT and LDH release at 3 h after incubation, and ROS generation was evaluated by measuring fluorescence intensity (Figure 1(e)). Apocynin, a specific Nox inhibitor [14], was used to further investigate the role of Nox2 in AEA-induced protection against H2O2. The cells were divided into five groups, including control, H2O2, AEA + H2O2, apocynin + AEA + H2O2, and apocynin + AEA + H2O2; western blotting, MTT assay, and LDH release were used to measure Nox2 expression and cell injury (Figure 1(f)).
2.4. Cell Viability
HT22 cells were plated at a density of 1 × 104 cells/well in 96-well plates. After treatment with different drugs for 3 h, cell injury was evaluated by MTT assay. 20 μL of 5 mg/mL MTT solution was added to each well, and after 4 h incubation at 37°C the supernatant of each well was carefully removed. Then 150 μL DMSO was added per well to solubilize the formazan product. The plate was then shaken for 10 min to ensure the formazan had completely dissolved. Absorbance at 490 nm was evaluated using a spectrophotometer (TECAN, CH).
2.5. LDH Release
HT22 cells were plated at a density of 2 × 104 cells/well into a 24-well plate. After the treatments, the supernatants of each well were removed for assessment of LDH release, which was measured according to the manufacturer's instructions for the LDH assay kit. In brief, 100 μL of cell-free supernatant, 250 μL of buffer, and 50 μL of coenzyme were mixed homogeneously and the supernatant was incubated with this reaction mixture for 15 min at 37°C. Next, 250 μL of 2,4-dinitrophenylhydrazine was added into the mixture and incubated for an additional 15 min at 37°C in the dark. Finally, 2.5 mL of 400 mM of NaOH was added into the mixture to stop the reaction. After 3 min, the absorbance of the mixture was determined at 440 nm by spectrophotometry. The absorbance of the sample blank, standard, and standard blank was measured at the same time. LDH activity was calculated according to the following formula:
| (1) |
2.6. Apoptotic Rate
HT22 cells were seeded into a 6-well plate at a density of 1 × 105 cells/well. After treatment with H2O2, AEA, and/or AM251, cells were centrifuged at 1000 rpm for 5 min. After two washes with ice-cold phosphate buffered saline (PBS), cells were resuspended in binding buffer at a density of 1 × 106 cells/mL. And 5 μL of fluorescein 5-isothiocyanate [2-(3,6-dihydroxy-9H-xanthen-9-yl)-5-isothiocyanatobenzoic acid] FITC-conjugated anti-annexin-V staining antibody and 2 μL of propidium iodide (PI) solution were added to 100 μL of the binding buffer. After thorough mixing, and 15 min incubation at room temperature in the dark, the apoptotic rate was assessed by flow cytometry (BD, USA).
2.7. Immunocytochemistry
HT22 cells were seeded into five confocal microscopy special dishes at a density of 2 × 104 cells/dish. After 24 h, the cells were incubated with drug-free medium, 200 μM H2O2, 10 μM AEA + 200 μM H2O2, 10 μM AM251 + 10 μM AEA + 200 H2O2, and 10 μM AM251 + 200 μM H2O2, respectively, for 3 h before the dishes were washed three times with PBS, then fixed with 4% paraformaldehyde solution for 1 h, and blocked with 50 mg BSA/mL in PBS for 30 min. Cells were incubated with primary anti-CB1 antibody (1 : 50) for 24 h at 4°C and then washed three times with PBS, before incubation with Cy3-labeled secondary antibody (1 : 200) for 1 h at room temperature. 200 μL of DAPI staining solution was added into each dish for 5 min; then the dishes were washed thrice with PBS. CB1 expression was assessed by a confocal microscope (FV10i, Olympus, Japan) (excitation = 550 nm; emission = 570 nm).
2.8. Intracellular ROS
Intracellular ROS was detected by a ROS reagent kit utilizing the ability of intracellular ROS to oxidize nonfluorescent and colorless 2′,7′-dichlorofluorescin diacetate (DCFH-DA) into a fluorescent dichlorofluorescein (DCF). Cells were incubated with 10 μM DCFH-DA at 37°C for 10 min and then rinsed twice with PBS, and ROS level was immediately assessed under a Leica DMI6000B, fluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany) (excitation = 488 nm and emission = 525 nm). Images were analyzed using Image Pro-Plus software (IPP 6.0, Media Cybernetics, Silver Spring, MD, USA).
2.9. Intracellular SOD, GSH, and GSSG
HT22 cells were seeded into 6-well plates at a density of 5 × 105 cells/well. After treatment with H2O2, AEA, and/or AM251 (Figure 1(c)), cells were harvested and homogenized in 0.5 mL of 0.1 M phosphate buffer (pH 7.4). The mixture was centrifuged at 3000 rpm for 10 min at 4°C, and supernatants were used for SOD, GSH, and GSSG activity assessments with the corresponding reagent kits by spectrophotometry.
2.10. Short Interfering RNA
The sequence of mouse-CB1 short interfering RNA (CB1-siRNA, sc-39911) and scrambled short interfering RNA (SC-siRNA, sc-37007) were purchased from Santa Cruz Biotechnology. HT22 cells were seeded in 6-well plates at a density of 2 × 105 cells/well in 2 mL DMEM without FBS. When the cell density reached 60–80% confluence, cells were washed with 2 mL siRNA Transfection Medium (sc-36868). CB1-siRNA and SC-siRNA were added to the cells and incubated for 5 h at 37°C before 1 mL of normal growth medium containing 20% FBS was added into each well without removing the transfection mixture. After an additional incubation of 24 h, cells were harvested.
2.11. Immunoblot Analysis
HT22 cells were lysed with modified RIPA-buffer containing a protease inhibitor-cocktail and 100 μM phenylmethanesulfonyl fluoride on ice for 30 min. The total protein content was qualified by a bicinchoninic acid kit. Total protein lysates were subjected to 12% sodium dodecyl SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Membranes were incubated with rabbit anti-mouse primary antibody (CB1, 1 : 1000; Nox2/gp91phox, 1 : 1000; Abcam, UK; cleaved caspase-3, 1 : 500; Santa Cruz, USA) in PBS with 0.1% Tween-20 overnight at 4°C and then incubated for 1 h at room temperature with anti-rabbit IgG. β-Actin, tubulin, and GADPH served as the control, respectively. Expression was visualized by enhanced chemiluminescence. The signal was quantified by densitometry by an immunoblotting detection system (Alpha Innotech, USA).
2.12. Real-Time PCR
Total RNA was isolated from HT22 cells with TRIzol reagent (Invitrogen, USA) according to the manufacturer's instructions. Total RNA (5 μg) was used for first strand cDNA synthesis using the cDNA synthesis kit (TaKaRa, Japan). The PCR conditions for Nox2 were as follows: after initial denaturation at 95°C for 5 min, 40 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 1 min were performed, followed by a 10 min extension at 72°C. The Nox2 RNA primers were as follows: 5′-CAAGATGGAGGTGGGACAGT-3′ (sense) and 5′-CAGGAGCAGAGGTCAGTGTG-3′ (antisense); and for β-actin, 5′-GAT GAG ATTGGC ATG GCT TT-3′ (sense) and 5′-GAG AA G TGGGGT GGC TT-3′ (antisense). Quantification of Nox2 mRNA was normalized to β-actin. The specificity of the PCR amplification products was assessed by dissociation melting curve analysis. Relative multiples of changes in mRNA expression were determined with the relative comparative threshold method [15].
2.13. Statistical Analysis
SPSS 11.0 (SPSS Inc, Chicago, IL) was used to conduct statistical analysis. Values were expressed as means ± standard deviation (SD). Results were compared by one-way ANOVA, followed by Tukey's Multiple Comparison Test. P < 0.05 was considered statistically significant.
3. Results
3.1. AEA Protected HT22 Cells Exposed to H2O2 in a Dose-Dependent Manner
HT22 cells were exposed to H2O2 for 3 h, which decreased the cell metabolic activity in a dose-dependent manner. Exposure to 200 μM H2O2 decreased the cell metabolic activity by roughly 50% (Figure 2(a)), and we used this condition for the subsequent experiments.
Figure 2.
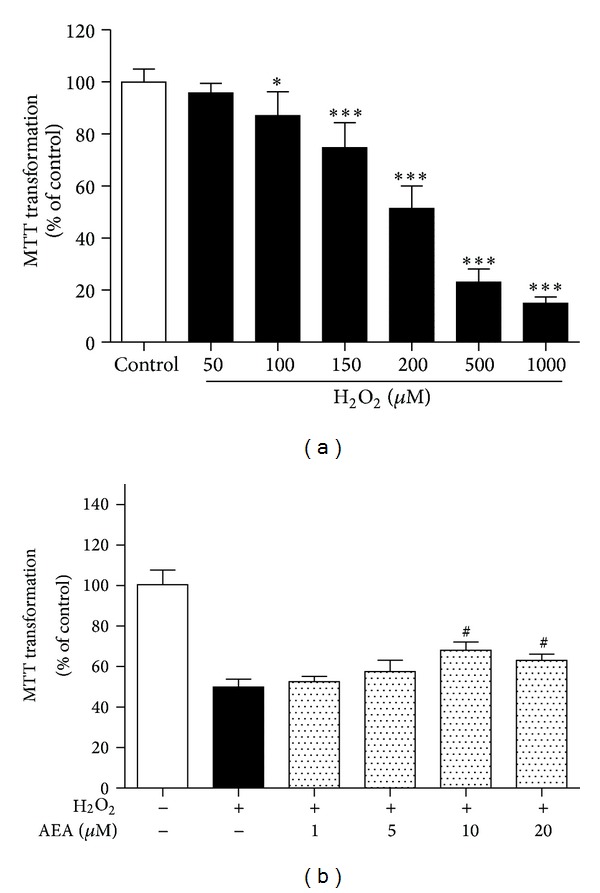
AEA increased the metabolic activity of HT22 cells exposed to H2O2 in a dose-dependent manner. (a) The correlation between the H2O2 concentration and cell metabolic activity. HT22 cells were exposed to different concentrations of H2O2 for 3 h (n = 8). (b) AEA increased the cell metabolic activity of HT22 cells exposed to 200 μM H2O2 for 3 h (n = 8). Results are expressed as means ± SD, *P < 0.05, ***P < 0.001 versus the control (no H2O2, and no AEA), # P < 0.05 versus the cells exposed to H2O2 alone.
HT22 cells were exposed to 1 to 20 μM of AEA in the presence of 200 μM H2O2. 10 μM and 20 μM AEA significantly ameliorated the cytotoxic effect of H2O2 (Figure 2(b)). We used 10 μM of AEA for subsequent experiments.
3.2. AEA Upregulated CB1 Expression in HT22 Cells
We used immunofluorescence and western blotting to assess whether AEA could up-regulate CB1 expression in HT22 cells. We observed CB1 staining in the cell membrane and cytoplasm of HT22 cells, consistent with a previous study [11]. Treatment with 10 μM AEA induced a significant up-regulation of CB1 expression (P < 0.05), and the selective CB1 antagonist AM251 reversed the AEA-induced up-regulation of CB1 expression (Figure 3).
Figure 3.
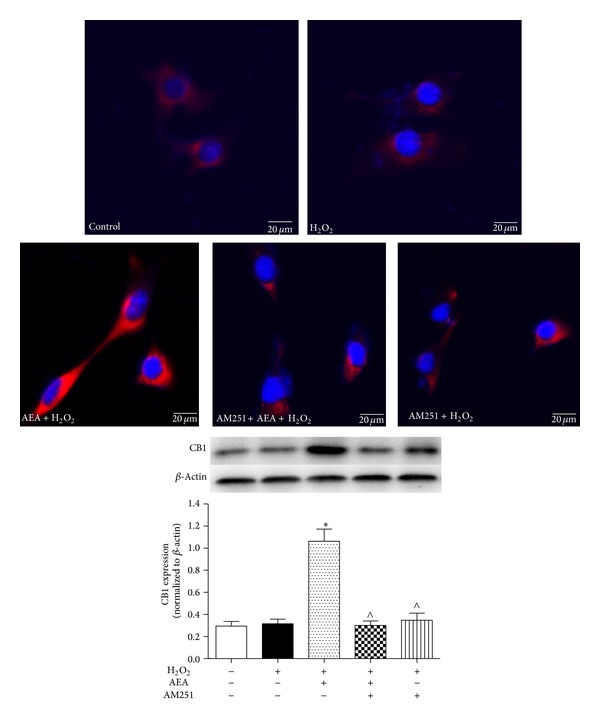
AEA upregulated the expression of CB1 in HT22 cells. Immunofluorescence staining and western blotting were used to investigate the AEA-induced effect on CB1 protein expression in HT22 cells. The cells were divided into five groups, Control: cells cultured in drug-free medium; H2O2: cells exposed to 200 μM H2O2 for 3 h; AEA + H2O2: cells exposed to 10 μM AEA plus 200 μM H2O2 for 3 h; AM251 + AEA + H2O2: cells exposed to 10 μM AEA plus 10 μM CB1 antagonist AM251 at the presence of 200 μM H2O2 for 3 h; AM251 + H2O2: cells exposed to 10 μM AM251 plus 200 μM H2O2 for 3 h. CB1 protein (red) was expressed in HT22 cells. AEA upregulated the expression of CB1 receptor; however CB1 antagonist AM251 reversed the CB1 upregulation in HT22 cells. Nuclei were counter-stained with DAPI (blue). Results are expressed as means ± SD (n = 4). *P < 0.05 versus control (no H2O2, no AEA, and no AM251), ∧ P < 0.05 versus the cells exposed to AEA plus H2O2. Bar = 20 μm.
3.3. Protection of AEA against Oxidative Stress in HT22 Cells Involved CB1
In the absence of AEA, AM251 did not affect the cytotoxic impact of H2O2 (Figure 4(a)); however AM251 abolished the AEA-induced protection of HT22 cells, reducing the cell metabolic activity from 66.9 ± 2.4% to 49.5 ± 7.1% (P < 0.05). AM251 also reversed the influence of AEA on LDH release, increasing the LDH release from 29.1 ± 7.6 U/L to 51.2 ± 7.9 U/L (P < 0.05) (Figure 4(b)). We also evaluated cleaved caspase-3 expression and apoptotic rate by western blotting (Figure 4(c)) and flow cytometry (Figures 4(d)–4(i)), respectively, to assess the apoptosis of HT22 cells. AEA significantly decreased the expression of cleaved caspase-3 and the apoptotic rate of HT22 cells in response to H2O2 (P < 0.05). And AM251 abolished these effects caused by AEA. In addition, AEA ameliorated the changes in cellular morphology elicited by H2O2 and maintained the integrity of HT22 cells, and AM251 reversed this effect (Figure 5). These results indicated AEA protected HT22 cells from the damage caused by H2O2, and AM251 reversed this protection, suggesting that the protective effects of AEA may be mediated via CB1.
Figure 4.
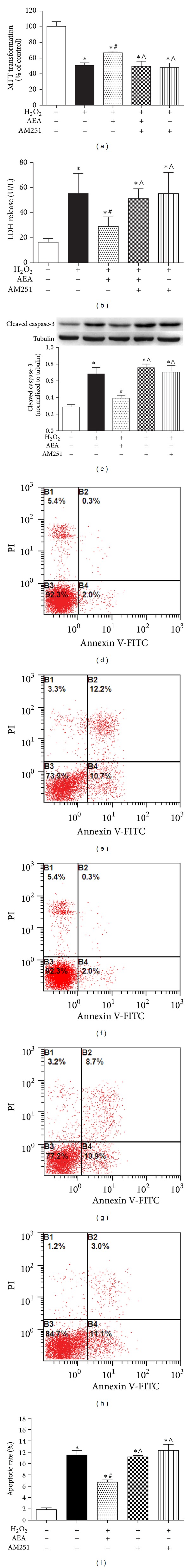
AEA protected HT22 cells exposed to H2O2 via CB1. (a) CB1 antagonist AM251 reversed AEA-induced protection on cell metabolic activity (n = 8). (b) AM251 reversed AEA-induced protection on LDH release (n = 6). (c) AM251 reversed AEA-induced reduction of cleaved caspase-3 expression (n = 4). (d)–(h) Apoptotic rates assessed by flow cytometry. (d) Control cells cultured in drug-free medium. (e) Cells exposed to 200 μM H2O2 for 3 h. (f) Cells exposed to 10 μM AEA plus 200 μM H2O2 for 3 h. (g) Cells exposed to 10 μM AEA plus 10 μM AM251 in the presence of 200 μM H2O2 for 3 h. (h) Cells exposed to CB1 antagonist AM251 of 10 μM plus 200 μM H2O2 for 3 h. (i) Statistical results of (c)–(g). Results are expressed as means ± SD (n = 4). *P < 0.05 versus the control (no H2O2, no AEA, and no AM251), # P < 0.05 versus the cells exposed to H2O2 alone, and ∧ P < 0.05 versus the cells exposed to AEA plus H2O2.
Figure 5.

AEA ameliorated the morphology of HT22 cells exposed to H2O2 via CB1. (a) Control cells cultured in drug-free medium. (b) Cells exposed to 10 μM AEA for 3 h. (c) Cells exposed to 200 μM H2O2 for 3 h. (d) Cells exposed to 10 μM AEA plus 200 μM H2O2 for 3 h. (e) Cells exposed to 10 μM AEA plus 10 μM AM251 at the presence of 200 μM H2O2 for 3 h. (f) Cells exposed to CB1 antagonist AM251 of 10 μM plus 200 μM H2O2 for 3 h. H2O2 markedly damaged the cell morphology and hindered the growth of neurites. AEA attenuated the H2O2-induced injury of HT22 cells whereas CB1 antagonist AM251 reversed the AEA-induced protective effect on cell morphology. Bar = 50 μm.
3.4. AEA Decreased Intracellular ROS and Maintained Intracellular Redox Status via CB1
Exposure of HT22 cells to H2O2 led to accumulation of intracellular ROS, and simultaneous treatment with AEA markedly reduced the generation of ROS (Figure 6). SOD plays a vital role in protecting cells against oxidative injury. H2O2 treatment sharply decreased SOD activity in HT22 cells to 32.2 ± 5.0% (Figure 7(a)), and AEA restored SOD activity to 70.3 ± 4.0% of baseline (P < 0.05). GSH is also an important cellular antioxidant. H2O2 treatment sharply decreased GSH activity in HT22 cells from 18.8 ± 2.7 μM to 6.1 ± 1.0 μM (Figure 7(b)). Simultaneous application of AEA partially restored GSH levels to 12.1 ± 1.4 μM (P < 0.05). GSSG levels were increased in response to H2O2 treatment from 1.5 ± 0.3 μM to 2.9 ± 0.3 μM (Figure 7(c)), and this effect was almost entirely abolished by AEA, which reduced GSSH levels to 1.6 ± 0.4 μM (P < 0.05). The GSH/GSSG ratio was reduced from 12.3 ± 1.1 to 2.2 ± 0.3 by H2O2 treatment, and AEA partially restored this balance, increasing the ratio to 7.3 ± 0.7 (Figure 7(d)). The influences of AEA on intracellular ROS, SOD, GSH, GSSG, and GSH/GSSG ratio were abolished by the CB1 antagonist AM251, indicating that the antioxidative effects of AEA may be mediated via CB1 of HT22 cells.
Figure 6.
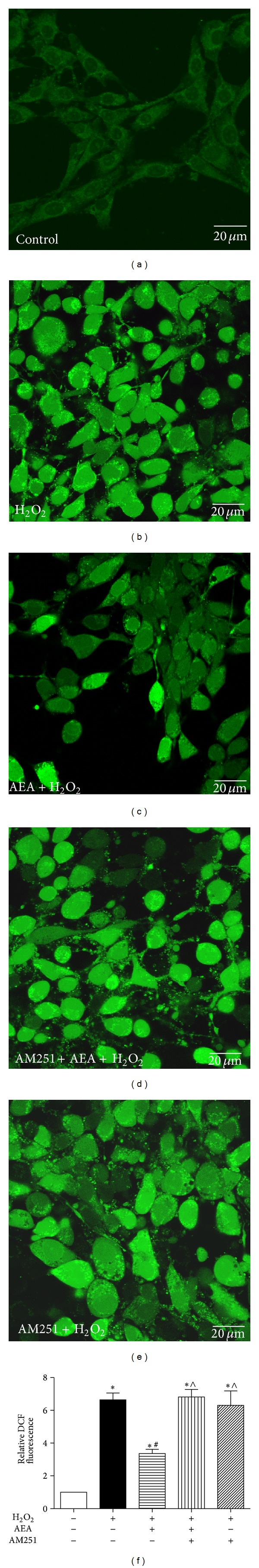
AEA decreased intracellular ROS generation via CB1. The intracellular ROS levels were assessed by ROS reagent kit. (a)–(e) indicate the fluorescence intensity of ROS. (a) Control cells cultured in drug-free medium. (b) Cells exposed to 200 μM H2O2 for 3 h. (c) Cells exposed to 10 μM AEA plus 200 μM H2O2 for 3 h. (d) Cells exposed to 10 μM AEA plus 10 μM AM251 in the presence of 200 μM H2O2 for 3 h. (e) Cells exposed to 10 μM CB1 antagonist AM251 plus 200 μM H2O2 for 3 h. (f) Statistical results of (a)–(e). Results are expressed as means ± SD (n = 6). *P < 0.05 versus the control (no H2O2, no AEA, and no AM251), # P < 0.05 versus the cells exposed to H2O2 alone, and ∧ P < 0.05 versus the cells exposed to AEA plus H2O2.
Figure 7.
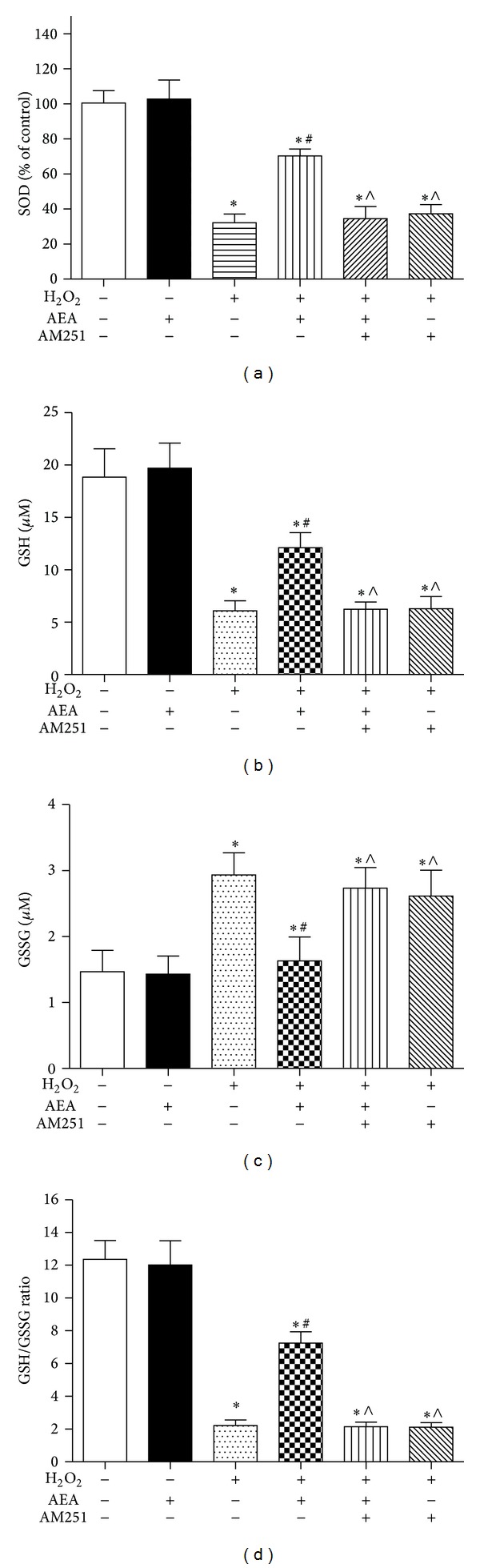
AEA increased intracellular SOD and ameliorated GSH/GSSG ratio. The cells were divided into six groups, Control: cells cultured in drug-free medium; AEA: cells exposed to 10 μM AEA for 3 h; H2O2: cells exposed to 200 μM H2O2 for 3 h; AEA + H2O2: cells exposed to 10 μM AEA plus 200 μM H2O2 for 3 h; AM251 + AEA + H2O2: cells exposed to 10 μM AEA plus 10 μM CB1 antagonist AM251 in the presence of 200 μM H2O2 for 3 h; AM251 + H2O2: cells exposed to 10 μM AM251 plus 200 μM H2O2 for 3 h. The intracellular SOD, GSH, and GSSG levels were assessed by the corresponding reagent kit, and the GSH/GSSG ratio was calculated according to the GSH and GSSG levels. (a) Intracellular SOD level. (b) Intracellular GSH level. (c) Intracellular GSSG level. (d) Intracellular GSH/GSSG ratio. Results are expressed as means ± SD (n = 6). *P < 0.05 versus control (no H2O2, no AEA, and no AM251), # P < 0.05 versus the cells exposed to H2O2 alone, and ∧ P < 0.05 versus the cells exposed to AEA plus H2O2.
3.5. CB1 Knockdown Reversed the Beneficial Effects of AEA on HT22 Cells Exposed to H2O2
To further determine whether AEA-induced antioxidative ability was mediated by CB1 in HT22 cells, we used CB1-siRNA to knock down the expression of CB1. CB1-siRNA was effective in reducing the expression of CB1 (Figure 8(a)) and reversed AEA-induced cytoprotection, leading to a significant reduction of cell metabolic activity (Figure 8(b)), an increase of LDH release (Figure 8(c)), and a rise of intracellular ROS level (Figures 8(d)–8(i)).
Figure 8.
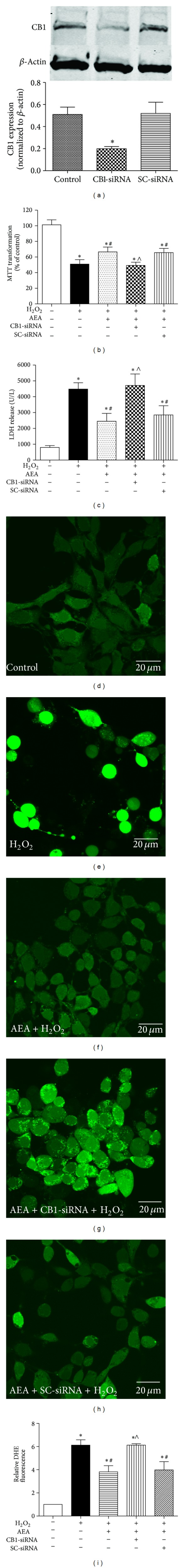
CB1-siRNA reversed AEA-induced protection against oxidative stress. The cells were divided into five groups, Control: cells cultured in drug-free medium; H2O2: cells exposed to 200 μM H2O2 for 3 h; AEA + H2O2: cells exposed to 10 μM AEA plus 200 μM H2O2 for 3 h; CB1-siRNA + AEA + H2O2: cells incubated with CB1-siRNA for 5 h and then exposed to 10 μM AEA plus 200 μM H2O2 for 3 h; scrambled siRNA (SC-siRNA) + AEA + H2O2: cells incubated with SC-siRNA for 5 h and then exposed to 10 μM AEA plus 200 μM H2O2 for 3 h. CB1-siRNA abolished the AEA-induced protection against 200 μM H2O2 in HT22 cells; SC-siRNA did not affect the protection. (a) CB1-siRNA significantly downregulated the expression of CB1, assessed by western blotting. (b) Cell metabolic activity, assessed by MTT (n = 8). (c) LDH release, assessed by reagent kit and spectrophotometry (n = 6). (d)–(h) The fluorescence intensity of ROS. (i) Statistical results of (d)–(h) (n = 6). Results are expressed as means ± SD, *P < 0.05 versus the control (no H2O2, no AEA, and no siRNA), # P < 0.05 versus the cells exposed to H2O2 alone, and ∧ P < 0.05 versus the cells exposed to AEA plus H2O2.
3.6. Nox2 Was Involved in AEA-Induced Cytoprotection via CB1
The expression of Nox2 in HT22 cells was upregulated in the presence of 200 μM H2O2 in a time-dependent manner (Figure 9(a)). AEA decreased the Nox2 protein expression (Figure 9(b)) and mRNA transcription (Figure 9(c)). However, CB1 antagonist AM251 or CB1-siRNA (Figure 9(d)) abolished AEA-induced influence on Nox2 protein expression and mRNA transcription, suggesting that the Nox2 may be involved in AEA-induced cytoprotection against H2O2 via CB1.
Figure 9.
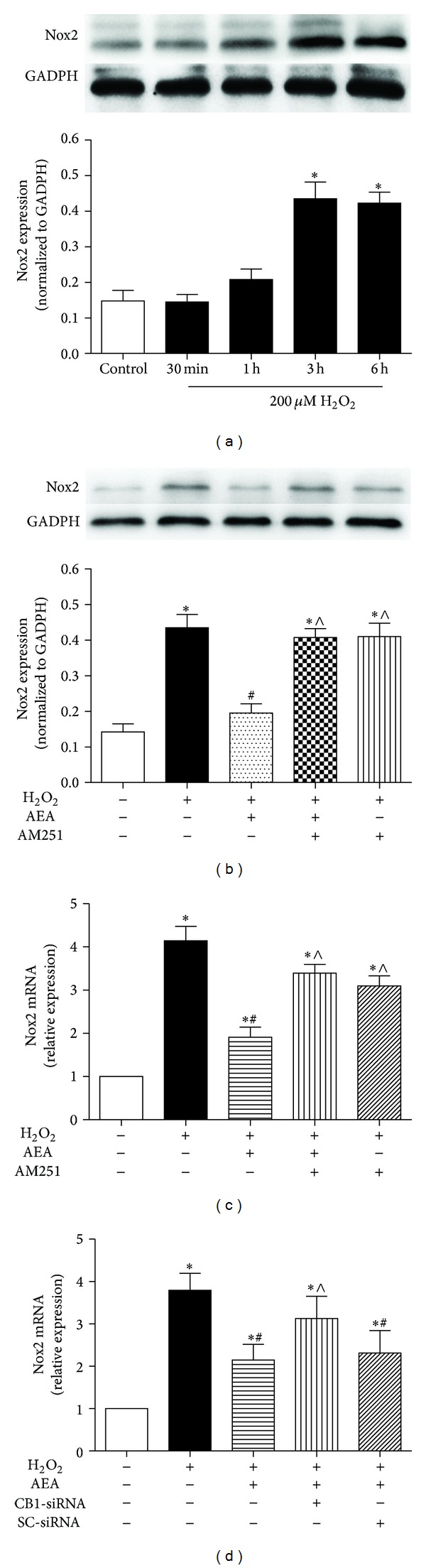
Nox2 expression was inhibited in the presence of AEA via CB1. (a) Nox2 expression was increased in HT22 cells exposed to H2O2 in a time-dependent manner. Then the cells were divided into five groups, Control: cells cultured in drug-free medium; H2O2: cells exposed to 200 μM H2O2 for 3 h; AEA + H2O2: cells exposed to 10 μM AEA plus 200 μM H2O2 for 3 h; AM251 + AEA + H2O2: cells exposed to 10 μM AEA plus 10 μM CB1 antagonist AM251 in the presence of 200 μM H2O2 for 3 h; AM251 + H2O2: cells exposed to 10 μM AM251 plus 200 μM H2O2 for 3 h. Nox2 protein expression (b) and mRNA transcription (c) were evaluated by western blotting and real-time PCR, respectively. (d) Incubation with CB1-siRNA for 5 h abolished the AEA-induced inhibition of Nox2 mRNA transcription. Results are expressed as means ± SD (n = 4). *P < 0.05 versus the control (no H2O2, no AEA, and no AM251 or siRNA), # P < 0.05 versus the cells exposed to H2O2 alone, and ∧ P < 0.05 versus the cells exposed to AEA plus H2O2.
To further investigate the role of Nox2 in AEA-induced protection in HT22 cells exposed to H2O2, we used apocynin, a specific Nox inhibitor. We found that the presence of 50 μM apocynin decreased the expression of Nox2 protein significantly (Figure 10(a)), and there was no significance between AEA and apocynin alone on Nox2 expression in H2O2-treated HT22 cells. In addition, a combination of AEA and apocynin did not cause a more significant reduction of Nox2 expression than either AEA or apocynin used alone (P > 0.05). Similarly, we noticed a combination of AEA and apocynin did not induce a more significant increase of cell metabolic activity (Figure 10(b)) and reduction of LDH release (Figure 10(c)) than either AEA or apocynin alone (P > 0.05), indicating that Nox2 inhibition may be involved in AEA-induced cytoprotection against H2O2.
Figure 10.
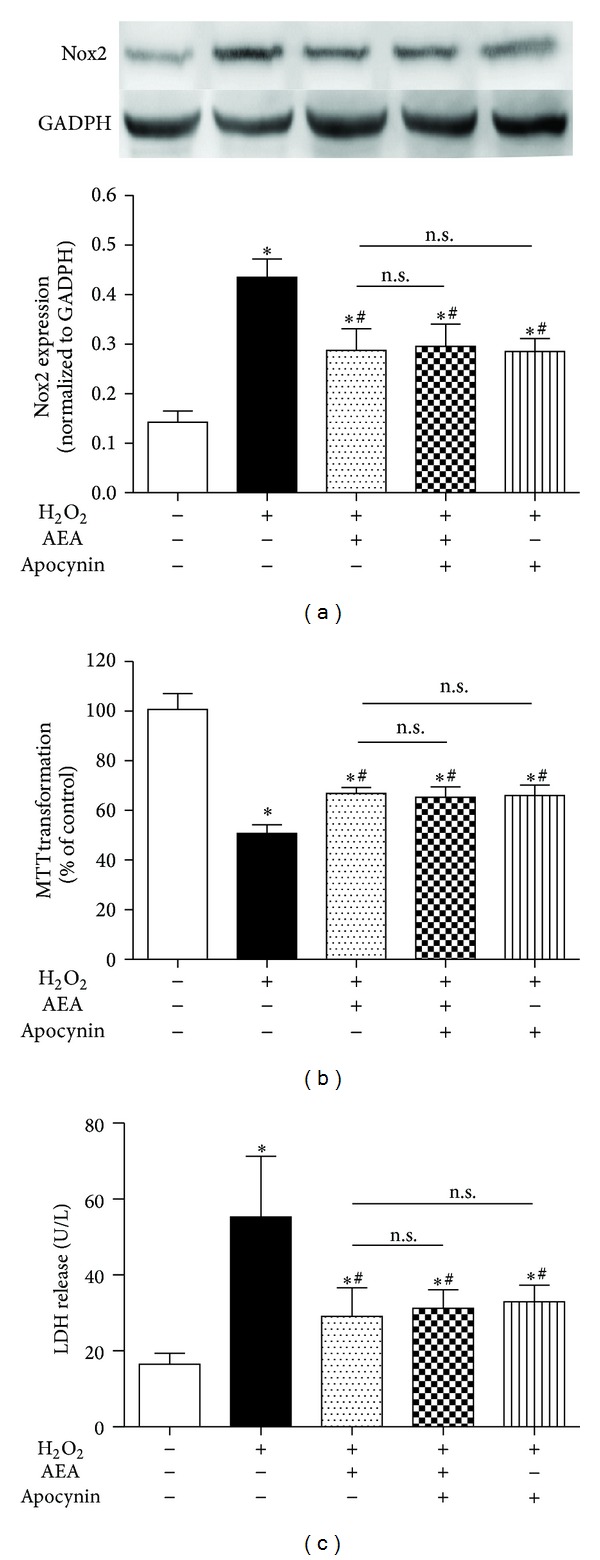
Nox inhibitor did not induce a more significant reduction of Nox2 expression than AEA alone. The cells were divided into five groups, Control: cells cultured in drug-free medium; H2O2: cells exposed to 200 μM H2O2 for 3 h; AEA + H2O2: cells exposed to 10 μM AEA plus 200 μM H2O2 for 3 h; Apocynin + AEA + H2O2: cells exposed to 10 μM AEA plus 50 μM Nox inhibitor AM251 in the presence of 200 μM H2O2 for 3 h; Apocynin + AEA + H2O2: cells exposed to 50 μM apocynin plus 10 μM AEA in the presence of 200 μM H2O2 for 3 h. Nox2 protein expression (a) was evaluated by western blotting (n = 4). (b) Cell metabolic activity and (c) LDH release were determined by MTT (n = 8) and reagent kit (n = 6), respectively. Results are expressed as means ± S.D. *P < 0.05 versus the control (no H2O2, no AEA, and no apocynin), # P < 0.05 versus the cells exposed to H2O2 alone, n.s.: no significance.
4. Discussion
In this study, we found that 10 μM AEA treatment of a murine hippocampal neuron cell line, HT22, significantly improved cell injury, decreased apoptosis, and ameliorated the morphological changes induced by oxidative stress in the form of 200 μM H2O2. Treatment with AEA reduced intracellular ROS and Nox2 expression in HT22 cells exposed to H2O2, and these effects were reversed by application of CB1 antagonist AM251 or CB1-siRNA. In addition, Nox inhibitor apocynin plus AEA did not induce a more significant downregulation of Nox2 or neuroprotection than apocynin or AEA used alone. These findings indicate that Nox2 inhibition is involved in AEA-induced neuroprotection against H2O2 through CB1 activation in HT22 cells.
Oxidative stress is involved in the pathophysiology of many CNS diseases [1–3]. Overaccumulation of intracellular ROS causes oxidative stress, which can damage cellular membranes, injure the mitochondria, and induce cell death. Thus enhancement of the cellular processes that suppress ROS generation or remove excess ROS may be effective in treating oxidative stress-induced diseases. Recently Nox proteins have been demonstrated to be major producers of ROS in CNS cells such as neurons, astrocytes, and microglia under pathophysiological conditions [16, 17]. Thus, inhibition of Nox proteins may present an effective mechanism to limit oxidative stress in the CNS. The Nox family includes Nox 1, 2, 3, 4, and 5, dual oxidase (DUAX) 1, and DUAX2 [18]. Nox2 appears to be the most important Nox in cerebral injury [19]. The infarct volume of Nox2 deficient mice is smaller than that of the wild-type and Nox2 deficient mice experience less blood-brain barrier injury than wild-type mice in a stroke model [20, 21].
Cannabinoids, such as AEA, appear to protect neurons against excitotoxicity, oxidative stress, and hypoxia through the activation of CB1 [12, 22]. Moldzio et al. reported that the cannabinoid THC protected dopaminergic neurons against 1-methyl-4-phenylpyridinium (MPP+) induced oxidative injury [23]. Chung et al. reported that cannabinoids WIN55, 212-2, and HU210 decreased lipopolysaccharide-induced activation of Nox in microglia [24]. We, therefore, hypothesize that inhibition of Nox may be involved in cannabinoid-induced protection from oxidative stress. In one of our previous studies, we observed that electroacupuncture induced neuroprotection against cerebral ischemia by increasing cerebral levels of the endogenous cannabinoid AEA [25]. However, AEA-mediated neuroprotection which is mediated via inhibition of Nox remained unknown. Thus, we investigated the protective effects of AEA against H2O2-induced neuronal injury.
The cannabinoid receptor CB1 is a G protein-coupled receptor present in both the cellular membrane and cytoplasm of neurons [26]. Nox2 is also mostly localized on the cellular membrane of neurons [19]. We found that antagonism of CB1 or silencing of CB1 expression reversed the AEA-induced inhibition of Nox2 protein expression and Nox2 mRNA transcription. In addition, Nox inhibitor apocynin alone or plus AEA did not induce a significant downregulation of Nox2 expression compared with AEA alone in the HT22 cells exposed to H2O2. Therefore, we inferred that AEA may activate CB1, which, in turn, inhibits Nox2 expression resulting in a reduction of cellular ROS.
Intracellular redox balance reflects the level of oxidative stress levels in cells and is crucial to cell function and survival. Accumulation of oxidants or overconsumption of antioxidants will damage cellular metabolism and even result in cell death [27]. In this study, we found that when in conditions of oxidative stress AEA raised intracellular SOD and GSH, reduced GSSG, and increased the GSH/GSSG ratio, and these effects were reversed by CB1 antagonist AM251, indicating that AEA could restore the balance of intracellular antioxidative and oxidative substances via CB1 receptor.
The ability of the endocannabinoid AEA to protect HT22 cells against H2O2-induced injury recommends AEA as a candidate therapy for oxidative stress-related neurological disorders. Endogenous agents have specific inactivation systems and therefore may run less risk of interfering with ongoing developmental profiles than artificial ligands. This is vital, as low levels of AEA can affect embryonic implantation, neural development, and suckling [28, 29]. Furthermore, cannabis use has been associated with the onset of schizophrenia [30, 31]. Exposure to synthetic cannabinoids WIN55, 212-2 caused disruption of learning and decreased emotional reactivity [32]. Thus interventions targeting the cannabinoid system need to be minimal during development [33] and endogenous agonists may be less deleterious.
However, further work must be done to determine the relevance of our findings in vivo. We have not fully elucidated the precise antioxidative mechanisms induced by AEA. In this study we investigated only the role of Nox2 in AEA efficacy, and whether other components of Nox are associated with AEA activities is not yet known.
5. Conclusions
This study provides evidence that the endocannabinoid AEA protects the mouse hippocampal neuron cell line HT22 against H2O2-induced oxidative injury through CB1-mediated inhibition of Nox2.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant 81072888), the Overseas, Hong Kong & Macao Scholars Collaborated Researching Fund (Grant 81228022), and the Program for Changjiang Scholars and Innovative Research Team in University (Grant IRT 1053).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Ji Jia and Lei Ma contributed equally to this work.
References
- 1.Zhao ZY, Luan P, Huang SX, et al. Edaravone protects HT22 neurons from H2O2-induced apoptosis by inhibiting the MAPK signaling pathway. CNS Neuroscience and Therapeutics. 2013;19(3):163–169. doi: 10.1111/cns.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen S, Lin M, Chang C. Ischemic and oxidative damage to the hypothalamus may be responsible for heat stroke. Current Neuropharmacology. 2013;11(2):129–140. doi: 10.2174/1570159X11311020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun L, Jin Y, Dong L, Sumi R, Jahan R, Li Z. The neuroprotective effects of Coccomyxa gloeobotrydiformis on the ischemic stroke in a rat model. International Journal of Biological Sciences. 2013;9(8):811–817. doi: 10.7150/ijbs.6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gheysarzadeh A, Yazdanparast R. Inhibition of H2O2-induced cell death through FOXO1 modulation by EUK-172 in SK-N-MC cells. European Journal of Pharmacology. 2012;697(1–3):47–52. doi: 10.1016/j.ejphar.2012.09.036. [DOI] [PubMed] [Google Scholar]
- 5.Lu X, Xu H, Sun B, Zhu Z, Zheng D, Li X. Enhanced neuroprotective effects of resveratrol delivered by nanoparticles on hydrogen peroxide-induced oxidative stress in rat cortical cell culture. Molecular Pharmaceutics. 2013;10(5):2045–2053. doi: 10.1021/mp400056c. [DOI] [PubMed] [Google Scholar]
- 6.Sajad M, Zargan J, Zargar MA, et al. Quercetin prevents protein nitration and glycolytic block of proliferation in hydrogen peroxide insulted cultured neuronal precursor cells (NPCs): implications on CNS regeneration. NeuroToxicology. 2013;36:24–33. doi: 10.1016/j.neuro.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Ano Y, Sakudo A, Kimata T, et al. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neuroscience Letters. 2010;469(1):39–43. doi: 10.1016/j.neulet.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 8.Ye Q, Huang B, Zhang X, Zhu Y, Chen X. Astaxanthin protects against MPP+-induced oxidative stress in PC12 cells via the HO-1/NOX2 axis. BMC Neuroscience. 2012;13(1, article 156) doi: 10.1186/1471-2202-13-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo F, Wang H, Li L, et al. A novel domain of amino-nogo-A protects HT22 cells exposed to oxygen glucose deprivation by inhibiting NADPH oxidase activity. Cellular and Molecular Neurobiology. 2013;33(3):443–452. doi: 10.1007/s10571-013-9911-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veldhuis WB, van der Stelt M, Wadman MW, et al. Neuroprotection by the endogenous cannabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: role of vanilloid receptors and lipoxygenases. Journal of Neuroscience. 2003;23(10):4127–4133. doi: 10.1523/JNEUROSCI.23-10-04127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt W, Schäfer F, Striggow V, Fröhlich K, Striggow F. Cannabinoid receptor subtypes 1 and 2 mediate long-lasting neuroprotection and improve motor behavior deficits after transient focal cerebral ischemia. Neuroscience. 2012;227:313–326. doi: 10.1016/j.neuroscience.2012.09.080. [DOI] [PubMed] [Google Scholar]
- 12.Shouman B, Fontaine RH, Baud O, et al. Endocannabinoids potently protect the newborn brain against AMPA-kainate receptor-mediated excitotoxic damage. British Journal of Pharmacology. 2006;148(4):442–451. doi: 10.1038/sj.bjp.0706755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Li X, Chen Y, et al. Activation of epsilon protein kinase C-mediated anti-apoptosis is involved in rapid tolerance induced by electroacupuncture pretreatment through cannabinoid receptor type 1. Stroke. 2011;42(2):389–396. doi: 10.1161/STROKEAHA.110.597336. [DOI] [PubMed] [Google Scholar]
- 14.Carnevale R, Biondi-Zocai G, Peruzzi M, et al. New insights into the steen solution properties: breakthrough in antioxidant effects via NOX2 downregulation. Oxidative Medicine and Cellular Longevity. 2014;2014:10 pages. doi: 10.1155/2014/242180.242180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Deng R, Lu Y, et al. Gambogic acid as a non-competitive inhibitor of ATP-binding cassette transporter B1 reverses the multidrug resistance of human epithelial cancers by promoting ATP-binding cassette transporter B1 protein degradation. Basic and Clinical Pharmacology and Toxicology. 2013;112(1):25–33. doi: 10.1111/j.1742-7843.2012.00921.x. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Yoshioka H, Kim GS, et al. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxidants and Redox Signaling. 2011;14(8):1505–1517. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Páramo B, Montiel T, Hernández-Espinosa DR. Calpain activation induced by glucose deprivation is mediated by oxidative stress and contributes to neuronal damage. International Journal of Biochemistry & Cell Biology. 2013;45(11):2596–2604. doi: 10.1016/j.biocel.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Wei X, Liu K, et al. NOX2 deficiency ameliorates cerebral injury through reduction of complexin II-mediated glutamate excitotoxicity in experimental stroke. Free Radical Biology and Medicine. 2013;65C:942–951. doi: 10.1016/j.freeradbiomed.2013.08.166. [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. Journal of Cerebral Blood Flow and Metabolism. 2009;29(7):1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahles T, Luedike P, Endres M, et al. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38(11):3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 22.Duncan RS, Chapman KD, Koulen P. The neuroprotective properties of palmitoylethanolamine against oxidative stress in a neuronal cell line. Molecular Neurodegeneration. 2009;4(1, article 50) doi: 10.1186/1750-1326-4-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moldzio R, Pacher T, Krewenka C, et al. Effects of cannabinoids Δ(9)-tetrahydrocannabinol, Δ(9)-tetrahydrocannabinolic acid and cannabidiol in MPP+ affected murine mesencephalic cultures. Phytomedicine. 2012;19(8-9):819–824. doi: 10.1016/j.phymed.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Chung ES, Bok E, Chung YC, Baik HH, Jin BK. Cannabinoids prevent lipopolysaccharide-induced neurodegeneration in the rat substantia nigra in vivo through inhibition of microglial activation and NADPH oxidase. Brain Research. 2012;1451:110–116. doi: 10.1016/j.brainres.2012.02.058. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Peng Y, Chen S, et al. Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke. 2009;40(6):2157–2164. doi: 10.1161/STROKEAHA.108.541490. [DOI] [PubMed] [Google Scholar]
- 26.Ahn KH, Scott CE, Abrol R, Goddard WA, Kendall DA. Computationally-predicted CB1 cannabinoid receptor mutants show distinct patterns of salt-bridges that correlate with their level of constitutive activity reflected in G protein coupling levels, thermal stability, and ligand binding. Proteins: Structure, Function and Bioinformatics. 2013;81(8):1304–1317. doi: 10.1002/prot.24264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ha JS, Lim HM, Park SS. Extracellular hydrogen peroxide contributes to oxidative glutamate toxicity. Brain Research. 2010;1359:291–297. doi: 10.1016/j.brainres.2010.08.086. [DOI] [PubMed] [Google Scholar]
- 28.Fride E. The endocannabinoid-CB receptor system: importance for development and in pediatric disease. Neuroendocrinology Letters. 2004;25(1-2):24–30. [PubMed] [Google Scholar]
- 29.Fride E. The endocannabinoid-CB 1 receptor system in pre- and postnatal life. European Journal of Pharmacology. 2004;500(1–3):289–297. doi: 10.1016/j.ejphar.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 30.Zammit S, Allebeck P, Andreasson S, Lundberg I, Lewis G. Self reported cannabis use as a risk factor for schizophrenia in Swedish conscripts of 1969: historical cohort study. British Medical Journal. 2002;325(7374, article 1199) doi: 10.1136/bmj.325.7374.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solowij N, Walterfang M, Lubman DI, et al. Alteration to hippocampal shape in cannabis users with and without schizophrenia. Schizophrenia Research. 2013;143(1):179–184. doi: 10.1016/j.schres.2012.10.040. [DOI] [PubMed] [Google Scholar]
- 32.Antonelli T, Tomasini MC, Tattoli M, et al. Prenatal exposure to the CB1 receptor agonist WIN 55,212-2 causes learning disruption associated with impaired cortical NMDA receptor function and emotional reactivity changes in rat offspring. Cerebral Cortex. 2005;15(12):2013–2020. doi: 10.1093/cercor/bhi076. [DOI] [PubMed] [Google Scholar]
- 33.Bernard C, Milh M, Morozov YM, Ben-Ari Y, Freund TF, Gozlan H. Altering cannabinoid signaling during development disrupts neuronal activity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(26):9388–9393. doi: 10.1073/pnas.0409641102. [DOI] [PMC free article] [PubMed] [Google Scholar]