

Abstract
Background
A methyl derivative natural triterpenoid amooranin (methyl-25-hydroxy-3-oxoolean-12-en-28-oate, AMR-Me) has been found to possess antiproliferative, proapoptotic and anti-inflammatory effects against established tumor cells. Large-scale synthesis of pure AMR-Me has eliminated the need of the natural phytochemical for further development of AMR-Me as an anticancer drug. Metastatic melanoma is a fatal form of cutaneous malignancy with poor prognosis and limited therapeutic options. It was hypothesized that antitumor pharmacological effect of AMR-Me could be linked to AMR-Me-mediated suppression of the metastatic potential of B16F10 murine melanoma.
Methods
AMR-Me was assessed for its antimetastatic efficacy by cell adhesion, migration and invasion assays in B16F10 cells. The signaling crosstalk was explored by gelatin zymography, Western blot, ELISA and immunocytochemistry.
Results
The results elicited that AMR-Me was successful in restricting the adhesion, migration and invasion of highly metastatic cells. The antimetastatic potential of this compound may be attributed to the reduced expression of membrane type 1 metalloproteinase (MT1-MMP) and matrix metalloproteinases (MMP-2 and MMP-9). AMR-Me was found to downregulate vascular endothelial growth factor (VEGF)/prosphorylated forms of focal adhesion kinase (pFAK397)/Jun N-terminus kinase (pJNK)/extracellular signal-regulated kinase (pERK). This, in turn, inhibited transcription factor nuclear factor-κB (NF-κB) and transactivation of MMPs. Moreover the activation of tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2) might have influenced the downmodulation of MT1-MMP, MMP-2 and MMP-9.
Conclusion
AMR-Me suppresses the activity of MT1-MMP, MMP-2 and MMP-9 by downregulation of VEGF/pFAK397/pJNK/pERK/NF-κB and activation of TIMP-1 and TIMP-2 in metastatic melanoma cell line, B16F10.
General significance
AMR-Me has the potential as an effective anticancer drug for metastatic melanoma which is a dismal disease.
Keywords: Methyl amooranin, melanoma, migration, invasion, metastasis, MMPs, NF-κB
Introduction
Terpenoids, also known as terpenes or isoprenoids, represent the largest group of phytochemicals and are classified according to the number of isoprene units [1]. More than 20,000 triterpenoids are believed to exist in nature and a large number of these phytochemicals find use in Asian medicine [2]. Pentacyclic triterpenoids are the most potent natural compounds endowed with anti-inflammatory and antitumorigenic activities [3,4]. Oleanolic acid is a ubiquitous pentacyclic triterpenoid in plant kingdom, including various dietary and medicinal plants [4,5]. Olive pulp (obtained following the oil is pressed from the fruit) as well as leaves represent rich sources of oleanolic acid [6]. Accumulating evidence shows that oleanolic acid and its derivatives possess broad spectrum biological and pharmacological activities, including cancer preventive and anticancer potential [7–9].
To improve pharmacological potency, a series of new synthetic oleanane triterpenoids have been designed and synthesized based on oleanolic acid as a starting material [10]. More than 300 derivatives of oleanolic acid have been developed and the most active and useful derivatives include 2-cyano-3,12-dioxoolena-1,9(11)-dien-28-oic acid (CDDO), methyl ester (CDDO-Me), imidazolide (CDDO-Im), dinitrile (di-CDDO), methyl amide (CDDO-MA), ethyl amide (CDDO-EA) and trifluoroethyl amide (CDDO-TFEA) [11]. Several of these synthetic oleanane triterpenoids are considered to be the most potent anti-inflammatory and anticarcinogenic triterpenoids known to mankind [2]. Synthetic oleanane triterpenoids are known as potential “multifunctional” drugs that target cellular networks that mediate anti-inflammatory, antioxidative, antiproliferative, differentiative, and proapoptotic activities. Several known signaling pathways modulated by synthetic oleanane triterpenoids include nuclear factor-E2-related factor 2/Kelch-like erythroid cap’n’Collar homologue-associated protein 1/antioxidant response element, nuclear factor-κB (NF-κB)-inhibitor of κB kinase (IKK), transforming growth factor-β, janus-tyrosine kinase/signal transducer and activator of transcription, peroxisome proliferator-activated receptor γ, phosphatase and tensin homolog, human epidermal growth factor receptor 2, and phosphatidylinositol 3-kinase (PI-3K)/protein kinase B (AKT) pathway [11]. Emerging studies show that synthetic oleanane triterpenoids inhibit proliferation and induce apoptosis in a wide variety of human and rodent cancer cells as well as exert chemopreventive and anti-tumor effects in various rodent cancer models (reviewed in ref. [11]). At least two phase I clinical trials evaluated the antitumor activities of CDDO (NCT00322140) and CDDO-Me (NCT00508807) in adult patients with solid tumors and lymphomas (http://clinicaltrials.gov/).
Amooranin (AMR), a triterpene acid (25-hydroxy-3-oxoolean-12-en-28-oic acid) isolated from the stem bark of Indian medicinal plant Amoora rohituka, has been found to possess anticancer effects in vitro [12–16] and in vivo [14]. A methyl derivative of the natural triterpenoid compound AMR, namely methyl-25-hydroxy-3-oxoolean-12-en-28-oate (AMR-Me), has been found to possess superior cytotoxic effect against MCF-7 human breast cancer cells to the parent compound AMR [12]. Subsequently, AMR-Me has been shown to inhibit the proliferation of human breast cancer cells MCF-7 and MDA-MB-231 and human acute lymphoblastic leukemia cells by apoptosis-inducing mechanisms [17–19] as well as was found to improve the survival of mice bearing Dalton’s ascites tumor cells [18]. Recently, we have developed a novel method of large-scale synthesis of pure AMR-Me, eliminating the need of using plant material and hence the dependency on the nature [20]. Oral administration of AMR-Me dose-dependently reduced the incidence and burden of 7,12-dimethylbenz(a)anthracene-induced rat mammary tumors through antiproliferative, proapoptotic and anti-inflammatory mechanisms mediated through modulation of multiple signaling pathways, including estrogen receptors, Wnt/β-catenin and NF-κB [20–22].
Metastatic melanoma has been the most devastating form of cutaneous malignancy. Once metastasized to remote sites, it is characteristically unresponsive to treatment. Only 14% of the metastatic melanoma patients survive for 5 years [23]. At present, prevention and early detection are the most effective measures against melanoma-related mortality.
Invasion and metastasis have been postulated as the “sixth hallmark” of cancer [24] and represent interrelated processes characterized by cell growth, cell adhesion, cell migration, and proteolytic degradation of tissue components, including extracellular matrix (ECM) and basement membrane. Metastatic process initiates with alterations of the cell–cell and cell–matrix attachment followed by a proteolytic degradation of the ECM and migration of tumor cells. Such degradation of the ECM is catalyzed by matrix metalloproteinases (MMPs). They are a family of zinc-dependent endopeptidases responsible for degradation of ECM including basement membrane collagen, interstitial collagens, fibronectin (FN) and various proteoglycans both in physiological and pathological conditions [25]. Membrane type 1 metalloproteinase (MT1-MMP) is a key ECM-degrading enzyme that acts as part of the invasion machinery [26]. MT1-MMP was the first membrane type MMP identified and characterized as a specific activator of pro-MMP-2 at the cell surface [27]. Gelatinases including gelatinase A (MMP-2) and gelatinase B (MMP-9) are found to be over expressed in several malignant cancers, including melanoma and fibrosarcoma [28,29], and are postulated to play a critical role in tumor invasion and angiogenesis. A complicated signaling network is believed to mediate the expression and activity of MMP-2 and MMP-9. Tissue inhibitors of metalloproteinases (TIMPs) were reported as endogenous inhibitors of the proteolytic activity of activated MMP by forming a 1:1 stoichiometric inhibitory complex with the enzyme [30].
Cell adhesion to one of the ECM components, such as FN, has been evidenced to regulate MMP secretion in normal and tumor cell systems [31]. Mitogen-activated protein kinases (MAPKs), namely extracellular signal-regulated kinases (ERKs), Jun N-terminus kinases (JNKs) and p38 MAPKs apart from their well-characterized role in cell proliferation/differentiation and cell survival/apoptosis, have been reported for active participation in cell migration. It has been evidenced that FN activates MMP-9 secretion via the MAPK/PI3K-Akt pathways in ovarian cancer cells [31]. Interaction of α5β1 with FN increases MMP-9 expression through activation of c-Fos via ERK and PI3K pathways in human lung carcinoma cells [32]. Phosphorylation of focal adhesion kinase (FAK), a non receptor tyrosine kinase and PI3K and subsequent nuclear translocation of ERK and NF-κB upon FN binding demonstrated possible participation of the FAK/PI-3K/ERK signaling pathways in the regulation of MMP-2 activity [33]. MMP-2 and MMP-9 have a key role in angiogenesis due to their collagenolytic activity which enables endothelial cells to invade the basement membrane of vascular structures, where type IV collagen forms the principal component. MMP-9 is known to enhance the angiogenic process not only by mediating the digestion of ECM, but also by releasing ECM-bound angiogenic factors such as vascular endothelial growth factor (VEGF) and transforming growth factor-β1, cleaving endothelial cell–cell adhesion and generating promigratory ECM fragments. FN- and vitronectin-induced MMP-9 was shown to mediate later phases of angiogenesis [34].
With this background, we have hypothesized that the antitumor pharmacological effect of AMR-Me could be involved in AMR-Me-mediated suppression of adhesion, invasion and migration of established tumor cells which, in turn, would underscore the therapeutic potential of this compound. Accordingly, the present study was designed to investigate the antimetastatic effect of AMR-Me on highly metastatic murine melanoma cell line, B16F10, and the plausible implication of the compound on the signaling network of the gelatinases which are the key players of the metastatic process.
Materials and methods
Chemicals
AMR-Me has been synthesized following a proprietary method described elsewhere [35]. Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) was purchased from Gibco Life Technologies Corporation (Grand Island, NY). FN (440 kDa), protease inhibitor cocktail tablets (complete, mini, and EDTA-free), were purchased from Roche (Mannheim, Germany). Gelatin sepharose 4B beads were purchased from GE Healthcare Bio-Sciences AB (Uppsala, Sweden). 3-(4,5-Dimethylthiazol-2-yl)-2-5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Amresco (Solon, OH). All primary antibodies (monoclonal and polyclonal), secondary antibodies were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Super Signal West Femto Maximum sensitivity substrate for enhanced chemiluminiscence was purchased from Thermo Scientific (Rockford, IL). RNAqueousR-4PCR (DNA-free™ RNA isolation for RT-PCR) kit, RETROscript Kit (for RT-PCR) and Super Taq Plus polymerase was purchased from Ambion, Life Technologies, Grand Island, NY. Primers for NF-κB were synthesized by Integrated DNA Technologies, Inc. (Corallville, IA). Costar transwell plates were purchased from Corning Inc. (Corning, NY).
Cell culture
Highly metastatic murine melanoma cell line B16F10 was obtained from National Centre for Cell Sciences (Pune, India). Cells were cultured and maintained in DMEM supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C in a humidified atmosphere of 5% CO2 incubator.
Cytotoxicity assay
Cytotoxicity was assessed by the MTT assay. Exponentially growing cells (1×104) were seeded in 96-well plates. After 24 h of growth, the cells were treated with a series of concentrations of AMR-Me. Doxorubicin was used as the positive control. Incubation was carried out at 37°C for 24 and 48 h. A separate lane was also used as a vehicle control, i.e. dimethyl sulfoxide (DMSO); however, apart from the cytotoxicity assays the DMSO concentration was not allowed to exceed 1% for other experiments. The viability of B16F10 cells exposed to 1% DMSO varied between 98 and 99%. MTT solution was added to each well (1.2 mg/ml) and incubated for 4 h. The reaction resulted in the reduction of MTT by the mitochondrial dehydrogenases of viable cells to a purple formazan product. The MTT-formazan product dissolved in DMSO was estimated by measuring absorbance at 570 nm in a multiwall microplate reader (Infinite M200, TECAN, Mannedrof, Switzerland).
Treatment of cells for various assays
During cells cycle analysis, two concentrations (10 μM AMR-Me with 24 h treatment and 5 μM with 48 h treatment) were selected from cytotoxicity assay which were observed with more than 70% cell viability. 1.0% and 0.5% DMSO was used as the solvent control for 24 h and 48 h treatment respectively. All the experiments for cell cycle analysis, cell adhesion assays, cell migration assays, cell invasion assays and zymography were conducted with both the treatment protocols. Subsequently, the experiments for the signaling pathway were performed following single treatment protocol of 5 μM AMR-Me for 48 h which appeared to be the most effective.
Cell cycle analysis
2×104 B16/F10 melanoma cells both for treated and control were harvested, washed with PBS, resuspended in sterile PBS. Cells were fixed in ice cold 100% methanol −20°C for 1 h. The cells were centrifuged at 5,000 rpm for 10 min and the cell pellets were suspended in 1 ml of hypotonic buffer (0.5% Triton X-100 in PBS and 0.5 mg/ml RNase), and incubated at 37°C for 30 min. Subsequently, propidium iodide solution (50 mg/ml) was added, and the mixture was allowed to stand for 1 h in darkness. Fluorescence that emitted from the propidium iodide-DNA complex was quantitated after excitation of the fluorescent dye by (FACS Calibur with sorter, BD Biosciences, San Jose, CA) using Cell Quest software (BD Biosciences, San Jose, CA) equipped with a 488 nm argon laser and a 525±10 nm band pass emission filter. Fluorescence was captured on a FL2H channel with linear amplification.
Cell adhesion assay
Cell adhesion assay was performed with both treated and control B16F10 cells. Microtitre plate wells were coated in triplicate with ECM ligands FN (120 kDa chymotryptic fragment) at various concentrations of 5, 10, and 20 mg/ml. Ligands were allowed to bind for 1.5 h at 37°C. The wells were then blocked with Buffer C (1% BSA, 1 mM CaCl2 and 1 mM MgCl2 in PBS) for 1 h at 37°C. Cells were collected from culture flasks by trypsinization, washed and suspended in Buffer C. Approximately 5×104 cells were added to each well and allowed to bind for 1.5 h at 37°C. The wells were washed thrice with Buffer C. The attached cells were trypsinized and counted on a haemocytometer. Percentage of adhesion was calculated (considering control as 100%).
Wound healing assay
For cell motility determination, wound healing assay was performed [36]. The monolayer was scratched with a sterile pipette tip in both treated and control sterile petridishes followed by washing with serum free culture media (SFCM) for three times to remove cellular debris. The cells were maintained in fresh SFCM and the wound closure was monitored and photographed at 0, 6, 12, 24 h using an inverted microscope and camera (Nikon, Tokyo, Japan). To quantify the migrated cells, pictures of the initial wounded monolayers were compared with the corresponding pictures of cells at the end of the incubation. Artificial lines fitting the cutting edges were drawn on pictures of the original wounds and overlaid on the pictures of cultures after incubation. Cells that had migrated across the black lines were counted in six random fields from each triplicate treatment.
Transwell invasion assay
Cells (both treated and control) were added to Boyden’s invasion chamber (2×105 cells/chamber in triplicate) and grown for 24 h in SFCM with the chamber inserted in MEM containing 2% FBS as chemoattractant. Cells were then allowed to grow and after 24 h of incubation, media was pipetted out from membrane. SFCMs from lower chambers were collected and centrifuged at 3,000 rpm for 3 min. The membranes of the inserts were washed thrice with PBS. Cells were then fixed with 4% formaldehyde solution, followed by washing with PBS. Cells were then stained with Gill’s hematoxylin for 10 min. Membranes were then washed thoroughly in running water. Cells spread on top of the Matrigel coated membrane were removed using cotton swab. The cells which had migrated to the opposite surface of the Matrigel were observed under inverted microscope. Cells from at least 5 different fields were counted at 40× magnification and the means were calculated. The results were expressed as percentage of control.
Gelatin zymography
Gelatin zymography was performed with both treated and control B16F10 cells. MMPs were concentrated from conditioned SFCM by binding to gelatin sepharose 4B beads for 2 h at 4 °C. The beads were washed 3× with Tris-buffered saline with Tween-20 (TBST) and suspended in 50 μl 1× sample buffer (0.075 g Tris, 0.2 g SDS in 10 ml water, pH 6.8). The suspension was incubated for 30 min at 37°C then subjected to zymography on 10% SDS-PAGE copolymerized with 0.1% gelatin. Gels were washed in 2.5% Triton-X100 for 30 min to remove SDS and then incubated overnight in reaction buffer (50 mM Tris–HCl pH 7, 4.5 mM CaCl2, 0.2 M NaCl). After incubation, the gel was stained with 0.5% Coomassie Blue in 30% methanol and 10% glacial acetic acid. The bands were visualized by destaining the gel with 30% methanol and 10% glacial acetic acid.
Extraction of cellular and nuclear proteins and their estimation
Cell extraction of both treated and control B16F10 cells was performed with RIPA buffer (Tris HCl 50 mM, pH 7.5; NaCl 150 mM; NP40 10%; sodium deoxycholate 0.5%; SDS 0.1%; protease inhibitor cocktail; NaF 0.01%; Na3VO4 0.01%) at −80°C for 1 h. Subsequently, it was followed with centrifugation at 12,000 rpm for 15 min. Cell supernatant was used for protein estimation. Protein estimation was done from the cell lysate using the Lowry’s method [37].
The pellet was subsequently used to prepare the nuclear protein extract for the NF-κBp65 assay. Nuclear proteins were isolated following the technique of Dignam et al. [38] with modifications. In short, the pellet was resuspended in ice-cold low-salt buffer (pH 7.9) containing 1.0 mM DTT, 0.2 mM EDTA, 25% (v/v) glycerol, 20 mM Hepes, 20 mM KCl, 1.5 mM MgCl2 and 0.2 mM PMSF. The release of nuclear proteins was achieved by adding a high-salt buffer (pH 7.9) containing 1.0 mM DTT, 0.2 mM EDTA, 25% (v/v) glycerol, 20 mM Hepes, 1.2 M KCl, 1.5 mM MgCl2 and 0.2 mM PMSF drop by drop to a final KCl concentration of 0.4 M [36]. Following 30 min incubation on ice with smooth shaking, the soluble nuclear proteins were recovered by centrifugation at 25,000 g for 30 min at 4°C. The samples were estimated for protein using the Lowry’s method [37] and stored at −80°C until assayed.
Protein expression by ELISA
The wells of microtitre plate were coated in triplicate with 10 μg of protein from cell lysate of both control and experimental set and kept at 4°C overnight (under moist condition to prevent evaporation). Blank wells (only with buffer in which samples are suspended) were also prepared. Next day, wells were washed with blocking buffer (1% BSA in PBS) to block non-specific binding sites and incubated for 1 h at 37°C. Then the wells were washed thrice with Washing Buffer (0.5% NP-40 & 0.5% BSA dissolved in PBS). Anti-MMP-9 antibody (1:1000, 1 μg antibody in 1 ml buffer) was added to the wells and incubated at 37°C for 1 h. Wells were washed thrice with Washing Buffer. Respective second antibody solution (1:1000 dilution buffer) was added to wells and incubated at 37°C for 1 h. Wells were washed six times with Washing Buffer (3–5 min per wash). Substrate (TMB) was added to the wells (in darkness) till color developed. Then 1 M H2SO4 stop solution was added and reading was recorded by using an ELISA reader at 450 nm.
Western blot analysis
B16F10 cells (3×105 cells/ml) were grown with vehicle control (0.5% DMSO) and AMR-Me (5 μM) for 48 h. In case of MMP-2 and MMP-9, SFCM was collected and gelatinase was extracted from it by Gelatin Sepharose beads and then eluted at 37°C for 30 min. The cells (and SFCM elute in case of MMP-9) were collected; extracted and equal amount of total protein or nuclear proteins as in case of NFκBp65 (70 μg) were suspended in Laemmli’s buffer containing β-mercaptoethanol for 5 min at 90°C. The samples were run on SDS-PAGE (7.5%) and blotted onto polyvinylidene difluoride membranes. The membranes were blocked using 5% BSA in Tris-buffered saline with Tween-20 (TBS-T; 50 mM Tris, 150 mM NaCl, and 0.05% Tween-20), incubated with anti-MMP-2, anti-MMP-9, anti-TIMP-1, anti-TIMP-2, anti-VEGF, anti-FAK polyclonal antibody, anti-phospho FAK Try 397 (p-FAK Try 397), anti-JNK), anti-phospho JNK (p-JNK), anti-ERK1/2, anti-phospho ERK1/2 Thr 202/Tyr 204 (pERK1/2 Thr 202/Tyr 204); anti NF-κBp65 monoclonal antibodies [1:1000 (1 μg antibody in 1 ml buffer) dilution] for 90 min at 37°C, washed thrice in TBS-T, incubated with horse peroxidase coupled goat-anti rabbit secondary antibody for 90 min at 37°C and washed thrice with TBS-T. Bands were visualized using enhanced chemiluminiscence method.
Immunocytochemical localization of NF-κB
B16F10 cells were allowed to grow overnight on coverslips in DMEM containing 10% FBS. After washing, the cells on the coverslips were incubated with AMR-Me (5 μM) for 48 h and 0.5% DMSO vehicle control at 37°C in a CO2 incubator. The coverslips were then washed in PBS, fixed with 3.5% formaldehyde, treated with 0.5% Triton-X100 and nonspecific sites were blocked with 1% BSA. The cells were then treated with anti-NF-κB (1:1000 dilution) for 1.5 h at 37 °C followed by FITC-coupled secondary antibody at 37°C (1:1000 dilution) for 1.5 h at 37 °C in a humidified chamber. After washing 5× in PBS, the coverslips were mounted on glass slides and observed under a fluorescence microscope using the software Fluorescent Workstation 500 (Nikon, Tokyo, Japan).
Semi-quantitative RT-PCR
RNA was extracted from 1 × 106 cells/ml B16F10 cells grown in absence and in presence of AMR-Me (5 μM) for 48 h. Isolation of total RNA from cells was performed using RNAqueous-4PCR kit (Ambion, Life Technologies, Grand Island, NY) according to manufacturer’s instructions. cDNA was synthesized from 2 μg of total RNA using RetroScript kit (Ambion/Applied Biosystem). The cDNA was then amplified by polymerase chain reaction for 30 cycles with an initial hot start followed by denaturation, annealing and extension at (94°C for 30 sec, 55°C for 30 sec, 72°C for 90 sec; 28–30 cycles) using primers (forward primer sequence 5′-GTGGAGGCATGTTCGGTAGT and reverse primer sequence 5′-GATGGAATGTAATCCCACCG; gene bank accession number: NM 8689.2) designed for NF-κBp105 gene. Positive control was supplied with the kit which was a constitutively expressed “housekeeping” gene rig/S15, encoding a small ribosomal subunit protein (forward primer sequence 5′-TTCCGCAAGTTCCACCTACC and reverse primer sequence 5′-CGGGCCGGCCATGCTTTACG). PCR product was then analyzed by electrophoresis on 2% agarose gel along with a 50 bp DNA ladder and visualized under Gel Documentation System (Bio-Rad, Molecular Image Chemi Doc™ XRS+ with Image Lab™ Software, Hercules, CA).
Quantification of the results
The gel pictures were obtained from the Gel Documentation System (Bio-Rad, Molecular Image Chemi Doc™ XRS+ with Image Lab™ Software, Hercules, CA) in JEPG or TIFF format and analyzed with a computer (HP Compaq dx2480 Business Desktop) equipped with the Image J Launcher (version 1.4.3.67) software. The blots were scanned with the scanner HPscanjet G2410 and further analyzed with computer equipped with the Image J Launcher (version 1.4.3.67) software.
Statistical analysis
The statistical significance between treated and control groups were determined using the one way ANOVA followed by Dunnett t-test, where P value was set at 0.05 to check the statistical difference between groups. Dunnett t-test treats one group as a control and treats all other groups against it. All results were computed and analyzed using the SPSS statistical software package 10.0 (SPSS, Chicago, IL).
Results
AMR-Me reduces melanoma cell viability
The cytotoxic effect of AMR-Me on exponentially growing B16F10 cells was assessed by MTT assay. AMR-Me was tested for its cytotoxic effect at concentrations of 2.5, 5, 10, 20 and 40 μM for two different time intervals – 24 and 48 h. The percentage of surviving cells over untreated control cells was calculated and plotted against drug concentrations (Fig. 1A). The IC50 value for AMR-Me was calculated from the dose response curve which was 24 μM for 24 h and 21 μM for 48 h treatment. From the cell viability results, two AMR-Me concentrations were used for further experiments which were 10 μM for 24 h treatment (10/24) and 5 μM for 48h treatment (5/48). The cell viability for 10/24 was 64.62% and for 5/48 it was 73.6%. The vehicle control at these doses did not induce significant cytotoxicity.
Fig. 1.

Dose-dependent and time-dependent effects of AMR-Me on viability and cell cycle progression of B16F10 murine melanoma cells. (A) Cytotoxicity studies as assessed by MTT assay depicting effect of various doses AMR-Me on percentage of cell viability at two different time intervals – 24 and 48 h. (B) FACS analysis showing effect of AMR-Me on B16F10 cells along with the respective graphical representation of the percentages of cells at different stages of cell cycle. (a,b) Comparison of cell cycle analysis of B16F10 cells with vehicle control and AMR-Me (10 μM) for 24 h treatment (P>0.05); (c,d) Comparison of cell cycle analysis of B16F10 cells with vehicle control and AMR-Me (5 μM) for 48 h treatment (P>0.05).
AMR-Me exerts marginal effect on cell cycle distribution and apoptosis
In order to find out the effect of AMR-Me on the cell cycle and apoptosis, we had performed cell cycle analysis by fluorescence activated cell sorter (FACS) analysis (Fig. 1B). Both concentrations selected for AMR-Me (10/24 and 5/48) did not induce any apoptotic effects on the melanoma cells which is depicted by the negligible sub G1 peaks (Fig. 1B–b and 1B–d). The FACS analytical representation of the cell counts at different phases of the cell cycle with two different doses of AMR-Me (10/24 and 5/48) did not show any significant effect of the compound (Fig. 1B–b, 1B–d) compared to the control (Fig. 1B–a and1B–c) on B16F10 cells. The percentages of cells at different phases of cell cycle (Fig. 1B–e and 1B–f) revealed similar trend of AMR-Me effect on melanoma cells.
AMR-Me reduces adhesion of B16F10 cells with the extracellular ligand FN
From cell adhesion assay, it was observed that the number of adherent B16F10 cells (vehicle control) increased in a dose dependent manner with enhancement in the dose of extra cellular ligand FN (5, 10 and 20 μg/ml). On the other hand, both the treatment modes of AMR-Me (10/24 and 5/48) significantly inhibited (P<0.01) the number of adhered cells with increasing doses of FN (Fig. 2A). The reduction of cell adhesion was more effective with 5 μM AMR-Me treatment for 48 h where the number of adhered cells reached below 5,000 with 20 μg/ml FN (Fig. 2A).
Fig. 2.

Effect of AMR-Me on adhesion and migration of B16F10 cells. (A) Inhibition of FN-induced cell adhesion by AMR-Me (10 μM) for 24 h and AMR-Me (5 μM) for 48 h. Inhibition of cell adhesion by AMR-Me was significant (P<0.001) compared to vehicle control with increasing doses of FN. (B) Inhibition of cell migration by treatment with AMR-Me (10 μM) for 24 h and AMR-Me (5 μM) for 48 h compared to vehicle control as evidenced at different time intervals (0, 6 and 24 h). After 24 h incubation migrated cells across the black lines were counted in six random fields from each treatment and the mean number of cells in the marked zone was quantified by three independent experiments. The graphical representation in terms of % of control exhibited AMR-Me (5 μM) for 48 h compared to vehicle control caused significant inhibition (*P<0.05) of cell migration.
AMR-Me reduces the migration of B16F10 cells
In order to investigate the effect of AMR-Me on migration of B16F10 cells, wound-healing assay was performed. The cells were treated with the two concentrations of AMR-Me for varying time intervals - 10 μM for 24 h and 5 μM for 48h. After treatment the cell migration was observed at different time points up to 24 h. As depicted in Fig. 2B, the cell migration was significantly inhibited with the 5/48 treatment mode (13%; P<0.05) than the 10/24 treatment (9%).
AMR-Me suppresses B16F10 cell invasion
B16F10 cells are well-known for strong invasive properties in Matrigel. In this study, we investigated the effect of AMR-Me on cell invasion using transwell microplate. The observations clearly demonstrated that both treatment modes of AMR-Me (10/24 and 5/48) had significant inhibitory effects (P<0.01) on cell invasion (Fig. 3A). Based on quantitative analysis, the comparison between the two treatments showed that 5/48 treatment reduced cell invasion by 72% and was more effective than 10/24 mode of treatment which reduced cell invasion by 38% (Fig. 3B).
Fig. 3.

Effect of AMR-Me on invasion of B16F10 cells. (A) Inhibition of cell invasion by treatment with AMR-Me (10 μM) for 24 h and AMR-Me (5 μM) for 48 h compared to vehicle control. (B) Graphical representation showing significant reduction (P<0.001) in the number of invading cells with AMR-Me (10 μM) for 24 h and AMR-Me (5 μM) for 48 h compared to vehicle control.
AMR-Me reduces gelatinolytic activity of B16F10 cells
In order to determine the gelatinolytic activity of gelatinase A (MMP-2) and B (MMP-9) following AMR-Me treatment, we performed gelatin zymography with SFCM collected from the treated petri plates as well as from vehicle controls. The zymogram (Fig. 4A) and quantitative representation of respective band intensities (Fig. 4B) showed that with AMR-Me (10 μM) treatment for 24 h had no significant effect on the activity of MMP-2 and MMP-9 in comparison to the vehicle control. However, the zymogram (Fig. 4A) as well as the calculated band intensities (Fig. 4B) with AMR-Me (5 μM) treatment for 48 h showed that there was an appreciable reduction of both the MMP-2 (72 kD) and MMP-9 (92 kD) activity in comparison to the control (P<0.01).
Fig. 4.

Effects of AMR-Me on MMP activities in B16F10 cells. (A) Gelatin zymography showing no effect on the activity of MMP-2 (72kD) and MMP-9 (92kD) with AMR-Me (10 μM) for 24 h compared to vehicle control whereas treatment with AMR-Me (5 μM) for 48 h exhibited inhibition of MMP-2 and MMP-9 activity compared to vehicle control. (B) The respective band intensities of the zymograms as calculated by Image J Launcher (version 1.4.3.67) reflected similar profile of no effect on the activity of MMP-2 and MMP-9 with AMR-Me (10 μM) for 24 h compared to vehicle control (P>0.05) and inhibition of MMP-2 and MMP-9 activity by treatment with AMR-Me (5 μM) for 48h compared to vehicle control (P< 0.01)
AMR-Me modulates MT-MMP-1, MMP9, MMP2, TIMP1, and TIMP2 expressions
The investigation of the signaling pathway was followed with single treatment mode AMR-Me (5 μM for 48 h) due to its effectiveness in the previous experiments. The protein expression of MT-MMP1, MMP-2 and MMP-9 as well as the tissue inhibitors TIMP-2 and TIMP-1 were analyzed with ELISA and Western blot. The protein expression of MMP-9 (P<0.01) as well as that of MMP-2 and MT-MMP1 (P<0.05) were significantly reduced with respect to control as evidenced from the Western blot pictures (Fig. 5A) and quantitative analysis of their respective band intensities (Fig. 5B). The expression of tissue inhibitors TIMP-1 (P<0.01) and TIMP-2 (P<0.05) were increased with AMR-Me treatment (Figs. 5A and 5B). The ELISA results reflected a similar modulation of protein profile which was significant (P<0.01 or 0.05) with respect to vehicle control (Fig. 5C).
Fig. 5.
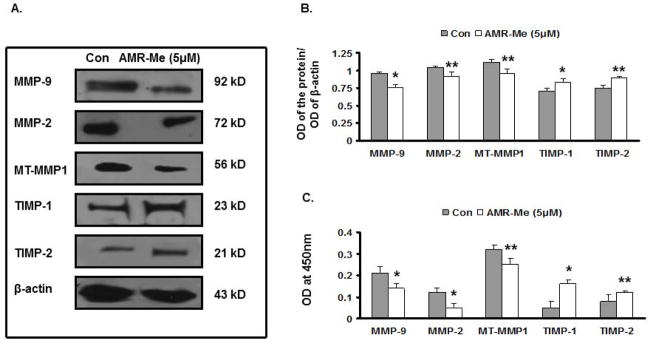
Effect of AMR-Me on expression of metastasis-related genes in B16F10 cells. (A) Western blot showing inhibition in expression of MT-MMP1, MMP-2 and MMP-9 and increased expression of TIMP-1 and TIMP-2 with AMR-Me (5 μM) for 48 h compared to vehicle control. (B) The respective band intensities of the blots as calculated by Image J Launcher (version 1.4.3.67) reflected similar profile of significant inhibition in expression of MMP-9, MMP-2 and MT-MMP1 (P<0.01–0.05) and increased expression of TIMP-1 and TIMP-2 (P<0.01–0.05) with AMR-Me (5 μM) for 48 h compared to vehicle control. (C) Graphical representation of ELISA showing inhibition of protein expression for MMP-9, MMP-2 and MT-MMP1 (P<0.01–0.05) and increased expression of TIMP-1 and TIMP-2 (P<0.01–0.05) with AMR-Me (5 μM) for 48 h compared to vehicle control.
AMR-Me affects various signaling cascades
The Western blot analyses (Fig. 6A) as well as quantitative assessment of the band intensities (Fig. 6B) exhibited that treatment of B16F10 cells with AMR-Me (5 μM) for 48 h considerably downregulated the expression of VEGF, FAK, pFAK397, JNK, pJNK, ERK1/2, pERK1/2 compared to that of the control cells. The band intensities represented significant inhibition of VEGF, pFAK397, pJNK, pERK1/2 (P<0.01) as well as that of FAK, JNK and ERK1/2 (P<0.05). The ELISA results reflected a similar modulation of protein profile which reflected significant reduction of VEGF, pFAK397, pJNK, pERK1/2 (P<0.01) as well as that of FAK, JNK and ERK1/2 (P<0.05) with respect to vehicle control (Fig. 6C).
Fig. 6.

Effect of AMR-Me on regulation of metastasis-related signaling network in B16F10 cells. (A) Western blot analyses showing reduced expression of VEGF, FAK, pFAK397, JNK, pJNK, ERK1/2, pERK1/2 compared to that of the control cells by treatment with AMR-Me (5 μM) for 48 h. (B) The respective band intensities of the blots as calculated by Image J Launcher (version 1.4.3.67) reflected similar profile of significant inhibition in expression of VEGF, pFAK397, pJNK, pERK1/2 (P<0.01) as well as that of FAK, JNK and ERK1/2 (P<0.05) with AMR-Me (5 μM) for 48 h compared to vehicle control. (C) Graphical representation of ELISA showing significant reduction of VEGF, pFAK397, pJNK, pERK1/2 (*P<0.01) as well as that of FAK, JNK and ERK1/2 (P<0.05) with AMR-Me (5 μM) for 48 h with respect to vehicle control.
In order to investigate the localization and changes of expression profile of the transcription factor NF-κB following AMR-Me treatment (5 μM for 48 h) compared to that of control cells, we performed immunocytochemistry. It was observed that the nuclear expression of the NF-κB was markedly reduced in case of treated cells compared to that of control where the fluorescence with FITC was much more intense (Fig. 7A). The protein profile of NF-κB was further validated with Western Blot analysis where the nuclear extracts exhibited reduced expression of NFκBp65 in AMR-Me (5 μM for 48 h) treated cells than control cells (Fig. 7B). As presented in Fig 7C, the band intensity also elicited a similar profile where NF-κB in AMR-Me treated cells was significantly (*P<0.05) reduced compared to control. The mRNA profile (Fig. 7D) and the corresponding band intensity of NF-κB (Fig. 7E) revealed that the transactivation of NF-κBp105 was significantly repressed with AMR-Me (5 μM for 48 h) treatment in B16F10 cells than the vehicle control (*P<0.01).
Fig. 7.

Inhibition of nuclear translocation and expression of NF-κB following AMR-Me treatment. (A) Immunocytochemistry showing inhibition of nuclear localization of NFkBp65 with AMR-Me (5 μM) for 48 h with respect to vehicle control. (B) Western blot showing reduced expression of NF-κBp65 in nuclear fraction of B16F10 melanoma cells treated with AMR-Me (5 μM) for 48 h with respect to vehicle control. (C) The respective band intensities of NF-κBp65 as calculated by Image J software exhibiting significant inhibition (*P<0.05) of pNF-κBp65 in nuclear fraction of B16F10 melanoma cells treated with AMR-Me (5 μM) for 48 h with respect to vehicle control. (D) RT-PCR showing downregulated expression of NF-κBp105 in B16F10 melanoma cells treated with AMR-Me (5 μM) for 48 h with respect to vehicle control. (E) The respective band intensities as calculated by Image J software exhibiting significant inhibition (*P<0.01) of NF-κBp105 in melanoma cells treated with AMR-Me (5 μM) for 48 h with respect to vehicle control.
Discussion
Uncontrolled cell proliferation and metastasis are the primordial events of cancer progression. The presence of metastasis is the main cause of morbidity and mortality in millions of patients with cancer. Melanoma is a fatal form cutaneous malignancy that arises from the transformation of melanocytes. Metastatic melanoma is a serious public health burden due to poor prognosis and limited therapeutic options [39]. An agent that could effectively retard the growth, migration and invasion of cancer cells would be a potential candidate to suppress cancer progression and metastasis.
The present study exhibited for the first time the cytotoxic effects of AMR-Me against B16F10 melanoma cells. Our results are in line with several prior reports with oleanolic acid and its synthetic derivatives. The synthetic oleanane triterpenoid CDDO has been reported to inhibit the proliferation of various human skin cancer cells [40,41]. According to a recent study, CDDO-Me inhibited the viability of various stages of human melanoma cells [42]. Another CDDO analog, namely CDDO-Im, has been shown to arrest proliferation of B16F1 mouse melanoma cells in vitro and decrease liver metastasis of injected B16F1 cells in mice [43].
Our laboratory has recently reported that AMR-Me exerted antiproliferative, proapoptotic and anti-inflammatory activities against rat mammary tumors mediated through modulation of multiple signaling pathways, including estrogen receptors, Wnt/β-catenin and NF-κB [20–22]. However, the results obtained from the cell cycle analysis of the present study revealed that the compound did not exert any apoptotic effect on the murine melanoma cells. At the same time, it was interesting to note that doses of AMR-Me far below the IC50 value efficiently suppressed adhesion, migration and invasion of the highly metastatic B16F10 melanoma cells. This finding prompted us to further investigate the underlying crosstalk of the signaling pathway which might have been responsible for the antimetastatic potential of the compound.
Cell migration, ECM invasion and epithelial mesencymal transition are the hallmarks of malignancy. MMPs are a family of zinc-dependant, structurally and functionally related endopeptidases responsible for degradation of extracellular matrix components (e.g. basement membrane collagen, FN and proteoglycans etc.) and thus play a critical role in invasion and metastasis of tumor cells. The MMPs are over expressed in a variety of malignant tumor types and their over expression is associated with tumor aggressiveness and metastatic potential [44,45]. MT1-MMP is an important component of the MMP-2 activation complex (integrin αvβ3/TIMP-2/MT1-MMP/MMP-2) on the surface of tumor cells [46]. MMP-2 and MMP-9 are reported to be involved with progression, metastasis and poor prognosis of melanoma [47, 48]. In addition to maintaining control over the activation of MMPs, the tissue inhibitors of MMPs, TIMP-1 and TIMP-2, are able to bind directly to the hemopexin domain of MMP-9 and MMP-2 respectively [33]. Our results indicated that AMR-Me could successfully inhibit the expression of MT-1 MMP, MMP-2 and MMP-9 in B16F10 melanoma cells and at the same time could activate the expression of TIMP-1 and TIMP-2, the inhibitors of MMP-2 and MMP-9. Interestingly, another synthetic triterpenoid CDDO-Me has been found to suppress the secretion of MMP-9 from PyMT murine mammary tumor cells [49] and inhibit TNF-induced MMP-9 expression in KBM-5 chronic myelogenous leukemia cells [50].
A number of signaling pathways, such as mitogen activated protein kinase-1 (MEK1)-MAPK, PI-3K-Akt, FAK/PI-3K/ERK, PI-3K-integrin linked kinase (ILK), have been reported to be involved with regulation of gelatinases [31,36]. In this context, we were interested to investigate the influence of AMR-Me on the signaling crosstalk which might have been responsible for the downregulation of MMP-2 and MMP-9 and subsequent inhibition of migration and invasion of the highly metastatic melanoma cells.
Cell adhesion to the ECM regulates many cellular functions, including cell differentiation, proliferation, apoptosis, and migration [51]. Expression of the β6 integrin subunit in cancer enhances the motility and MMP secretion, especially MMP-2 and MMP-9 by epithelial cells in response to the ECM protein, FN (120 kDa fragment). These endopeptidases can subsequently further degrade FN to generate additional fragments, leading to a positive feedback loop [52]. FN on the other hand is an important chemoattractant which is required for cell adhesion. In the present study, it was observed that even in presence of FN, AMR-Me could reduce cell adhesion which might have been due to inhibition of MMP-2 and MMP-9.
Cell migration is a complex process that requires the concerted action of focal adhesion complexes and it has been conclusively established that FAK, a non-receptor cytoplasmic tyrosine kinase, is activated in response to both the extracellular matrix and soluble signaling factors like FN. FAK plays a key role in the regulation of cytoskeletal reorganization, cellular adhesion, growth, survival, and migration [53,54]. There are at least three families of MAPKs, namely ERKs, JNKs and p38 MAPKs which apart from their well-characterized role in cell proliferation/differentiation and cell survival/apoptosis, actively participate in cell migration. Integrin stimulation by extracellular matrix proteins such as FN leads to activation of MAPKs, including JNK and ERK in a variety of cell types. Among the types of MAPK signaling transmitted by integrins, JNK activation is believed to correlate particularly with increased cell migration and invasion. Stimulation of cells with FN is known to activate JNK through a FAK-mediated signaling crosstalk [55]. Cell adhesion to extracellular matrix causes autophosphorylation of FAK at Tyr-397 and thereby associates with Src family kinases, leading to enhancement of its tyrosine phosphorylation and kinase activity. Binding of FAK to c-Src also induces the formation of a multimolecular signaling complex in which FAK functions as a scaffold [56]. Notably, in congruence with the above studies, it was observed that cell adhesion and migration were both inhibited by AMR-Me which involved suppression JNK activation and FAK autophosphorylation at Tyr-397 in B16F10 cells.
Cell motility has been also associated with VEGF expression which is up regulated under hypoxic conditions [57]. VEGF-stimulated FAK tyrosine phosphorylation in endothelial cells is related with increased recruitment of FAK to new focal adhesions and increased endothelial cell migration [58]. Our results clearly demonstrated that downregulation of VEGF in response to AMR-Me treatment reduced recruitment of FAK as well as phosphorylation of FAK at the Tyr397 residue. This, in turn, again contributed towards reduced cell adhesion and migration of B16F10 cells. Thus these results are in conjunction with other studies which reported that synthetic oleanane compounds down-regulated VEGF in various human cancer cells [50,59].
ERK is another important factor for the migration of numerous cell types [60]. The ERK-Jun signaling cascade is required for c-Jun-mediated transcription of cyclin D1, which is often found to be over expressed in human melanomas [61]. In the present study, AMR-Me was found to effectively inhibit the phosphorylated form of ERK1/2 which might have been a result of inhibition of upstream molecules of pFAKTry397 and pJNK. Thus, the AMR-Me regulated the expression and activity of the gelatinases by downregulating VEGF which, in turn, suppressed JNK activation, FAK autophosphorylation at Tyr-397 and inhibited pERK1/2 in B16F10 cells.
NF-κB is a dimeric transcription factor that has been reported to be associated with the regulation of MMPs [62]. The constitutive activation of ERK has been associated with MMP-9 transactivation by increasing the DNA binding activity of important transcription factors like NF-κB, AP-1 and Sp1 and consequently metastasis in certain human tumors [63]. Recent reports suggested that AMR-Me was successful in degradation of IκB which, in turn, retarded the translocation of NFκB from cytoplasm to the nucleus and suppressed rat mammary carcinogenesis [22]. AMR-Me also inhibited NF-κB binding activity in MDA-MB-231 human breast adenocarcinoma cells [19]. The results obtained in the present study indicated that AMR-Me may have effect on several NF-κB subunits. In addition to blocking nuclear translocation of NF-κBp65, it may also downregulate NF-κBp105 which, in turn, might have been responsible for reduced expression of the downstream targets, such as MMPs in the melanoma cells. Similar suppression of NF-κB and down-modulation of target genes, including cyclooxygenase-2, MMP-9 and VEGF, has been shown by CDDO-Me in human leukemic cells [50].
Conclusion
It can be summarized that AMR-Me was successful in restricting the migration and invasion of the highly metastatic murine melanoma cells to an appreciable extent. The antimetastatic potential of this novel synthetic compound may be attributed to the reduced expression of MMP-2 and MMP-9 which might have been due to downregulation of VEGF/pFAK/pJNK/pERK/NF-κB signaling axis. The coordinated activation of TIMPs might have orchestrated along with the signaling network to downregulate the expression of MMP-2 and MMP-9. The present findings may be further validated in in vivo metastasis assay using syngenic mouse model for establishing more convincing evidences of the antitumor potential of AMR-Me against metastatic melanoma.
Acknowledgments
A portion of this work was supported by a grant (R03CA136014) from National Institutes of Health (NIH)/National Cancer Institute (NCI) to A.B. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of NCI or NIH.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Rabi T, Bishayee A. Terpenoids and breast cancer prevention. Breast Cancer Res Treat. 2009;115:223–239. doi: 10.1007/s10549-008-0118-y. [DOI] [PubMed] [Google Scholar]
- 2.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–369. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 3.Petronelli A, Pannitteri G, Testa U. Triterpenoids as new promising anticancer drugs. Anticancer Drugs. 2009;20:880–892. doi: 10.1097/CAD.0b013e328330fd90. [DOI] [PubMed] [Google Scholar]
- 4.Yadav VR, Prasad S, Sung B, Kannappan R, Aggarwal BB. Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins. 2010;2:2428–2466. doi: 10.3390/toxins2102428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J. Oleanolic acid and ursolic acid: research perspectives. J Ethnopharmacol. 2005;100:92–94. doi: 10.1016/j.jep.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 6.Jäger S, Trojan H, Kopp T, Laszczyk MN, Scheffler A. Pentacyclic triterpene distribution in various plants - rich sources for a new group of multi-potent plant extracts. Molecules. 2009;14:2016–2031. doi: 10.3390/molecules14062016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishayee A, Ahmed S, Brankov N, Perloff M. Triterpenoids as potential agents for the chemoprevention and therapy of breast cancer. Front Biosci. 2011;16:980–996. doi: 10.2741/3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thoppil RJ, Bishayee A. Terpenoids as potential chemopreventive and therapeutic agents in liver cancer. World J Hepatol. 2011;3:228–249. doi: 10.4254/wjh.v3.i9.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shanmugam MK, Nguyen AH, Kumar AP, Tan BK, Sethi G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: potential role in prevention and therapy of cancer. Cancer Lett. 2012;320:158–170. doi: 10.1016/j.canlet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 10.Sporn MB, Liby KT, Yore MM, Fu L, Lopchuk JM, Gribble GW. New synthetic triterpenoids: potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J Nat Prod. 2011;74:537–545. doi: 10.1021/np100826q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev. 2012;64:972–1003. doi: 10.1124/pr.111.004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabi T, Karunagaram D, Nair MK, Bhattathiri VN. Cytotoxic activity of amooranin and its derivatives. Phytother Res. 2002;16:S84–S86. doi: 10.1002/ptr.803. [DOI] [PubMed] [Google Scholar]
- 13.Ramachandran C, Rabi T, Fonseca HB, Melnick SJ, Escalon EA. Novel plant triterpenoid drug amooranin overcomes multidrug resistance in human leukemia and colon carcinoma cell lines. Int J Cancer. 2003;105:784–789. doi: 10.1002/ijc.11180. [DOI] [PubMed] [Google Scholar]
- 14.Ramachandran C, Nair PK, Alamo A, Cochrane CB, Escalon E, Melnick SJ. Anticancer effects of amooranin in human colon carcinoma cell line in vitro and in nude mice xenografts. Int J Cancer. 2006;119:2443–2454. doi: 10.1002/ijc.22174. [DOI] [PubMed] [Google Scholar]
- 15.Rabi T, Ramachandran C, Fonseca HB, Nair RP, Alamo A, Melnick SJ, Escalon E. Novel drug amooranin induces apoptosis through caspase activity in human breast carcinoma cell lines. Breast Cancer Res Treat. 2003;80:321–330. doi: 10.1023/A:1024911925623. [DOI] [PubMed] [Google Scholar]
- 16.Rabi T, Wang L, Banerjee S. Novel triterpenoid 25-hydroxy-3-oxoolean-12-en-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Breast Cancer Res Treat. 2007;101:27–36. doi: 10.1007/s10549-006-9275-z. [DOI] [PubMed] [Google Scholar]
- 17.Rabi T, Banerjee S. Novel synthetic triterpenoid methyl 25-hydroxy-3-oxoolean-12-en-28-oate induces apoptosis through JNK and p38 MAPK pathways in human breast adenocarcinoma MCF-7 cells. Mol Carcinog. 2008;47:415–423. doi: 10.1002/mc.20399. [DOI] [PubMed] [Google Scholar]
- 18.Rabi T, Banerjee S. Novel semisynthetic triterpenoid AMR-Me inhibits telomerase activity in human leukemic CEM cells and exhibits in vivo antitumor activity against Dalton’s lymphoma ascites tumor. Cancer Lett. 2009;278:156–163. doi: 10.1016/j.canlet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Rabi T, Huwiler A, Zangemeister-Wittke U. AMR-Me inhibits PI3K/Akt signaling in hormone-dependent MCF-7 breast cancer cells and inactivates NF-κB in hormone-independent MDA-MB-231 cells. Mol Carcinog. 2013 doi: 10.1002/mc.22012. in press. [DOI] [PubMed] [Google Scholar]
- 20.Bishayee A, Mandal A, Thoppil RJ, Darvesh AS, Bhatia D. Chemopreventive effect of a novel oleanane triterpenoid in a chemically induced rodent model of breast cancer. Int J Cancer. 2013;133:1054–1064. doi: 10.1002/ijc.28108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mandal A, Bhatia D, Bishayee A. Simultaneous disruption of estrogen receptor and Wnt/β-catenin signaling is involved in methyl amooranin-mediated chemoprevention of mammary gland tumorigenesis in rats. Mol Cell Biochem. 2013;384:239–250. doi: 10.1007/s11010-013-1803-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandal A, Bhatia D, Bishayee A. Suppression of inflammatory cascade is implicated in methyl amooranin-mediated inhibition of experimental mammary carcinogenesis. Mol Carcinog. 2013 doi: 10.1002/mc.22067. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 24.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2008;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 25.Ilic D, Kovacic B, Johkura K, Schlaepfer DD, Toma-sevic N, Han Q, Kim JB, Howerton K, Baumbusch C, Ogiwara N, Streblow DN, Nelson JA, Dazin P, Shino Y, Sasaki K, Damsky CH. FAK promotes organization of fibronectin matrix and fibrillar adhesions. J Cell Sci. 2004;117:177–187. doi: 10.1242/jcs.00845. [DOI] [PubMed] [Google Scholar]
- 26.Koshikawa N, Minegishi T, Sharabi A, Quaranta V, Seiki M. Membrane-type matrix metalloproteinase-1 (MT1-MMP) is a processing enzyme for human laminin gamma 2 chain. J Biol Chem. 2005;280:88–93. doi: 10.1074/jbc.M411824200. [DOI] [PubMed] [Google Scholar]
- 27.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. Matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 28.Kirstein M, Sanz L, Quinones S, Moscat J, Diaz-Meco MT, Saus J. Cross-talk between different enhancer elements during mitogenic induction of the human stromelysin-1 gene. J Biol Chem. 1996;271:18231–18236. doi: 10.1074/jbc.271.30.18231. [DOI] [PubMed] [Google Scholar]
- 29.Chandler S, Miller KM, Clements JM, Lury J, Corkill D, Anthony DC, Adams SE, Gearing AJ. Matrix metalloproteinases, tumor necrosis factor and multiple sclerosis: an overview. J Neuroimmunol. 1007;72:155–161. doi: 10.1016/s0165-5728(96)00179-8. [DOI] [PubMed] [Google Scholar]
- 30.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circulation Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 31.Thant AA, Nawa A, Kikkawa F, Ichigotani Y, Zhang Y, Sein TT, Amin AR, Hamaguchi M. Fibronectin activates matrix metalloproteinase-9 secretion via the MEK1-MAPK and the PI-3K-Akt pathways in ovarian cancer cells. Clin Expt Metastasis. 2000;18:423–428. doi: 10.1023/a:1010921730952. [DOI] [PubMed] [Google Scholar]
- 32.Han S, Ritzenthaler JD, Sitaraman SV, Roman J. Fibronectin increases matrix metalloproteinase 9 expression through activation of c-Fos via extracellularregulated kinase and phosphatidylinositol 3-kinase pathways in human lung carcinoma cells. J Biol Chem. 2006;281:29614–29624. doi: 10.1074/jbc.M604013200. [DOI] [PubMed] [Google Scholar]
- 33.Das S, Banerji A, Frei E, Chatterjee A. Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium. Life Sci. 2008;82:467–476. doi: 10.1016/j.lfs.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 34.Jin YJ, Park I, Hong IN, Byun H-J, Choi J, Kim Y-M, Lee H. Fibronectin and vitronectin induce AP-1-mediated matrix metalloproteinase-9 expression through integrin α5β1/αvβ3-dependent Akt, ERK and JNK signaling pathways in human umbilical vein endothelial cells. Cell Signal. 2011;23:125–134. doi: 10.1016/j.cellsig.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Bishayee A, Martirosian A, Yeranosyan A. Amooranin compounds and analogs thereof, and related methods of use. US Provisional Patent Application No. 61/580. 2011;449 [Google Scholar]
- 36.Sen T, Dutta A, Maity G, Chatterjee A. Fibronectin induces matrix metalloproteinase-9 (MMP-9) in human laryngeal carcinoma cells by involving multiple signaling pathways. Biochimie. 2010;92:1422–1434. doi: 10.1016/j.biochi.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 38.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acid Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perrot CY, Javelaud D, Mauviel A. Insights into the transforming growth factor-β signaling pathway in cutaneous melanoma. Ann Dermatol. 2013;25:135–144. doi: 10.5021/ad.2013.25.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suh N, Wang Y, Honda T, Gribble GW, Dmitrovsky E, Hickey WF, Maue RA, Place AE, Porter DM, Spinella MJ, Williams CR, Wu G, Dannenberg AJ, Flanders KC, Letterio JJ, Mangelsdorf DJ, Nathan CF, Nguyen L, Porter WW, Ren RF, Roberts AB, Roche NS, Subbaramaiah K, Sporn MB. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999;59:336–341. [PubMed] [Google Scholar]
- 41.Hail N, Jr, Konopleva M, Sporn M, Lotan R, Andreeff M. Evidence supporting a role for calcium in apoptosis induction by the synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) J Biol Chem. 2004;279:11179–11187. doi: 10.1074/jbc.M312758200. [DOI] [PubMed] [Google Scholar]
- 42.Qin Y, Deng W, Ekmekcioglu S, Grimm EA. Identification of unique sensitizing targets for anti-inflammatory CDDO-Me in metastatic melanoma by a large-scale synthetic lethal RNAi screening. Pigment Cell Melanoma Res. 2013;26:97–112. doi: 10.1111/pcmr.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Townson JL, Macdonald IC, Liby KT, Mackenzie L, Dales DW, Hedley BD, Foster PJ, Sporn MB, Chambers AF. The synthetic triterpenoid CDDO-Imidazolide suppresses experimental liver metastasis. Clin Exp Metastasis. 2011;28:309–317. doi: 10.1007/s10585-011-9374-z. [DOI] [PubMed] [Google Scholar]
- 44.Hidalgo M, Eckhardt SG. Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Inst. 2001;93:178–93. doi: 10.1093/jnci/93.3.178. [DOI] [PubMed] [Google Scholar]
- 45.Yang JM, Xu Z, Wu H, Zhu H, Wu X, Hait WN. Overexpression of extracellular matrix metalloproteinase inducer in multidrug resistant cancer cells. Mol Cancer Res. 2003;1:420–427. [PubMed] [Google Scholar]
- 46.Kurschat P, Zigrino P, Nischt R, Breitkopf K, Steurer P, Klein CE, Krieg T, Mauch C. Tissue inhibitor of matrix metalloproteinase-2 regulates matrix metalloproteinase-2 activation by modulation of membrane-type 1 matrix metalloproteinase activity in high and low invasive melanoma cell lines. J Biol Chem. 1999;274:21056–21062. doi: 10.1074/jbc.274.30.21056. [DOI] [PubMed] [Google Scholar]
- 47.Guruvayoorappan C, Kuttan G. Amentoflavone inhibits experimental tumor metastasis through a regulatory mechanism involving MMP-2, MMP-9, prolyl hydroxylase, lysyl oxidase, VEGF, ERK-1, ERK-2, STAT-1, nm23 and cytokines in lung tissues of C57BL/6 mice. Immunopharmacol Immunotoxicol. 2008;30:711–727. doi: 10.1080/08923970802278276. [DOI] [PubMed] [Google Scholar]
- 48.Väisänen AH, Kallioinen M, Turpeenniemi-Hujanen T. Comparison of the prognostic value of matrix metalloproteinases 2 and 9 in cutaneous melanoma. Hum Pathol. 2008;39:377–385. doi: 10.1016/j.humpath.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 49.Tran K, Risingsong R, Royce D, Williams CR, Sporn MB, Liby KT. The synthetic triterpenoid CDDO-methyl ester delays estrogen receptor-negative mammary carcinogenesis in polyoma middle T mice. Cancer Prev Res. 2012;5:726–734. doi: 10.1158/1940-6207.CAPR-11-0404. [DOI] [PubMed] [Google Scholar]
- 50.Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IκBβ kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor κB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12:1828–1838. doi: 10.1158/1078-0432.CCR-05-2044. [DOI] [PubMed] [Google Scholar]
- 51.Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006;99:35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- 52.Al-Hazmi N, Thomas GJ, Speight PM, Whawell SA. The 120 kDa cell-binding fragment of fibronectin up-regulates migration of αvβ6-expressing cells by increasing matrix metalloproteinase-2 and -9 secretion. Eur J Oral Sci. 2007;115:454–458. doi: 10.1111/j.1600-0722.2007.00481.x. [DOI] [PubMed] [Google Scholar]
- 53.Takino T, Nakada M, Miyamori H, Watanabe Y, Sato T, Gantulga D, Yoshioka K, Yamada KM, Sato H. Jsap1/jip3 cooperates with focal adhesion kinase to regulate c-jun n-terminal kinase and cell migration. J Biol Chem. 2005;280:37772–37781. doi: 10.1074/jbc.M505241200. [DOI] [PubMed] [Google Scholar]
- 54.Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res. 2006;17:606–616. doi: 10.1161/01.RES.0000207408.31270.db. [DOI] [PubMed] [Google Scholar]
- 55.Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 56.Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signalling. Mol Cell Biol. 2004;24:8113–8133. doi: 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fiedler J, Leucht F, Waltenberger J, Dehio C, Brenner RE. VEGF-A and PlGF-1 stimulate chemotactic migration of human mesenchymal progenitor cells. Biochem Biophys Res Commun. 2005;334:561–568. doi: 10.1016/j.bbrc.2005.06.116. [DOI] [PubMed] [Google Scholar]
- 58.Yun SP, Lee MY, Ryu JM, Song CH, Han HJ. Role of HIF-1 α and VEGF in human mesenchymal stem cell proliferation by 17β-estradiol: involvement of PKC, PI3K/Akt, and MAPKs. Am J Physiol Cell Physiol. 2009;296:C317–C326. doi: 10.1152/ajpcell.00415.2008. [DOI] [PubMed] [Google Scholar]
- 59.Jutooru I, Chadalapaka G, Abdelrahim M, Basha MR, Samudio I, Konopleva M, Andreeff M, Safe S. Methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription factors and inhibits pancreatic tumor growth: role of microRNA-27a. Mol Pharmacol. 2010;78:226–236. doi: 10.1124/mol.110.064451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 61.Lopez-Bergami P, Huang C, Goydos JS, Yip D, Bar-Eli M, Herlyn M, Smalley KS, Mahale A, Eroshkin A, Aaronson S, Ronai Z. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chase AJ, Bond M, Crook MF, Newby AC. Role of nuclear factor-κB activation in metalloproteinase-1, -3, and -9 secretion by human macrophages in vitro and rabbit foam cells produced in vivo. Arteriosclerosis Thrombosis Vascular Biol. 2002;22:765–771. doi: 10.1161/01.atv.0000015078.09208.92. [DOI] [PubMed] [Google Scholar]
- 63.O-charoenrat P, Wongkajornsilp A, Rhys-Evans PH, Eccles SA. Signaling pathways required for matrix metalloproteinase-9 induction by betacellulin in head-and-neck squamous carcinoma cells. Int J Cancer. 2004;111:174–183. doi: 10.1002/ijc.20228. [DOI] [PubMed] [Google Scholar]