

Abstract
Ataxia with oculomotor apraxia type 2 (AOA2) is an autosomal recessive cerebellar ataxia associated with mutations in SETX, which encodes the senataxin protein, a DNA/RNA helicase. We describe the clinical phenotype and molecular characterization of a Colombian AOA2 patient who is compound heterozygous for a c.994 C>T (p.R332W) missense mutation in exon 7 and a c.6848_6851delCAGA (p.T2283KfsX32) frameshift deletion in SETX exon 21.
Immunocytochemistry of patient-derived fibroblasts revealed a normal cellular distribution of the senataxin protein, suggesting that these mutations do not lead to lead to loss or mis-localization of the protein, but rather that aberrant function of senataxin underlies the disease pathogenesis. Furthermore, we used the alkaline comet assay to demonstrate that patient-derived fibroblast cells exhibit an increased susceptibility to oxidative DNA damage. This assay provides a novel and additional means to establish pathogenicity of SETX mutations.
Keywords: AOA2, Autosomal recessive cerebellar ataxia, DNA repair, Helicase, Senataxin
1. Introduction
Ataxia with oculomotor apraxia type 2 (AOA2) (Mendelian Inheritance in Man [MIM] #606002) is an autosomal recessive cerebellar ataxia (ARCA) characterized clinically by progressive cerebellar ataxia, sensorimotor peripheral neuropathy, oculomotor apraxia, as well as chorea and/or dystonia. Symptom onset is usually noted in adolescence or early adulthood.1–9 Laboratory testing of AOA2 patients frequently reveals elevated serum α-fetoprotein (AFP)10–12 and creatine kinase levels.12
ARCA with oculomotor apraxia13,14 also includes ataxia with oculomotor apraxia type 1 (AOA1) (MIM #208920), associated with mutations in the aprataxin (APTX) gene15–18, ataxia-telangiectasia (AT) (MIM #208900) due to mutations in the AT mutated (ATM) gene19,20 and ataxia-telangiectasia-like disorder (MIM #604391) caused by mutations in the MRE11A gene.21-22 Compared with AOA2, AOA1 usually manifests earlier in childhood16 and may be more distinctively associated with hypoalbuminemia and hypercholesterolemia. AFP levels are also elevated in AT patients, but in contrast to AT, individuals with AOA2 do not show increased sensitivity to ionizing radiation or susceptibility to cancer.19 Senataxin, the protein encoded by SETX, is a large 2677 amino acid DNA/RNA helicase17,22,23 functioning in the processing of non-coding RNA and in defense against DNA damage.17,25
Various missense, nonsense and frameshift mutations in SETX have been described in families mostly within Europe, North America, Japan, and North Africa (Supp. Table 1). Exonic or multiexonic deletions and duplications have also been reported.3,5 In general, missense mutations in the helicase domain (HD) of SETX seem to result in less severe phenotypes than deletions, truncation mutations, or missense mutations outside of the HD. Mutations in SETX are also responsible for a rare autosomal dominant form of juvenile amyotrophic lateral sclerosis (ALS), ALS4.26
In this study, we report a patient from Colombia who is compound heterozygous for two known AOA2 mutations in SETX. We used skin fibroblasts and the comet assay to demonstrate an alteration in recovery from DNA damage, providing a novel way to investigate the pathogenicity of senataxin mutations as well as providing support for oxidative DNA damage as the underlying mechanism for disease in AOA2.
2. Materials and methods
2.1. Patient and genetic testing
The patient provided informed consent to participate in this study under a clinical research protocol approved by the USA National Institutes of Health (NIH) Combined Neuroscience Institutional Review Board, and was evaluated at the NIH Clinical Research Center by board-certified neurologists. Mutations were identified in DNA isolated from peripheral blood through Clinical Laboratory Improvement Amendments-certified molecular genetic testing (Athena Diagnostics, Worcester, MA, USA).
2.2. Cell culture and immunocytochemistry
Fibroblasts from a forearm skin punch biopsy were prepared using standard procedures and maintained in Dulbecco's modified Eagle's medium (DMEM) media supplemented with 20% fetal bovine serum at 37°C in 5% CO2. For immunostaining, cells grown on a coverglass were washed three times with ice-cold phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 5 minutes at room temperature, permeabilized for 15 minutes in 0.1% NP-40, and blocked for 30 minutes in 5% normal goat serum. Slides where then incubated overnight with 1:200 anti-senataxin antibody (ab56984; Abcam, Cambridge, UK) at 4°C, washed three times with ice-cold PBS, incubated for 2 hours at room temperature with 1:500 goat anti-mouse Alexa Fluor 488 (A-11001; Life Technologies molecular probes, Carlsbad, CA, USA), washed once with ice-cold PBS, incubated for 10 minutes with 1:100 Alexa Fluor 555 Phalloidin (Invitrogen, Carlsbad, CA, USA) and washed once more, incubated for 10 minutes with 4′,6-diamidino-2-phenylindole (DAPI), washed three times with PBS, and finally mounted for imaging using Fluoromount-G (SouthernBiotech, Birmingham, AL, USA). Cells were imaged using a Zeiss LSM 710 confocal microscope with a 63 × 1.4 NA Plan-Apochromat oil objective, and image acquisition was performed using LSM 710 version 3.2SP2 software (all Carl Zeiss Microscopy GmbH, Jena, Germany).
2.3. Comet assay
We used the OxiSelect Comet Assay Kit (Cell Biolabs, San Diego, CA, USA). Fibroblasts (both AOA2 patient cells and normal controls) were treated with 2 mM H2O2 in serum-free DMEM for 45 minutes. Cells were then washed with PBS, and fresh medium (DMEM with serum) was added to the culture plates for different times (no recovery, 4 hours, and 24 hours); the “no recovery” set did not receive fresh media. After the recovery period, cells were trypsinized, washed, counted, and finally diluted to 105/ml in PBS. Cells were then added to low-melting temperature agarose (104/ml final). This cell suspension was plated (80 μl) on comet assay glass slides (Cell Biolabs) coated with normal-melting temperature agarose. After lysis, slides were placed in a horizontal gel electrophoresis chamber and covered with an alkaline buffer (5 mM NaOH and 200 mM Na2EDTA; pH >13). Following a 20 minute DNA “unwinding” period, electrophoresis was performed under standard conditions (20 V, 300 mA; distance between electrodes = 20 cm) for 25 minutes. Following neutralization to pH 7.5 using Trizma base (Sigma-Aldrich, St Louis, MO, USA), gels were stained with Vista Green DNA dye and stored at 4°C until analysis. Images were acquired with a Zeiss LSM 710 confocal microscope and LSM 710 version 3.2SP2 software. DNA damage was quantified per the manufacturer's instructions by calculating the extent tail moment: Extent Tail Moment = Tail DNA% × Length of Tail; where Tail DNA% = 100 × Tail DNA Intensity/Cell DNA intensity. For each time point, means ± standard error of the mean were calculated. Statistical analysis was performed using an unpaired Student's t-test.
3. Results
3.1. Identification of a compound heterozygous AOA2 patient from South America
The proband is a 47-year-old woman of Spanish ancestry originally from Colombia who started to develop difficulty walking in her early twenties. Her early motor development as a child was normal, and she walked at an appropriate age. She attended grade school and had no cognitive difficulties. Although she was not particularly athletic, she developed normally physically. Starting in her early twenties, she experienced a “sense of weakness,” with difficulty lifting objects as well as occasional falls. Concurrently, she developed paresthesias in her feet, “enveloping” her lower extremities. Over the past two decades her symptoms have steadily progressed. Examination revealed ocular apraxia, mild ophthalmoplegia, nystagmus in all directions of gaze, and hypermetric saccades. Overall muscle tone was increased, with mild proximal weakness in the upper and lower extremities. She had both appendicular and gait ataxia. Sensory examination revealed decreased vibratory sensation at the toes and decreased pinprick sensation to the mid-calf. She was non-ambulatory and required a wheelchair. Extensive laboratory studies were remarkable only for elevated serum α-fetoprotein level of 13.2 ng/mL (reference range: 0.6–6.6 ng/mL). MRI of the brain showed severe cerebellar atrophy, most notably within the vermis (Fig. 1A).
Fig. 1.
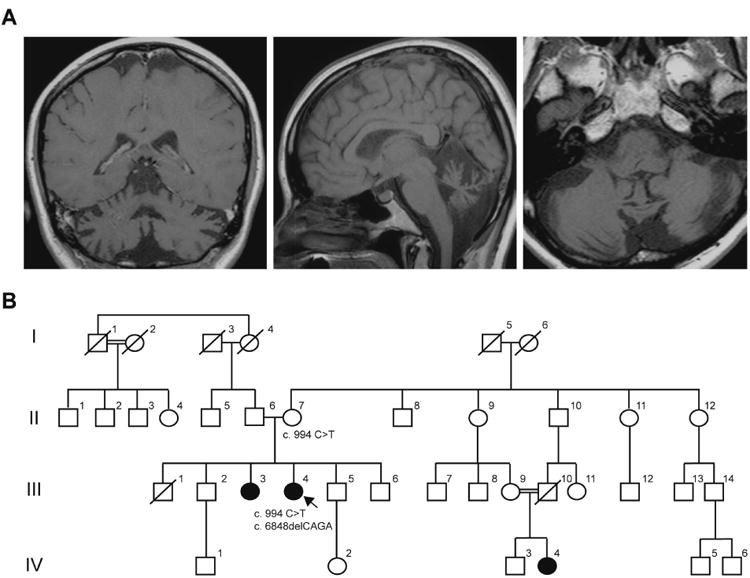
Identification of ataxia with oculomotor apraxia type 2 in a South American woman. (A) Coronal (left), sagittal (middle) and axial (right) T1-weighted brain MRI of III.4 showing severe cerebellar (particularly vermal) atrophy. (B) Pedigree of the reported family. The proband patient III.4 carries both c.994 C>T and c.6848delCAGA mutations. II.7 is a known carrier for the c.994 C>T mutation.
Family history revealed several similarly afflicted individuals (Fig. 1B). The proband (III. 4) has a sister (III.3) who displays similar, though less severe, symptoms by report. A distant cousin (IV.4) is the product of a consanguineous marriage (Fig. 1B), and has progressive ataxia as well. Neither III.3 nor IV.4 were available for evaluation.
Genetic studies for expansions in the FXN gene and mutations in the APTX, SIL1, POLG1 and TTPA genes revealed no abnormalities (Athena Diagnostics). However, sequencing of the SETX gene revealed two pathogenic changes: c.994 C>T (p.R332W) missense mutation in exon 7 and c.6848_6851delCAGA (p.T2283KfsX32) deletion in exon 21. Testing of the proband's mother confirmed that she is a carrier for only the c.994 C>T mutation, indicating that the two pathogenic changes are very likely on separate alleles.
3.2. Mutant senataxin localizes to the cell nucleus
Senataxin has been shown to localize in the cell nucleus, in agreement with its activity as a DNA/RNA helicase. We cultured skin fibroblasts from the proband and performed immunocytochemistry to determine whether there were detectable levels of senataxin. As a control we used skin fibroblasts from a healthy volunteer. Senataxin localized within the nucleus in control cells as well as in the AOA2 patient cells (Fig. 2). In both lines, there was a discrete pattern of senataxin immunoreactivity within the nucleus, with minimal, more diffuse staining within the cytoplasm. This distribution is in agreement with previous reports23,25. Thus, the mutations in this AOA2 patient did not appear to alter the cellular localization of senataxin.
Fig. 2.

Senataxin can be detected in ataxia with oculomotor apraxia type 2 fibroblasts. Patient (III.4) and control fibroblasts were cultured. Confocal images of immunocytochemical localization studies using antibodies against senataxin (green) and Alexa Fluor (Life Technologies molecular probes, Carlsbad, CA, USA) 555 Phalloidin (Invitrogen, Carlsbad, CA, USA) for actin (red) are shown. 4′,6-diamidino-2-phenylindole (DAPI) stains the nucleus blue. The merged image is at the right, and the boxed area is enlarged in the panels directly below. Scale bars = 25 μm.
3.3. SETX mutant cells are more susceptible to oxidative DNA damage
As a nucleic acid helicase, senataxin is involved in DNA damage repair.25 To determine whether this combination of mutations leads to increased sensitivity to oxidative damage, we performed a single cell gel electrophoresis assay (also known as comet assay).27,28 This assay uses H2O2 to induce oxidative damage, and recovery is measured over time. During electrophoresis, unrepaired DNA fails to travel as a compact unit; rather it forms a stream, or comet. If carried out under alkaline conditions, as in our study, single stranded breaks are preferentially made. Using this assay, we noted that after exposure to H2O2, but before repair had taken place (at time zero), there was more DNA damage in AOA2 patient cells (Extent Tail Moment = 2917 ± 67.9%) than in control cells (Extent Tail Moment = 2161 ± 64.5%, p < 0.0001) (Fig. 3). To evaluate the efficiency at which the DNA was repaired, we allowed a recovery period of 4 hours. After 4 hours there remained significantly more damage in the AOA2 patient cells (Extent Tail Moment = 1949 ± 70.2%) that in the control cells (Extent Tail Moment = 685 ± 83.8%, p < 0.0001). Furthermore, after normalizing the Extent Tail Moment to 100% at time zero for both lines, in the control cells only 31.7 ± 3.9% on the DNA remained unrepaired, which suggested a more efficient rate of repair than for the AOA2 cells where 66.8 ± 2.4% of the DNA remained un-repaired. Thus, the AOA2 patient cells had both increased sensitivity to oxidative DNA damage as well as slower recovery.
Fig. 3.
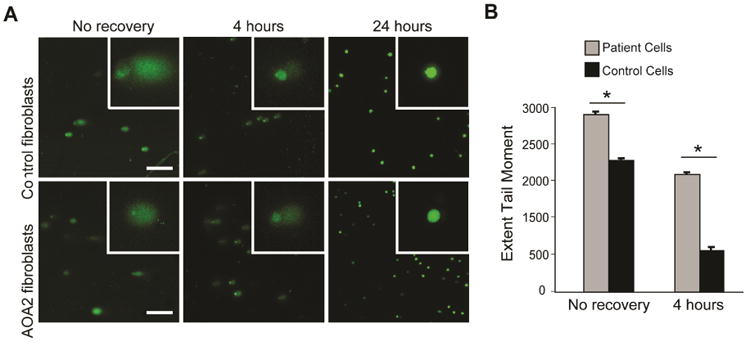
Ataxia with oculomotor apraxia type 2 (AOA2) fibroblasts have increased sensitivity to oxidative DNA damage and slower recovery. (A) Confocal images of comet assays for control and AOA2 cells, stained with Vista Green DNA dye. Insets are enlargements of individual cells. Scale bars,= 250 μm. (B) Extent Tail Moment for patient and control cells. Statistical analysis was performed using a two-sample t-test. Fifty cells were evaluated for each time point, with means ± standard error of the mean graphed.
*p < 0.0001.
4. Discussion
Recessive mutations in SETX causing AOA2 have been reported in a wide range of populations (Supp. Table 1). The c.6848_6851delCAGA allele has been previously reported in an Algerian AOA2 patient, while the c.994 C>T mutation has been identified in French AOA2 patients.1,3 Patient IV.4, a maternal cousin (once removed) of the proband, is reportedly affected with a similar syndrome, although this individual is most likely homozygous for the c.994 C>T (p.R332W) mutation given the known consanguinity in her parents (Fig. 1B; III.9 and III.10).
Immunocytochemical studies of fibroblasts from the patient were indistinguishable from those of a normal control with respect to senataxin localization. Most senataxin protein localized within the nucleus, with mild diffuse staining throughout the cytoplasm, in agreement with previous reports.23 This suggests that protein localization is unaffected in this AOA2 patient. Comet assays on fibroblasts showed that the patient cells were both more susceptible to oxidative damage and slower in repairing damage, supporting the prevailing view that SETX mutations lead to impaired DNA repair. This is also consistent with earlier studies evaluating AOA2 lymphoblastoid cell lines28, and indicates that such cellular studies may be an important adjunct in cases where pathogenicity of mutations in SETX is uncertain.
Supplementary Material
Acknowledgments
We thank Dr. Christopher Grunseich for performing the skin biopsy, Elizabeth Hartnett for assistance with the patient coordination and Dr. Benoît Renvoisé for assistance with the Comet assay. We also thank Dr. Sungyoung Auh for advice with the statistical analysis.
This research was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health.
Footnotes
Conflicts of Interest/Disclosures:The authors declare that they have no financial or other conflicts of interest in relation to this research and its publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anheim M, Fleury M, Monga B, et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11(1):1–12. doi: 10.1007/s10048-009-0196-y. [DOI] [PubMed] [Google Scholar]
- 2.Anheim M, Monga B, Fleury M, et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009;132(10):2688–98. doi: 10.1093/brain/awp211. [DOI] [PubMed] [Google Scholar]
- 3.Anheim M, Fleury MC, Franques J, et al. Clinical and molecular findings of ataxia with oculomotor apraxia type 2 in 4 families. Arch Neurol. 2008;65(7):958. doi: 10.1001/archneur.65.7.958. [DOI] [PubMed] [Google Scholar]
- 4.Criscuolo C, Mancini P, Saccà F, et al. Ataxia with oculomotor apraxia type 1 in Southern Italy: late onset and variable phenotype. Neurology. 2004;63(11):2173–2175. doi: 10.1212/01.wnl.0000145604.57000.36. [DOI] [PubMed] [Google Scholar]
- 5.Criscuolo C, Chessa L, Di Giandomenico S, et al. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology. 2006;66(8):1207–10. doi: 10.1212/01.wnl.0000208402.10512.4a. [DOI] [PubMed] [Google Scholar]
- 6.Duquette A, Roddier K, McNabb-Baltar J, et al. Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol. 2005;57(3):408–14. doi: 10.1002/ana.20408. [DOI] [PubMed] [Google Scholar]
- 7.Fogel BL, Perlman S. Novel mutations in the senataxin DNA/RNA helicase domain in ataxia with oculomotor apraxia 2. Neurology. 2006;67(11):2083–2084. doi: 10.1212/01.wnl.0000247661.19601.28. [DOI] [PubMed] [Google Scholar]
- 8.Le Ber I, Bouslam N, Rivaud-Péchoux S, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127(4):759–67. doi: 10.1093/brain/awh080. [DOI] [PubMed] [Google Scholar]
- 9.Tazir M, Ali-Pacha L, M'Zahem a, et al. Ataxia with oculomotor apraxia type 2: a clinical and genetic study of 19 patients. J Neurol Sci. 2009;278(1-2):77–81. doi: 10.1016/j.jns.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Asaka T, Yokoji H, Ito J, et al. Autosomal recessive ataxia with peripheral neuropathy and elevated AFP: novel mutations in SETX. Neurology. 2006;66(10):1580–1. doi: 10.1212/01.wnl.0000216135.59699.9b. [DOI] [PubMed] [Google Scholar]
- 11.Izatt L, Németh AH, Meesaq A, et al. Autosomal recessive spinocerebellar ataxia and peripheral neuropathy with raised alpha-fetoprotein. J Neurol. 2004;251(7):805–12. doi: 10.1007/s00415-004-0427-y. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe M, Sugai Y, Concannon P, et al. Familial spinocerebellar ataxia with cerebellar atrophy, peripheral neuropathy, and elevated level of serum creatine kinase, gamma-globulin, and alpha-fetoprotein. Ann Neurol. 1998;44(2):265–269. doi: 10.1002/ana.410440220. [DOI] [PubMed] [Google Scholar]
- 13.Le Ber I, Brice A, Dürr A. New autosomal recessive cerebellar ataxias with oculomotor apraxia. Curr Neurol Neuroscience Rep. 2005;5(5):411–417. doi: 10.1007/s11910-005-0066-4. [DOI] [PubMed] [Google Scholar]
- 14.Le Ber I, Rivaud-Péchoux S, Brice A, Dürr A. Autosomal recessive cerebellar ataxias with oculomotor apraxia. Rev Neurol (Paris) 2006;162(2):177–184. doi: 10.1016/s0035-3787(06)74997-9. [DOI] [PubMed] [Google Scholar]
- 15.Date H, Onodera O, Tanaka H, et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29(2):184–8. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- 16.Le Ber I, Moreira MC, Rivaud-Péchoux S, et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain. 2003;126(Pt 12):2761–2772. doi: 10.1093/brain/awg283. [DOI] [PubMed] [Google Scholar]
- 17.Moreira MC, Klur S, Watanabe M, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36(3):225–7. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- 18.Tranchant C, Fleury M, Moreira MC, et al. Phenotypic variability of aprataxin gene mutations. Neurology. 2003;60(5):868–870. doi: 10.1212/01.wnl.0000048562.88536.a4. [DOI] [PubMed] [Google Scholar]
- 19.Chun HH, Gatti Ra. Ataxia-telangiectasia, an evolving phenotype. DNA repair (Amst) 2004;3(8-9):1187–96. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 21.Hernandez D, McConville CM, Stacey M, et al. A family showing no evidence of linkage between the ataxia telangiectasia gene and chromosome 11q22-23. J Med Genet. 1993;30(2):135–140. doi: 10.1136/jmg.30.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart GS, Maser RS, Stankovic T, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99(6):577–87. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 23.Chen YZ, Hashemi SH, Anderson SK, et al. Senataxin, the yeast Sen1p orthologue: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol Dis. 2006;23(1):97–108. doi: 10.1016/j.nbd.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Ursic D, Chinchilla K, Finkel JS, Culbertson MR. Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res. 2004;32(8):2441–52. doi: 10.1093/nar/gkh561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suraweera A, Becherel OJ, Chen P, et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol. 2007;177(6):969–79. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74(6):1128–35. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mckelvey-martin VJ, Edwin TSH, Mckeown SR, et al. Emerging applications of the single cell gel electrophoresis (Comet) assay. I. Management of invasive transitional cell human bladder carcinoma. II. Fluorescent in situ hybridization Comets for the identification of damaged and repaired DNA sequence. Mutagenesis. 1998;13(1):1–8. doi: 10.1093/mutage/13.1.1. [DOI] [PubMed] [Google Scholar]
- 28.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175(1):184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 29.Airoldi G, Guidarelli A, Cantoni O, et al. Characterization of two novel SETX mutations in AOA2 patients reveals aspects of the pathophysiological role of senataxin. Neurogenetics. 2010;11(1):91–100. doi: 10.1007/s10048-009-0206-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.