

Abstract
Clinical and animal studies have documented that hearts of the elderly are more susceptible to ischemia/reperfusion damage compared to young adults. Recently we found that aging-dependent increase in susceptibility of cardiomyocytes to apoptosis was attributable to decrease in cytosolic glutaredoxin 1 (Grx1) and concomitant decrease in NF-κB-mediated expression of anti-apoptotic proteins. Besides primary localization in the cytosol, Grx1 also exists in the mitochondrial intermembrane space (IMS). In contrast, Grx2 is confined to the mitochondrial matrix. Here we report that Grx1 is decreased by 50–60% in the IMS, but Grx2 is increased by 1.4–2.6 fold in the matrix of heart mitochondria from elderly rats. Determination of in situ activities of the Grx isozymes from both subsarcolemmal (SSM) and interfibrillar (IFM) mitochondria revealed that Grx1 was fully active in the IMS. However, Grx2 was mostly in an inactive form in the matrix, consistent with reversible sequestration of the active-site cysteines of two Grx2 molecules in complex with an iron–sulfur cluster. Our quantitative evaluations of the active/inactive ratio for Grx2 suggest that levels of dimeric Grx2 complex with iron–sulfur clusters are increased in SSM and IFM in the hearts of elderly rats. We found that the inactive Grx2 can be fully reactivated by sodium dithionite or exogenous superoxide production mediated by xanthine oxidase. However, treatment with rotenone, which generates intramitochondrial superoxide through inhibition of mitochondrial respiratory chain Complex I, did not lead to Grx2 activation. These findings suggest that insufficient ROS accumulates in the vicinity of dimeric Grx2 to activate it in situ.
Abbreviations: GSH, reduced glutathione; GSSG, glutathione disulfide; Cys-SSG, l-cysteine–glutathione mixed disulfide; Mn-TMPyP, Mn(III) tetrakis (1-methyl-4-pyridyl) porphyrin; tetratosylate, hydroxide; DT, sodium dithionite; SSM, heart subsarcolemmal mitochondria; IFM, Heart interfibrillar mitochondria; t-Bid, caspase-8-cleaved human BID; Grx, glutaredoxin
Keywords: Aging, Glutaredoxin, Glutathionylation, Iron–sulfur cluster, Mitochondria, Reactive oxygen species (ROS), Redox regulation
Graphical abstract
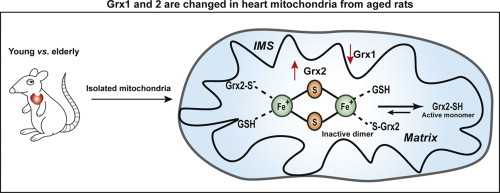
.
Highlights
-
•
Glutaredoxins play key roles in cellular redox regulation, which is sensitive to aging-dependent dysregulation.
-
•
Grx1 is diminished in the intermembrane space of mitochondria from aged heart; matrix Grx2 is increased but mostly in an inactive form.
-
•
The inactive Grx2 is selectively activated by superoxide.
-
•
Mitochondrial glutaredoxin changes may contribute to dysregulation of redox homeostasis during aging.
-
•
Changes in in situ activities of heart mitochondrial Grx1 and Grx2 with aging provide mechanistic insights for future studies.
Aging is a major risk factor for the development of ischemic cardiovascular disease [1]. Previous studies in humans and animals have shown that hearts of the elderly are more prone to oxidative damage after ischemia/reperfusion (I/R) than their younger counterparts [2–4]. Therefore, a better understanding is necessary of how changes with aging are linked to irreversible ischemic damage.
Reactive oxygen species (ROS) have been implicated in I/R injury, directing research efforts towards the mitochondria [5]. Mitochondria are the major source of ROS during cardiac I/R [6]. Accordingly, protective mechanisms have involved blockade of the electron transport chain (ETC) and modulation of the mitochondrial permeability transition pore (MPTP) and other ion channels [7–9]. Further, defects in mitochondrial activity have been associated with age-related functional decline of organs and tissues [2,10].
The heart is equipped with antioxidant systems to scavenge excess ROS and repair oxidative injury. Maintenance of sulfhydryl homeostasis is vital and two enzyme systems play the key roles. The thioredoxin (Trx)/thioredoxin reductase (TR) system and the glutaredoxin (Grx)/glutathione (GSH/GSSG) reductase (GR) systems maintain protein thiol status and regulate intracellular signal transduction [11]. Trx mainly catalyzes the reduction of protein intra-/inter-molecular disulfides, whereas Grx specifically catalyzes reduction of protein-SS-glutathione mixed disulfides (deglutathionylation). Thus, the Trx and Grx systems operate in a complementary fashion.
Two mammalian Grx isoforms (Grx1 and Grx2) catalyze deglutathionylation. Grx1 is mainly cytosolic, but also exists in the mitochondrial intermembrane space (IMS) [12]. Grx2 is localized in the mitochondrial matrix. Unlike Grx1, mammalian Grx2 forms a catalytically inactive dimer with an intercalating iron–sulfur cluster [13]. The purified Grx2 dimer can be dissociated with GSSG or sodium dithionite, yielding active monomers. Accordingly, Grx2 is proposed to serve as a redox sensor in response to oxidative stress [13], but evidence for this function in situ has not been reported.
The glutaredoxins have been implicated in cardiac health [14]. For example, H9c2 cells (model cardiomyocytes) that overexpress Grx1 showed diminished H2O2-induced apoptosis attributed to Grx1-mediated regulation of Akt activity [15]. S-glutathionylation has been demonstrated to modulate activity of several ion channels important for cardiac function, implicating regulation by Grx1 [16,17]. In addition we characterized an anti-apoptotic regulatory role for Grx1 in cardiomyocytes [18], as discussed further below.
As described, both Grx1 and Grx2 are present in the mitochondria [12]. The estimated concentration of Grx2 in the matrix is ~10-fold higher than the concentration of Grx1 in the IMS, potentially compensating for the ~10-fold lower catalytic activity of Grx2 [19,20]. Grx2 overexpression was reported to attenuate doxorubicin-induced cardiac injury, which is associated with increased S-glutathionylation of mitochondrial proteins including Complex I [21]. However, it remains to be determined whether overexpressed Grx2 is representative of natural abundance of Grx2 which is largely sequestered in an inactive dimer [55], and whether mitochondrial Grx1 plays a cardioprotective role.
Previously, our lab studied the effects of aging on cardiac redox homeostasis, finding that Grx1, was decreased 40% in cardiomyocytes isolated from elderly (24-mo) Fischer 344 rats (aging model animals), compared to young adults (6-mo), whereas the thioredoxin system was essentially unchanged. The decrease in Grx1 was correlated with decreased NF-κB transcriptional activity and diminution of expression of its anti-apoptotic gene product, Bcl-xL [18]. Analogous changes to NF-κB and its target genes were observed in H9c2 cells in which Grx1 was selectively knocked down to the same extent as observed in the hearts of the elderly rats [18].
Here we report that mitochondrial Grx1 and Grx2 are altered differently with aging. Whereas Grx1 content and corresponding activity is decreased in the intermembrane space of mitochondria from elderly Fischer 344 rat hearts, Grx2 content is increased in the matrix compared to young adults. However, the major portion of the Grx2 in situ is in an inactive state, consistent with sequestration as a dimeric iron–sulfur cluster complex. The inactive Grx2 in mitochondrial lysates could be reactivated by a superoxide generating system. However, the deglutathionylase activity of Grx2 was not increased by treatment of intact mitochondria with rotenone, which is known to stimulate matrix-directed intramitochondrial generation of ROS via inhibition of Complex I [22]. This finding is inconsistent with the concept that dimeric Grx2 serves as a redox sensor, dissociating into active monomers in response to oxidative stress.
Experimental procedures
General materials
l-Cysteine–glutathione mixed disulfide (Cys-SSG) was purchased from Toronto Research (Canada, C995500). Mn-TMPyP (Mn(III) tetrakis (1-methyl-4-pyridyl) porphyrin, tetratosylate, hydroxide) was purchased from EMD Millipore Chemicals. Xanthine oxidase was purchased from Roche Applied Science. Soybean trypsin and trypsin inhibitor were purchased from Worthington Biochemical Corporation. All other chemicals were purchased from Sigma (St. Louis, MO). Recombinant human BID, Caspase-8-cleaved/truncated BID (t-Bid) was purchased from R&D Systems, Minneapolis, MN. Anti-human Grx1 polyclonal antibody (1:1000 dilution) was generated and purified via an adaptation of the McKinney and Parkinson caprylic acid method [23]. Anti-human Grx2 polyclonal antibody was kindly provided by Dr. Vadim Gladyshev (1:1000 dilution). Rabbit anti-adenylate kinase 2 polyclonal antibody (1:1000 dilution) was purchased from Abcam and mouse anti-cytochrome c monoclonal antibody (1:1000 dilution) from BD Pharmingen, San Diego, CA. Anti-rabbit and anti-mouse secondary antibodies conjugated to HRP were purchased from Jackson Immuno Research Labs (1:10,000 dilutions).
Animals
Young adult (6-mo) and elderly (24-mo) male Fischer 344 rats were purchased from the National Institute on Aging's colonies (Harlan, Indianapolis, IN; Taconic, Germantown, NY) and housed in the animal facilities at Case Western Reserve University School of Medicine. Animals were acclimated for at least 1 week before use, and used for experiments within 3 months. The Institutional Animal Care and Use Committee at Case Western Reserve University approved all animal handling procedures and experiments.
Isolation and preparation of heart mitochondria
The isolation of subsarcolemmal mitochondria (SSM) and intrafibrillar mitochondria (IFM) from Fischer 344 rat hearts was performed as previously described [12] with slight modifications to improve the quality of the IFM. Briefly, the polytron homogenate was centrifuged at 500×g and the pellet saved in order to isolate the IFM fraction. The pellet was resuspended in Chappel–Perry buffer (100 mM KCl, 50 mM Mops, 5 mM MgSO4, 1 mM EGTA, 1 mM ATP, pH 7.4) containing trypsin (5 mg/g of heart tissue) and incubated for 10 min on ice. This solution was homogenized and then the trypsin activity was blocked by the addition of Chappel–Perry buffer plus 0.2% defatted BSA and trypsin inhibitor (2.5 mg/g of heart tissue). After centrifugation at 600×g to remove unbroken tissue and debris, the supernatant was centrifuged at 3000×g to sediment the IFM fraction. This fraction was washed twice thereafter by centrifugation at 3000×g for 10 min. Mitochondrial protein concentrations were determined either by the bicinchronic acid (BCA) method (using the micro BCA protein assay kit, Thermo-Scientific, Waltham, MA) or the Lowry method [24]. Mitochondria were analyzed within 8 h of their isolation from tissues.
Mitochondrial thioredoxin and thioredoxin reductase activity assays
Isolated heart mitochondria were homogenized at 4 °C in 1 mL buffer (10 mM potassium phosphate, pH 7.5; 5 mM EDTA; 50 μM PMSF; and 2 mM β-mercaptoethanol) using a glass homogenizer. The homogenates were centrifuged at 10,000×g for 10 min at 4 °C. Supernatants were dialyzed to remove free GSH at 4 °C against the same buffer using a ratio of 1 mL supernatant to 1000 mL dialysis buffer. Dialysis was performed overnight with one change of buffer, and then the dialyzed mitochondrial extract was assayed the next day for total dethiolase activity [25]. Briefly, the reaction was initiated by adding BSA-SSG [35S] (0.1 mM) to an aliquot of dialyzed mitochondrial extract in potassium phosphate buffer (0.1 M, pH 7.5), with a total volume of 500 μL. At 0, 2, 4 and 6 min, 100 μL aliquots were removed and mixed with 100 μL of ice-cold trichloroacetic acid (20%). After centrifugation at 10,000×g, the supernatants from each time point were analyzed for [35S] radioactivity by scintillation counting. Dethiolase activity is expressed as nmol [35S] GS-equivalents released per min/mg of heart mitochondrial protein, according to the specific radioactivity of the GS-moiety of BSA-SSG [35S] (~0.7 μCi/mmol GS-moiety). Thioredoxin reductase activity was measured using a modification of this dethiolase assay, as previously described [26]. Briefly, an aliquot of mitochondrial homogenates was transferred into the assay mixture containing 0.2 mM NADPH, 2.1 μM (0.04 unit) thioredoxin and 0.1 M potassium phosphate buffer, pH 7. The assay reaction was initiated by addition of 2 mM hydroxyethyldisulfide (HED).
Mitochondrial GSSG reductase activity assay
Mitochondrial GSSG reductase activity was determined based on the methods as described [26]. Briefly, mitochondrial lysate (40–100 μg) was incubated in reaction buffer containing Na/K phosphate buffer (0.1 mM, pH 7.5), and 0.2 mM NADPH at 30 °C for 5 min. The reaction was initiated by the addition of 0.1 mM GSSG and the oxidation of NADPH was monitored at 340 nm for 5 min using a ThermoMax® microplate reader (Molecular Devices Inc.).
Mitochondrial oxidative phosphorylation assays
Oxygen consumption was measured with a Clark-type electrode in respiration buffer (80 mM KCl, 50 mM Mops, 1 mM EGTA, 5 mM KH2PO4 and 1 mg/mL defatted BSA, pH 7.4 and final volume 0.5 mL) [27,28] at 30 ° C with glutamate (20 mM) as substrate. After depletion of endogenous substrates with 100 μM ADP, State 3 respiratory rate (ADP-dependent) was measured in the presence of 200 μM ADP and State 4 respiratory rate (ADP-limited) was recorded after ADP consumption. Then sequential additions of 2 mM ADP and 200 μM of the uncoupler 2,4-dinitrophenol (DNP) were made, as described [29]. Respiratory control ratios (RCR, defined as State 3 respiratory rate divided by State 4 respiratory rate) reflects the control of oxygen consumption by phosphorylation or “coupling”. The ADP/O ratio, defined as the number of ADP molecules added for each oxygen atom consumed, is an index of the efficiency of oxidative phosphorylation, and was calculated as described by Chance and Williams [30].
Western blot analysis of Grx1 and Grx2 content in F344 rat heart mitochondria
In order to release Grx1 from the intermembrane space of mitochondria of the SSM and IFM fractions isolated from Fischer 344 rats (6-mo and 24-mo), the mitochondrial preparations were washed once with cold KME buffer (100 mM KCl, 50 mM MOPS, 0.5 mM EGTA, pH 7.4) and centrifuged at 5400×g to remove any proteins released from non-intact mitochondria and cytosolic contamination in the mitochondrial preparations, then the mitochondria were diluted with KME buffer to a concentration of 20 mg/mL, and incubated at 37 °C with or without t-Bid (45 nM) for 1 h, and centrifuged at 3500×g for 10 min at 4 °C. The resulting supernatant containing the soluble intermembrane space proteins was saved as “supernatant”. The pellets were washed twice and resuspended in 100 µL of RIPA buffer (150 mM NaCl, 1 mM EGTA, 1% Triton X-100, 25.5 mM deoxycholic acid, 50 mM Tris–HCl, pH 7.5), incubated on ice for 10 min, and centrifuged at 10,000×g for 15 min. The resulting supernatant was saved as “mitochondrial extract.” The residual pellets were washed twice with RIPA buffer following a centrifugation at 12,000×g to precipitate insoluble proteins as “residue”. Equal volumes of each sample were analyzed by Western blot as previously described [12].
Electron microscopy
Cardiac tissues from young adult (6-mo F344) and elderly (24-mo F344) hearts were fixed for 5 min at 4 °C with 3% formaldehyde and 20 mM Methylacetimidate in HEPES-buffered saline (30 mM HEPES, 150 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2, 7.8 mM glucose, pH 7.3). These samples were washed, further fixed for 45 min in 3% formaldehyde containing 0.25% glutaraldehyde in HEPES-buffered saline at 4 °C, then dehydrated in ethanol and embedded in LR White resin (Polysciences, Inc., Warrington, PA). Thin sections were blocked with PBT (PBS containing 1% w/v BSA and 0.01% v/v Tween 20). Grids were then incubated with anti-human GRx1 polyclonal antibody at 1:5 and 1:25 dilution, respectively, in PBT for 12 h at 4 °C. After washing, grids were incubated for 1 h in 5 nM gold-conjugated anti-rabbit IgG (Amersham Life Sciences, Arlington, IL) diluted 1:30 in PBT, rinsed with PBS, and fixed with glutaraldehyde to stabilize the gold particles. Samples were stained with uranyl acetate and lead citrate, and then examined in a JEOL 1200EX electron microscope (Tokyo, Japan).
Standard Grx activity assay
The coupled spectrophotometric assay to determine Grx specific activity was performed as described previously [31,32]. Reaction mixtures containing Na/K phosphate buffer (0.1 mM, pH 7.5), NADPH (0.2 mM), yeast GSSG reductase (6 unit/mL), GSH (0.5 mM), and purified human Grx1 or mitochondrial lysates (2 mg of total protein) were added to a 96-well plate and incubated for 5 min at 30 °C. GSH-dependent deglutathionylation reactions were initiated by the addition of Cys-SSG (0.1 mM) as the substrate. The oxidation of NADPH was monitored at 340 nm for 4 min using a Thermo Max microplate reader (Molecular Devices). Non-enzymatic rates of NADPH oxidation (RIPA buffer with 1 mM GSH or 10 mM sodium dithionate) were subtracted from observed rates of NADPH oxidation using mitochondrial lysates to give the rates of deglutathionylation, corresponding to the specific activity of Grx. The number of nanomoles of NADPH oxidized per minute (nmol/min) was calculated using the standard extinction coefficient of NADPH (ε=6.22 A/mM/cm). This value was then divided by the total amount of protein added in the assay giving nmol/min/mg of mitochondrial protein, and corrected for differences in path length and extinction coefficient for the microplate reader compared to a standard spectrophotometer. For all analyses, Grx activity was calculated using an amount of mitochondrial lysate where the reaction rates fell within the linear range of dependence on the concentration of Grx.
Measurements indicative of in situ Grx activities in heart mitochondria of 6-mo and 24-mo Fischer 344 rats
In order to simplify the Grx activity assay to determine the specific Grx2 activity in mitochondrial lysates, isolated mitochondrial fractions (4 mg) without the t-Bid treatment were centrifuged at 5500×g at 4 °C to remove KME buffer, and the pellets lysed using RIPA buffer (400 μL), containing either 1 mM GSH or 10 mM sodium dithionate, to achieve a final protein concentration of 10 mg/mL. The reaction mixtures were incubated at room temperature for 40 min followed by centrifugation at 21,000×g for 10 min at 4 °C to precipitate mitochondrial debris. The resultant supernatants, containing Grx1 and Grx2 from the lysates, were saved. The pellets were then resuspended in RIPA buffer (150 μL, with either 1 mM GSH or 10 mM dithionite) in order to extract the remaining Grx from the mitochondrial debris. After a centrifugation at 21,000×g for 10 min at 4 °C, the insoluble pellets were removed. The saved supernatants were combined and 2 mg of mitochondrial protein from each sample were concentrated using centrifugal filter devices (Amicon-ultra 0.5 mL 10,000 MW, EMD Millipore) at 14,000×g for 10 min at 4 °C. For all analysis, the specific Grx2 activity was calculated as the observed activity after GSH or dithionite treatment minus the Grx1 activity which was calculated from previously established Grx1 contents for mitochondria from each age group of animals.
Several of the conditions necessary for successful isolation and analysis of the heart mitochondria were found to alter the Standard Grx Activity Assay. In order to account for this, several correction factors were necessary to normalize the results from these samples to previous Grx activity data. The addition of RIPA buffer (the medium in which mitochondrial extracts were prepared) was found to increase Grx activity by a factor of 1.67. Results were therefore divided by 1.67 to account for this difference. Sodium dithionite was found to inhibit GSSG reductase, thereby interfering with the Standard Grx Activity Assay. This was overcome by increasing the concentration of GSSG reductase 3-fold, with the residual effect adjusted by a factor of 1.1.
Reduction of mitochondrial cytochromes by differential ROS production system
Liver mitochondria were isolated from young adult Fischer 344 rats and prepared as described before [27]. Aliquots (4 mg) of frozen liver mitochondria were centrifuged at 5500×g to remove the KME buffer from the resulting pellets. The pellets were lysed with RIPA buffer (200 μL), and then supplemented with sodium cyanide (2 mM). After centrifugation at 21,000×g for 10 min at 4 °C to remove the mitochondrial debris, reduction of the cytochromes was carried out in ROS-generating mixtures containing 10 mg/mL mitochondrial lysates, 1 mM GSH, 10 μM xanthine oxidase and 2 mM xanthine, in the presence of either catalase (1 mg/mL), or Mn-TMPyP (SOD mimetic, varying concentration). After 10 min incubation at room temperature, each treated or untreated sample was subjected to spectrophotometric analysis by recording the absorption spectra between 500 and 660 nm (HP 8453 UV–VIS).
Dissociation of dimeric Grx2 by different ROS production systems
Freshly isolated mitochondria (4 mg) from young adult Fischer 344 rat hearts were centrifuged at 5500×g to remove the KME buffer. The pellets were lysed in RIPA buffer (400 μL) containing either 1 mM GSH, or 10 mM dithionite. After a 30 min incubation at 25 °C, the mitochondrial lysate was mixed with xanthine oxidase (10 μM) and xanthine (2 mM), in the presence of either catalase (1 mg/mL) or Mn-TMPyP (40 μM). This was incubated for an additional 10 min followed by centrifugation at 21,000×g at 4 °C to precipitate the debris. The pellets were rinsed and fully resuspended in of RIPA buffer (120 μL, with 1 mM GSH or 10 mM dithionite), and centrifuged again at 21,000×g. The supernatants from both 21,000×g spins were combined and concentrated through a centrifugal filter. A portion (2 mg) of the concentrated supernatants was subjected to the Standard Grx Activity Assay, using appropriate correction factors. The addition of RIPA buffer and xanthine oxidase were found to increase recombinant Grx1 activity by a factor of 2.08, and samples containing RIPA buffer, xanthine oxidase, and catalase increased the activity by a factor of 2.32.
Intramitochondrial superoxide anion production by rotenone-mediated inhibition of Complex I
To induce endogenous mitochondrial superoxide generation, heart mitochondria (8 mg) isolated from either young adult or elderly Fischer 344 rats were incubated with glutamate (20 mM) in the presence or absence of rotenone (7.5 μM) for 20 min at 37 °C. The samples were then centrifuged at 5500×g and the pellets were washed by the addition of KME buffer to remove excess glutamate and rotenone, and then lysed with RIPA buffer (volume adjusted to 10 mg/mL mitochondrial protein) containing either 1 mM GSH or 10 mM dithionite for 30 min. The mitochondrial lysates were then centrifuged at 21,000×g for 10 min at 4 °C to precipitate the mitochondrial debris. The insoluble pellets were then resuspended in RIPA buffer (150 μL) and re-centrifuged at 21,000×g. The supernatants were combined and concentrated through centrifugal filter devices at 14,000×g for 10 min at 4 °C. An equal amount (2 mg) of the concentrated proteins was analyzed using the Standard Grx Activity Assay.
Statistical analysis
Data are presented as mean±standard error (mean±SEM) of at least three independent experiments. On occasion when an individual data point was more than two standard deviations from the mean, it was considered to be an outlier and was omitted. Differences between data sets were tested for statistical significance using 2-tailed Student's t-test (Microsoft Excel), with p<0.05 considered statistically significant.
Results
Examination of heart mitochondria for changes in the thioredoxin and glutaredoxin systems in elderly animals
Our previous studies revealed that cytosolic Grx1 from heart tissues was significantly decreased in elderly rats (~50%) decrease in content and activity), compared to young adult rats. In contrast, little change was observed in cytosolic thioredoxin (Trx), Trx reductase (TR) or GSSG reductase [18]. Mitochondria are the primary source of ROS, and the ROS production is increased in mitochondria from elderly animals [9,33–35]. The goal of this project was to assess age-related changes to the two mitochondrial anti-oxidant systems responsible for thiol homeostasis, Trx and Grx. In the heart there are two populations of mitochondria, the subsarcolemmal mitochondria (SSM) and the interfibrillar mitochondria (IFM) characterized with distinct biochemical properties and responses to metabolic challenges [36]. We monitored the rates of oxidative phosphorylation in the mitochondria from both the SSM and IFM to verify the integrity and quality of the isolated mitochondria (Table 1). The uncoupled respiratory rates were similar to previous reports [36,37], indicating that the isolated mitochondria for the current study have normal maximal respiratory capacity. Moreover, unlike Complex I (Table 1B) the oxidative metabolism through Complex III was significantly decreased selectively in heart IFM from elderly rats at respiratory state 3, as we reported previously [38] (Table 1C), confirming the aging-associated defects in the electron transport chain that we had characterized.
Table 1.
Protein yield (A) and respiratory rates (B) and (C) in heart SSM and IFM from adult (6-mo) and elderly (24-mo) F344 rats.
| F344 rats (age) | 6-mo | 24-mo | ||
|---|---|---|---|---|
| A | ||||
| Heat mitochondrial fractions | SSM | IFM | SSM | IFM |
| Protein yield (mg/mL) | 20±0.9 | 19±0.7 | 18±0.7 | 15±0.4 |
| B | ||||||
|---|---|---|---|---|---|---|
| Glutamate-supported mitochondrial OXPHOS | State 3 | State 4 | RCR | ADP/O | 2 mM ADP | DNP |
| 6-mo | ||||||
| SSM | 159±12.2 | 13±3.5 | 15±2.6 | 2.86±0.11 | 169±18.3 | 164±13.1 |
| IFM | 240±20.3 | 21±3.5 | 13±2.6 | 2.87±0.08 | 259±25.0 | 251±31.8 |
| 24-mo | ||||||
| SSM | 162 ±9.4 | 14±2.0 | 13±2.0 | 2.84±0.10 | 169±13.5 | 160±14.7 |
| IFM | 195 ±10.8 | 19±2.4 | 11±1.3 | 2.90±0.10 | 210±15.7 | 211±21.0 |
| Pvalues (6-movs.24-mo) | ||||||
| SSM | 0.837 | 0.89 | 0.521 | 0.878 | 0.991 | 0.825 |
| IFM | 0.058 | 0.645 | 0.443 | 0.768 | 0.10 | 0.282 |
| C | ||||||
|---|---|---|---|---|---|---|
| DHQ-supported mitochondrial OXPHOS | State 3 | State 4 | RCR | ADP/O | 2 mM ADP | DNP |
| 6-mo | ||||||
| SSM | 382±20.7 | 88±4.6 | 4±0.2 | 1.65±0.08 | 354±16.9 | 443±34.4 |
| IFM | 549± 23.6 | 106±10.0 | 5±0.5 | 1.74±0.14 | 536±29.5 | 630±24.9 |
| 24-mo | ||||||
| SSM | 398±20.1 | 92±4.1 | 4±0.2 | 1.63±0.10 | 396±21.2 | 476±35.3 |
| IFM | 442±27.2 | 109±9.9 | 4±0.1 | 1.69±0.10 | 451±18.5 | 567±51.1 |
| Pvalues (6-movs.24-mo) | ||||||
| SSM | 0.572 | 0.459 | 0.893 | 0.872 | 0.125 | 0.476 |
| IFM | *0.009 | 0.788 | *0.018 | 0.756 | *0.024 | 0.251 |
SSM and IFM were isolated from F344 rats as described in Experimental procedures section. The uncoupled respiratory rates (B) were determined using glutamate (20 mM) as Complex I substrate, (C) and duroquinol (DHQ) (1 mM) as Complex III substrate. The protein concentration of isolated mitochondria is expressed as mg/g heart wet weight (mean±SEM, n=6); the respiratory rate is expressed as nAO per min/mg of mitochondrial protein (mean±SEM, n=6). RCR-respiratory control ratio; DNP-dinitrophenol uncoupled respiration.
The activities of Trx2 and Trx reductase-2 were assessed in both populations of heart mitochondria from young adult (6-mo) and elderly (24-mo) Fischer 344 rats. We found the activities of these enzymes in both IFM and SSM were not significantly different for the young adult and elderly rats (Table 2). Data for GSSG reductase (GR) are also shown in Table 1. Again we found the GR activities in both IFM and SSM were not significantly different for the young adult and elderly rats, although there was a trend toward a small increase in the SSM (Table 2).
Table 2.
Activities of thioredoxin, thioredoxin reductase and GSSG reductase in heart mitochondria of youn][g adult and elderly Fischer 344 rats.
| Activity |
SSM |
IFM |
||
|---|---|---|---|---|
| 6-mo | 24-mo | 6-mo | 24-mo | |
| Trx | 0.38±0.04 | 0.46±0.04 | 0.45±0.07 | 0.46±0.04 |
| Trx reductase | 10.2±1.9 | 11.3±0.7 | 9.9±1.7 | 9.6±1.1 |
| GSSG reductase | 7.4±0.9 | 8.9±1.4 | 8.9±1.6 | 9.2±0.6 |
The activities of thioredoxin (Trx), thioredoxin reductase and GSSG reductase were measured in the SSM and IFM from both young adult (6–8 mo) and elderly (24–28 mo) F344 rat hearts, as described under Experimental procedures section. Values represent the mean±SEM for duplicate assays of at least three separate biological samples (n>3) and are expressed as nmol/min/mg of mitochondrial protein.
Next we focused on the mitochondrial glutaredoxins. In mammalian cells two dithiol isoforms of Grx1 and Grx2 have been characterized and found to have distinct localizations within the subcompartments of the mitochondria [12,31]. Mitochondrial Grx1 is present exclusively in the intermembrane space (IMS), segregated from Grx2, which is located in the mitochondrial matrix. We used t-Bid to selectively release Grx1 from the IMS [12], leaving Grx2 in the mitochondrial matrix. This allowed us to quantify the content and activity of Grx1 and Grx2 separately. Accordingly, heart mitochondria from young adult and elderly rats were treated with t-Bid, fractionated, and then analyzed by Western blot to determine the protein content of Grx1 and Grx2, as shown for the SSM (Fig. 1A and B); analogous results were observed for the IFM (data not shown). We found that Grx1 was fully released from the mitochondria, with no Grx1 detectable in the mitochondrial extracts or residues after t-Bid treatment. In addition to Grx1, adenylate kinase 2 (AK-2), an established biomarker for IMS localization, was also fully released by t-Bid. As expected, cytochrome c which exists in bound and free forms in the IMS was only partially released as we observed before [12]. Grx2 was not released by the t-Bid treatment, remaining with the mitochondrial extract (matrix fraction). These results are consistent with our previous report of mitochondrial Grx1 as a t-Bid-releasable protein located in the IMS, and Grx2 localized to the matrix [12].
Fig. 1.

Western blot analysis of Grx1 and Grx2 content in the SSM from young adult and elderly Fischer 344 rat hearts. Heart SSM isolated from young adult and elderly F344 rats were treated with t-Bid to release the Grx1 localized in the mitochondrial intermembrane space. Subsequent mitochondrial fractionations resulted in “Supernatant,” “Mitochondrial Extract,” and “Residues” fractions, as described in detail previously [12]. Each fraction was analyzed for Grx1, Grx2, Ak-2 and cytochrome c by Western blot. The content of Grx1 and Grx2 was quantified by comparing the density of the bands to a standard curve generated on each gel using the corresponding purified human isozymes. Representative Western blots from (A) young adult and (B) elderly rat heart SSM fractions.
We found an age-associated decrease in Grx1 content in both the SSM (~50%) and the IFM (~60%) (Fig. 2A and B). Corroborating the diminution of Grx1 in the IMS in situ, we used immunocytochemistry with gold particles and electron microscopy to localize and quantify the Grx1 in isolated heart mitochondria (Fig. 2C and D). Thus, fewer gold particle-conjugated Grx1 antibodies were observed in the outer compartment and contiguous intracristal space (IMS) of the mitochondria isolated from an elderly rat as compared to young adult. The extent of diminution of Grx1 in the IMS is comparable to the diminution of Grx1 that we observed previously in the cytosol [18], suggesting that the content of Grx1 undergoes dynamic equilibration between the cytosol and IMS during aging (see Discussion section). In contrast, the content of Grx2 was increased in the SSM (1.4-fold) and more substantially in the IFM (2.6-fold) from elderly rats as compared to young adult rats (Fig. 3A and B), suggesting different regulatory mechanisms governing the contents of mitochondrial Grx1 and Grx2. We next investigated whether the changes in Grx1 and Grx2 content corresponded to changes in their catalytic deglutathionylase activities.
Fig. 2.
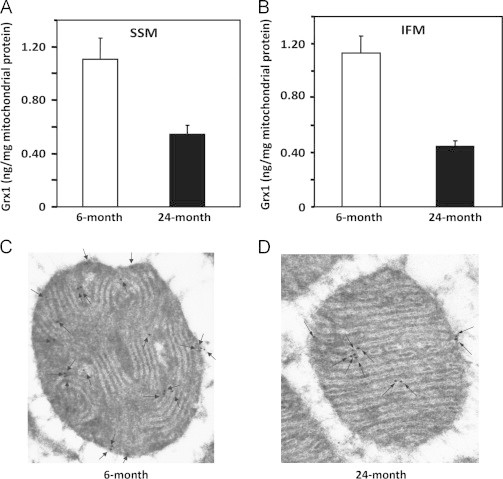
Mitochondrial Grx1 content is decreased in both populations of heart mitochondria from elderly Fischer 344 rats. Mitochondrial Grx1 contents in the SSM and IFM from young adult and elderly F344 rat heart tissues were determined by interpolation of the band density of t-Bid-released Grx1, using a standard curve relating the densities of the bands of several different amounts of purified human Grx1 protein that were analyzed on the same gels in each respective case, as described previously [12]. The values are reported as ng Grx1 per mg of mitochondrial protein (mean±SEM, n=5) for (A) SSM and (B) IFM. Diminutions of Grx1 content were observed in the IFM and SSM from the elderly rats compared to the young adult rats. The diminution of mitochondrial Grx1 content in the elderly F344 rat hearts was corroborated using gold labeling of Grx1 antibodies based on electron microscopy analysis (C) and (D). Black arrows indicate Grx1 localization in the outer mitochondrial compartment (intermembrane space) of heart mitochondria from (C) young adult and (D) elderly F344 rats. Twenty-two gold particles were counted in the mitochondrial sample from young adult rats, compared to ten that were observed in the sample from the elderly rats.
Fig. 3.
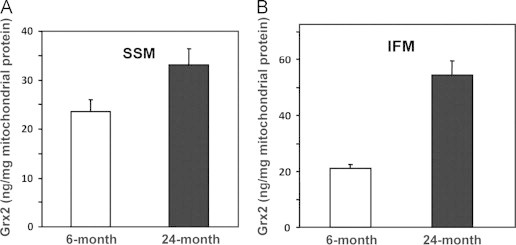
Grx2 content is increased in heart SSM and IFM from elderly Fischer 344 rats. Grx2 contents in heart (A) SSM and (B) IFM from 6-mo and 24-mo rats were processed and analyzed in a blinded fashion. Grx2 contents in the matrix fractions from the mitochondrial samples were analyzed by Western blot, using anti-Grx2 antibodies as described previously [12]. The reported values were determined by interpolation of the densities of Grx2 bands into standard curves relating the densities of the bands of several different amounts of purified human Grx2 protein that were analyzed on the same gels in each respective case. The values reported are expressed as ng Grx2 per mg mitochondrial protein (mean±SEM, n=5).
After separating Grx1 with t-Bid, the fraction containing Grx2 was then lysed, and both fractions were assayed for Grx activity. Based on their contents and their respective specific activities, the expected deglutathionylase activities for Grx1 and Grx2 were calculated (predicted), and compared to the observed activities (Table 3). The observed activities for Grx1 from SSM and IFM corresponded well to the predicted activities for both young adult and elderly rats. Thus, Grx1 activity was decreased in the heart mitochondria from the elderly rats in parallel to the decrease in content. The activities for Grx2 for the same mitochondria from elderly rats, however, corresponded to only a fraction of the predicted activities based on Grx2 protein content. This result suggested that the activity of Grx2 was partially inhibited or suppressed in the mitochondrial matrix.
Table 3.
Age-associated changes in mitochondrial Grx1 and Grx2 activities.
|
SSM |
||||
|---|---|---|---|---|
|
Predicted activity |
Observed activity |
|||
| 6-mo | 24-mo | 6-mo | 24-mo | |
| Grx1 | 0.1 | 0.05 | 0.10±0.003 | 0.062±0.004 |
| Grx2 | 0.23 | 0.34 | 0.13±0.002 | 0.16±0.004 |
|
IFM |
||||
|---|---|---|---|---|
| Predicted activity | Observed activity | |||
| 6-mo | 24-mo | 6-mo | 24-mo | |
| Grx1 | 0.115 | 0.042 | 0.10±0.004 | 0.065±0.014 |
| Grx2 | 0.21 | 0.55 | 0.15±0.005 | 0.17±0.025 |
The deglutathionylation activities (predicted according to specific protein content, and observed) of Grx1 and Grx2 in the (A) SSM and (B) IFM from young adult and elderly F344 rat hearts represent the mean±SEM for at least duplicate assays of at least three separate biological samples (n>3), expressed as nmol/min/mg of mitochondrial protein. The predicted activities of Grx1 and Grx2 were calculated based on the protein content of these enzymes in each mitochondrial population, see Fig. 1, and using the specific activity of each enzyme [31,32].
Grx2 activity can be fully realized with dithionite treatment
Previous studies have documented that a number of glutaredoxins, including mammalian Grx2, are capable of incorporating an iron–sulfur center [13,39]. Using structural protein analysis, it was determined that four cysteine residues contribute thiolate ligands to the iron–sulfur [Fe2S2] cluster; two are from GSH molecules and two from human Grx2 monomers (N-terminal catalytic Cys76) [40]. Biochemical characterizations of purified dimeric human Grx2 demonstrated that addition of GSH stabilizes but GSSG or sodium dithionite promote dissociation of the complex [13,41], as illustrated in Fig. 4 (panel A). We hypothesized that the less than predicted activity of Grx2 in the Grx Standard Assay is due to at least partial persistence of inactive Grx2 dimer bound to the iron–sulfur cluster, reflecting its natural existence in the mitochondrial matrix.
Fig. 4.
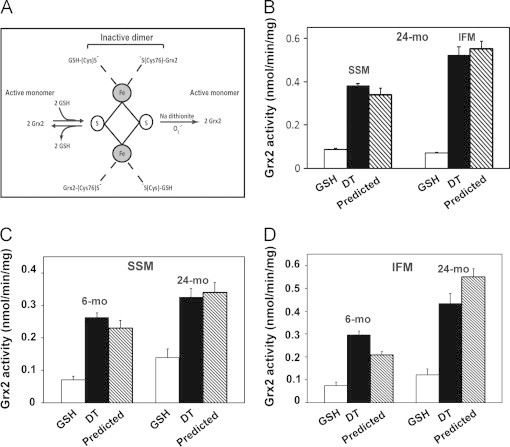
Grx2 is present as a dissociable dimeric iron-sulfur cluster complex. (A) Grx2 exists in an inactive dimeric form where the catalytic cysteine (Cys76) of each of the two monomeric Grx2 molecules is ligated reversibly to an iron moiety of the [Fe2S2] cluster. Two GSH molecules also serve as ligands. Diminution of GSH in the solution causes partial dissociation of the complex, and treatment with sodium dithionite (DT) results in full dissociation, resulting in release of two active Grx2 monomers. Superoxide radicals also are predicted to dissociate this complex, leading to activation of Grx2 under oxidative stress conditions, suggesting that the iron–sulfur cluster-bound Grx2 may function as a redox sensor in vivo[13,57]; (B) The Grx2 complex can be stabilized by mitochondrial lysis in the presence of GSH, or fully activated by sodium dithionite treatment. Following t-Bid treatment of mitochondria isolated from elderly F334 rats to release Grx1, lysates of t-Bid-treated mitochondria were analyzed for Grx2 activity (nmol/min/mg of mitochondrial protein). The mitochondrial lysates were adjusted to include either GSH (1 mM, white bars) or sodium dithionite (DT, 10 mM, black bars). The corresponding values were compared to the predicted Grx2 activity, calculated on the basis of the Grx2 content and its specific activity [31] (diagonal bar). Data represent mean±SEM, n=3; (C) and (D) Quantitative evaluation of the contribution of Grx2 to the deglutathionylation activities of (C) SSM and (D) IFM from the hearts of young adult and elderly F344 rats. Isolated heart SSM and IFM from young adult and elderly rats were lysed in RIPA buffer supplemented with either GSH (1 mM, white bar) or sodium dithionite (DT, 10 mM, black bar). The total deglutathionylation activity was then determined and the contribution of Grx2 in each case was calculated by subtracting the amount of activity attributable to Grx1 according to its known content and corresponding activity measured in previous separate experiments. Results for Grx2 are reported as mean (nmol/min/mg mitochondrial protein)±SEM (n=5 for young adult animals and n=8 for elderly animals). The values for observed Grx2 activities are compared to the predicted Grx2 activities (diagonally hatched bar), calculated from the previously determined Grx2 contents and the GRx2 specific activity.
To test this hypothesis, the heart mitochondria, after t-Bid treatment to remove Grx1, were lysed in RIPA buffer in the presence of either GSH (1 mM) to preserve the inactive complex, or sodium dithionite (10 mM) to dissociate the Grx2 monomers from the iron–sulfur cluster complex and reactivate the enzyme. These pre-treated mitochondrial lysates containing only Grx2 were then assayed for deglutathionylase activity by modifications of the Grx Standard Assay (see Experimental procedures section). Treatment with GSH maintained the limited Grx2 activity from the SSM (13%) and IFM (27%) matrix fractions from the hearts of the elderly animals. In contrast, when the lysates were treated with dithionite, the activity of Grx2 was essentially fully restored to the predicted values for both mitochondrial populations (Fig. 4B). These data suggest that most of the mitochondrial Grx2 in the hearts of elderly animals exists in the inactive dimeric complex, and this assessment is in good agreement with earlier results that used different techniques in vitro and in vivo to estimate the extent of sequestration of Grx2 in the iron–sulfur complex [13,42]. Our findings represent the first determinations of Grx2 activity in situ and provide a new analytical approach to assess the potential physiological roles of Grx2 in mitochondria.
Quantitative evaluations of in situ Grx2 monomer and dimer in mitochondria from young adult and elderly rat hearts
Selective quantification of Grx2 activity in situ was then used to estimate the monomeric and dimeric distribution of Grx2 in the heart mitochondria of young adult versus elderly rats. Consistent with the aforementioned results (Fig. 4B), limited Grx2 deglutathionylase activity was maintained by lysis in the presence of GSH, but essentially full activity was realized in most cases upon treatment with sodium dithionite (Fig. 4C and D), although the experimental variation between predicted and observed values was larger for the IFM (Fig. 4D).
Overall, both the content and the activity of mitochondrial Grx1 are decreased in the hearts of the elderly, whereas the content and activity of Grx2 are increased. However, in both young adults and elderly rats the majority of the heart mitochondrial Grx2 is inactive, likely sequestered in its dimeric iron–sulfur complex (see Discussion section).
Exogenous superoxide production by xanthine oxidase promotes reactivation of Grx2 complex
Previously, Lillig et al. demonstrated that high concentrations of hydrogen peroxide (10 mM) did not promote dissociation of the isolated complex of dimeric human recombinant Grx2 with the iron–sulfur cluster [13]. Likewise we found that treatment of mitochondrial lysates with H2O2 did not change the Grx activity (data not shown). On the other hand, the isolated complex can be dissociated by one-electron donors such as sodium dithionite only under aerobic conditions; or by one electron acceptors like ferricyanide [13]. Based on these observations and interpretation of voltametric dissociation of the complex [64], we hypothesized that the Grx2 complex could be dissociated also by superoxide, which can serve either as a one-electron donor or acceptor [43]. To test this hypothesis we used xanthine oxidase to generate superoxide radicals.
Xanthine oxidase catalyzes the oxidation of xanthine to uric acid, producing both superoxide and hydrogen peroxide [44], providing the opportunity to distinguish the effects of hydrogen peroxide and superoxide. To focus on hydrogen peroxide, liver mitochondrial lysates were treated with xanthine and xanthine oxidase, along with Mn-TMPyP (a chemical SOD mimetic). To focus on superoxide, the lysates were treated with xanthine and xanthine oxidase, along with catalase (H2O2 scavenger). To demonstrate the effectiveness of the xanthine/xanthine oxidase system+ROS scavengers, we first documented the reduction of cytochromes c and aa3 (Fig. 5) from liver mitochondrial lysates. These cytochromes are susceptible to reduction by superoxide (one-electron donor). We chose the concentration of xanthine oxidase by conducting separate experiments with isolated cytochrome c to find the minimum concentration of the enzyme (10 μM) that would facilitate rapid (≤3 min) and complete reduction of the cytochrome c (data not shown). As expected, reduction of cytochromes c and aa3 was observed when the mitochondrial lysates were treated with xanthine and xanthine oxidase, confirming production of adequate superoxide. This reduction was unchanged by the addition of catalase. Treatment with Mn-TMPyP, however, inhibited the reduction of the cytochromes, confirming diminished superoxide concentration.
Fig. 5.
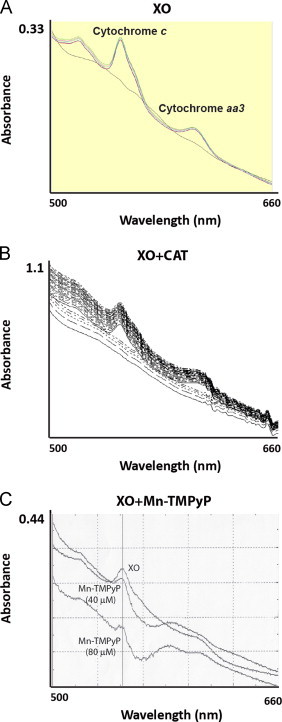
Mitochondrial cytochrome reductions in response to different ROS molecules. Isolated liver mitochondria from young F344 rats were lysed in RIPA buffer containing GSH (1 mM) and sodium cyanide (NaCN, 5 μM), a cytochrome c oxidase inhibitor that prevents the reduced form from being re-oxidized. Mitochondria samples lysed in the presence of GSH were then treated with xanthine (2 mM) and xanthine oxidase (XO, 10 μM) and either (A) vehicle, (B) catalase (CAT, 1 mg/mL) to scavenge H2O2 or (C) Mn-TMPyP (40 and 80 μM) to scavenge superoxide anions. Visible absorbance was recorded from 500 to 660 nm. The reduced forms of cytochrome c and aa3 have absorbance maxima at 550 and 600 nm, respectively.
Using this ROS-generation system, we then examined the effects of superoxide and hydrogen peroxide on the stability of the Grx2–iron–sulfur complex in heart mitochondria lysates. The activity of Grx2 in lysates of heart mitochondria was increased in the presence of xanthine oxidase to the same extent as treatment with sodium dithionite, and this activation was not inhibited by the addition of catalase (Fig. 6). These results indicate that superoxide mediates the dissociation of the inactive Grx2 dimer, and confirms that hydrogen peroxide does not.
Fig. 6.
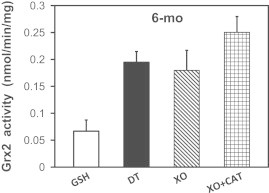
Modulation of Grx2 activity by ROS produced by the xanthine oxidase system in vitro. Isolated heart mitochondria from young adult rats were lysed in the presence of either GSH (1 mM) to maintain the inactive Grx2 complex (white bar), or sodium dithionite (10 mM, DT) to fully dissociate Grx2 (black bar). Mitochondrial samples lysed in the presence of GSH were then combined with xanthine oxidase (XO, 10 μM) to generate superoxide radicals and/or hydrogen peroxide upon metabolizing xanthine. Also added were either vehicle (diagonal bar), or catalase (checkered bar; CAT, 1 mg/mL) to selectively scavenge H2O2. The reactions were initiated by adding xanthine (2 mM), and then Grx2 activity was determined for each sample, with results reported as nmol/min/mg of mitochondrial protein (mean±SEM, n=6).
Intramitochondrial ROS generation does not promote the dissociation of Grx2 complex
Complexes I–III of the mitochondrial electron transport chain produce superoxide that is rapidly converted to hydrogen peroxide [45–47]. Inhibition of Complex I, such as by rotenone, leads to increased production of ROS particularly in the mitochondrial matrix [35]. Therefore we examined the ability of rotenone to activate Grx2 in situ by promoting intramitochondrial superoxide formation. The Grx2 activity was little affected by rotenone treatment of the mitochondria (Fig. 7). With heart mitochondria from young adult rats rotenone treatment had essentially no effect on the Grx2 activity (Fig. 7A). With the mitochondria from elderly rats, there appeared to be a slight increase in Grx2 activity with rotenone treatment (Fig. 7B), but this effect was not statistically significant (p=0.46). Treatment of the corresponding lysates with dithionite, however, still fully activated the Grx2 in both cases (Fig. 7A and B). These data challenge the concept that Grx2 deglutathionylase activity may be responsive to oxidative stress in vivo (see Discussion section).
Fig. 7.
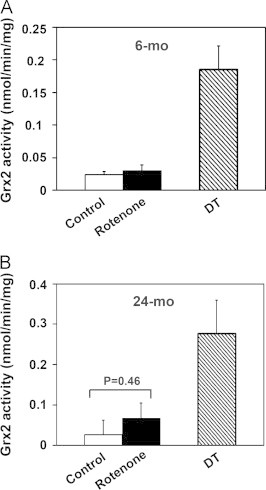
Inhibition of Complex I (intramitochondrial production of ROS) does not lead to Grx2 activation. Isolated intact heart mitochondria from (A) young adult and (B) elderly rats were incubated with glutamate (20 mM) in the absence (white bar) or presence (black bar) of rotenone (7.5 μM) for 25 min. The treated mitochondria were lysed in RIPA buffer with either GSH (1 mM, bar design) or sodium dithionite (10 mM, DT, diagonal bar). Grx2 activity in each sample was then determined, reported as nmol/min/mg of mitochondrial protein (mean±SEM, n=3–5).
Discussion
Changes in protein thiol-disulfide homeostasis in heart mitochondria from elderly animals
Intracellular redox homeostasis is tightly controlled by thiol-reducing enzymes, particularly two thiol-disulfide oxidoreductase enzyme systems: thioredoxin (Trx) and glutaredoxin (Grx). Both enzymes regulate thiol-disulfide status of cysteine residues on various proteins essential for many important cell functions, including the balance between cell survival and death [20,48,49]. Our previous studies found that diminution of cytosolic Grx1 in hearts of elderly rats contributes to an increase in the susceptibility of cardiomyocytes to oxidant-induced apoptosis through dysregulation of NF-κB activity [18]. We have now extended our study to learn that mitochondrial Grx1 and Grx2, but not thioredoxin and thioredoxin reductase, or GSSG reductase, are altered differently by age in Fischer 344 rat hearts. Another research group previously reported data for TR2 in this context [50]. Like our findings, they also reported no change in TR2 in IFM. Their value for TR2 activity in SSM from young adult rats also was very similar to our value (Table 2); however, in contrast to our finding of no change, they reported a doubling of TR2 activity in SSM from the elderly F344 rats. The basis for this discrepancy is unknown.
Diminution of Grx1 in the hearts of elderly animals
Glutaredoxin is the primary enzyme responsible for catalyzing deglutathionylation of protein-S-glutathione mixed disulfides in mammalian cells [51,52]. In the heart, the activity of Grx1 corresponds to its content in both the cytosol and the IMS, indicating that Grx1 exists in its active form throughout the cardiomyocytes. The contents of Grx1 in the cytosol and IMS decrease to about the same extent in elderly animals, suggesting that Grx1 in the IMS is equilibrated with that of the cytosolic counterpart across the outer membrane of mitochondria. The mechanism by which Grx1 is imported into the IMS is still unknown, but our current results suggest that similar or linked regulatory mechanisms control the level of Grx1 in both cellular compartments.
Since mitochondria are an essential organelle for initiation and activation of apoptosis [53,54], the age-associated diminution of Grx1 deglutathionylase activity in the IMS may contribute to the increased susceptibility to apoptosis of cardiomyocytes from the elderly [18]. Pertinent to this consideration, a number of proteins associated with the IMS have been reported to be susceptible to alterations in function by glutathionylation in vitro or in vivo [53,55]. For example, glutathionylation of exposed cysteines on components of Complex I (75 kDa subunit) and Complex II (succinate ubiquinone reductase) leads to aberrant generation of ROS and impaired respiratory function [55]. Moreover, the IMS appears to be under higher oxidative stress (compared to the cytosol and mitochondrial matrix) due to ROS from the electron transport chain and limited diffusion of GSH across the outer membrane [56]. Thus, loss of Grx1, apparently the sole deglutathionylase in the IMS, could lead to alterations in mitochondrial membrane potential, initiation of apoptotic signaling due to inhibition of respiratory chain or modification of proteins associated with the mitochondrial permeability transition pore [55], increased production of ROS, and interference with normal mitochondrial signaling. Additional studies are necessary to learn how Grx1 is decreased with aging and to distinguish among the many potential consequences of Grx1 diminution within the IMS.
Increased Grx2 in heart mitochondrial matrix of elderly animals
In contrast to Grx1, the content of Grx2 was increased in the matrix fraction of heart mitochondria from the elderly rats (Fig. 3). The mechanism by which the nuclear-encoded Grx2 is upregulated as a result of aging is unknown, providing an intriguing topic for future studies. The mitochondrial matrix deglutathionylase activity was much less than predicted according to the content of Grx2 and the known specific activity of the isolated monomeric enzyme. An inactive dimeric Grx2 complex with an iron–sulfur cluster has been characterized [13,41,57], but the in situ activity of Grx2 has not been assessed previously. We hypothesized that the lower than predicted activity of Grx2 is due to its involvement in the inactive dimeric complex in the mitochondrial matrix. To test this hypothesis, we treated mitochondrial lysates with sodium dithionite, which has been shown to dissociate human recombinant Grx2 from the iron–sulfur cluster complex in vitro [13]. We observed that the predicted activity of Grx2 in mitochondrial lysates was fully recovered by this treatment. In contrast, lysis of the mitochondria in the presence of GSH gave a preparation with low deglutathionylase activity—presumably by preserving the dimeric Grx2/iron–sulfur complex as it exists in situ. Accordingly, we conclude that a majority of the Grx2 exists as the inactive complex in the mitochondrial matrix. Nevertheless, there is a net increase in Grx2 deglutathionylase activity in the mitochondria from the elderly compared to the young adult animals (Fig. 4), suggesting an adaptive response to the oxidative stress associated with aging. However, the increase in activity is substantially smaller for the IFM because less of the total Grx2 in the IFM from the elderly rats is in the active form compared to the young adult animals. The relative diminution in deglutathionylase activity of Grx2 in the IFM may be a contributing factor to the greater sensitivity of IFM from the elderly to ischemia–reperfusion damage [6]. As discussed above for Grx1, discerning the relationship between the net deglutathionylase activity of Grx2 in the matrix and the status of S-glutathionylation of particular matrix proteins is a keen objective of future studies.
Potential role of ROS in activating Grx2 in the mitochondrial matrix
To examine whether the inactive Grx2 complex could be dissociated and reactivated in response to ROS production both in vitro and in vivo, we first tested the effects of xanthine oxidase, which generates superoxide and hydrogen peroxide. Adding catalase or a superoxide scavenger allowed us to distinguish the individual effects of superoxide or H2O2, respectively. The full activity of Grx2 could be realized with superoxide, but not with hydrogen peroxide. Since inhibition of Complex I of the mitochondrial electron transport chain is known to generate superoxide into the matrix [22,58], we tested the effect of treatment of the mitochondria with the Complex I inhibitor rotenone on the deglutathionylase activity of the matrix fraction (Grx2). Little to no activation of Grx2 was seen with rotenone treatment (Fig. 7). Either insufficient superoxide is formed in response to rotenone, or the superoxide is converted too rapidly to hydrogen peroxide (spontaneously or via Mn-SOD which is localized to the mitochondrial matrix [35,48,59]. These results suggest that the inactive Grx2 dimer is unlikely to be dissociated to enhance the deglutathionylase in response to ROS generation in mitochondria under stress conditions in vivo, unless the insult (e.g., ischemia/reperfusion) generates more superoxide in a sustained fashion than what rotenone treatment does. It is conceivable that unique spatial and temporal regulatory strategies may exist to ensure tight regulation of dimeric Grx2 by superoxide. This would allow superoxide to function locally even in the presence of antioxidant enzymes and GSH, as previously discussed [60–62].
Mechanism of dissociation of the dimeric Grx2–iron–sulfur complex
Previous studies of other iron–sulfur proteins have demonstrated dissociation of the complex through a one-electron oxidation of Fe2+ (reduced) to Fe 3+ (oxidized) [63,64]. We hypothesize that dissociation of the dimeric Grx2 complex by superoxide occurs analogously (Scheme 1).
Scheme 1.
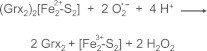
Proposed mechanism of dissociation of the dimeric Grx2–iron–sulfur complex by superoxide.
This hypothesis is supported by previous observations. As noted, Lillig et al. [13] reported that ferricyanide (one-electron oxidant) and dithionite (one-electron reductant) both dissociated the dimeric Grx2 complex under aerobic conditions. This action of dithionite appears confusing at first because the Fe(II) of the cluster would not be expected to be reduced further; i.e., Fe(I) is not a typical redox state of iron. The interpretation is clarified by another study that showed dithionite does not dissociate an analogous dimeric Grx–iron–sulfur complex under anaerobic conditions [39].Therefore the most likely mechanism by which dithionite promotes dissociation of the dimeric Grx2 complex is by donating an electron to molecular oxygen to form superoxide which then acts as a one electron oxidant as shown in Scheme 1. This conclusion is supported also by previous studies that documented formation of radicals (superoxide) when dithionite was added to aqueous solutions under aerobic conditions [65–67].
Our results suggest that the slightly increased concentration of GSSG that would be induced by rotenone does not promote the dissociation of the Grx2 complex, likely due to the relatively high GSH content in the mitochondrial matrix which stabilizes the cluster complex. Previous studies have shown that high concentration of GSSG alone can lead to the dissociation of the isolated iron–sulfur cluster complex with recombinant human Grx2 [13]. However, the physiological relevance of these experimental conditions is questionable, because even under extreme oxidative stress conditions, the ratio of GSH and GSSG among all intracellular compartments likely does not fall below 1:1, as compared to ~100:1 under normal conditions [68,69].
Alternative functions of Grx2
A role for Grx2 in cellular redox control during cell apoptosis, particularly under oxidative stress conditions, has been documented in previous studies. For example, depletion of Grx2 by selective knockout or knockdown in various cell types increases their susceptibility to apoptosis in response to oxidant insults [70,71]. In comparison, the overexpression of Grx2 in HeLa cells, cardiomyocytes and neurons has been found to be protective against apoptotic stimuli [21,72,73]. Therefore it is important to understand how Grx2 regulates specific apoptotic signaling pathways under oxidative stress conditions. Grx2 has dual functionality: deglutathionylase activity and the ability to bind an iron–sulfur cluster. For example, Grx2 is shown to catalyze deglutathionylation of the 75 kDa subunit of Complex I and restore activity, which is important for maintaining the integrity of the electron transport chain [70]. However, our results revealed that the majority of Grx2 in situ is inactive as a deglutathionylase under basal conditions and does not appear to be reactivated in response to a model oxidative stress insult (rotenone). These observations suggest that the primary physiological function of Grx2 may not relate to its enzymatic deglutathionylase activity. Accordingly, siRNA knockdown of Grx2 leads to decreased incorporation of iron into mitochondrial [Fen–Sn] proteins and corresponding loss of activity, e.g., Complex I and aconitase [74], suggesting that the Grx2 iron–sulfur cluster complex may function as a shuttle to transfer [FenSn] clusters to apo-targeted proteins in mitochondria. In our current study a significant increase of the Grx2 dimer was observed in the elderly, corresponding to a concomitant increase in associated iron–sulfur clusters. This finding suggests that the increased level of Grx2-linked iron–sulfur clusters may contribute in part to iron accumulation in the hearts of elderly rodents reported previously [75,76]. Further studies are necessary to distinguish whether mitochondrial Grx2 plays a more important role in iron–sulfur homeostasis than in sulfhydryl homeostasis.
Concluding remarks
A link between mitochondrial dysfunction and age-associated oxidative stress has been proposed previously [77], but how this dysregulation of mitochondrial redox hemostasis contributes to, or results from, the aging process remains unresolved [78]. Previous studies have shown that the IFM and SSM have distinct biochemical and functional differences, particularly with age [6]. The results of our current study demonstrate that both subpopulations exhibit a similar diminution of Grx1 content and activity with age, concomitant with an analogous diminution in cytosolic Grx1. The loss of cytosolic Grx1 was linked to diminution of NF-κB activity and decreased anti-apoptotic gene expressions [18]. The concomitant loss of Grx1 in the IMS may further exacerbate sensitivity to oxidant induced apoptosis by decreasing homeostatic regulation of the thiol status of proteins associated with the mitochondrial permeability transition pore, leading to cytochrome c release. In contrast to Grx1, we found Grx2 content was increased in the matrix of both SSM and IFM from the hearts of elderly rats, but the decreased deglutathionylase activity of the IFM matrix fraction indicated higher levels of dimeric Grx2–iron–sulfur cluster complex as compared to the SSM. This finding suggests an adaptation of iron homeostasis in the IFM correlated with age. However, accumulation of iron in mitochondria can induce ROS production via Fenton chemistry, which results in an increase in production of hydroxyl radical (OH·−), the most damaging free radical capable of oxidizing macromolecules and damaging cells [79]. This may explain in part why the IFM from the elderly rats display higher levels of oxidative stress and age-related heart dysfunction [33].
Acknowledgements
This work was supported by NIH grants R01 AG024413 (JJM) and P01 AG15885 (CLH, EJL, JK, JJM), and a Department of Veterans Affairs Merit Review Grant BX000290 (JJM). The authors are grateful to Dr. Erin M.G. Allen for reviewing and contributing to the editing of the manuscript before submission.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Levitsky J.M.a.S. In: New Solutions for the Heart. Podesser B., editor. Springer Verlag; 2011. p. 73. [Google Scholar]
- 2.Lesnefsky E.J., Chen Q., Moghaddas S., Hassan M.O., Tandler B., Hoppel C.L. Blockade of electron transport during ischemia protects cardiac mitochondria. J. Biol. Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 3.Lesnefsky E.J., Lundergan C.F., Hodgson J.M., Nair R., Reiner J.S., Greenhouse S.W., Califf R.M, Ross A.M. Increased left ventricular dysfunction in elderly patients despite successful thrombolysis: the GUSTO-I angiographic experience. J. Am. Coll. Cardiol. 1996;28:331–337. doi: 10.1016/0735-1097(96)00148-9. [DOI] [PubMed] [Google Scholar]
- 4.Lesnefsky E.J., Minkler P., Hoppel C.L. Enhanced modification of cardiolipin during ischemia in the aged heart. J. Mol. Cell. Cardiol. 2009;46:1008–1015. doi: 10.1016/j.yjmcc.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Wojtovich A.P., Nadtochiy S.M., Brookes P.S., Nehrke K. Ischemic preconditioning: the role of mitochondria and aging. Exp. Gerontol. 2012;47:1–7. doi: 10.1016/j.exger.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lesnefsky E.J., Hoppel C.L. Ischemia–reperfusion injury in the aged heart: role of mitochondria. Arch. Biochem. Biophys. 2003;420:287–297. doi: 10.1016/j.abb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 7.Garlid K.D., Dos Santos P., Xie Z.J., Costa A.D., Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K(+) channel in cardiac function and cardioprotection. Biochim. Biophys. Acta. 2003;1606:1–21. doi: 10.1016/s0005-2728(03)00109-9. [DOI] [PubMed] [Google Scholar]
- 8.Cao Y., Zhang S.Z., Zhao S.Q., Bruce I.C. The mitochondrial Ca(2+)-activated K(+) channel contributes to cardioprotection by limb remote ischemic preconditioning in rat. Life Sci. 2011;88:1026–1030. doi: 10.1016/j.lfs.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka-Esposito C., Chen Q., Lesnefsky E.J. Blockade of electron transport before ischemia protects mitochondria and decreases myocardial injury during reperfusion in aged rat hearts. Transl. Res. 2012;160:207–216. doi: 10.1016/j.trsl.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choksi K.B., Papaconstantinou J. Age-related alterations in oxidatively damaged proteins of mouse heart mitochondrial electron transport chain complexes. Free Radical Biol. Med. 2008;44:1795–1805. doi: 10.1016/j.freeradbiomed.2008.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer Y., Buchanan B.B., Vignols F., Reichheld J.P. Thioredoxins and glutaredoxins: unifying elements in redox biology. Annu. Rev. Genet. 2009;43:335–367. doi: 10.1146/annurev-genet-102108-134201. [DOI] [PubMed] [Google Scholar]
- 12.Pai H.V., Starke D.W., Lesnefsky E.J., Hoppel C.L., Mieyal J.J. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid Redox Signal. 2007;9:2027–2033. doi: 10.1089/ars.2007.1642. [DOI] [PubMed] [Google Scholar]
- 13.Lillig C.H., Berndt C., Vergnolle O., Lonn M.E., Hudemann C., Bill E., Holmgren A. Characterization of human glutaredoxin 2 as iron–sulfur protein: a possible role as redox sensor. Proc. Nat. Acad. Sci. U.S.A. 2005;102:8168–8173. doi: 10.1073/pnas.0500735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berndt C., Lillig C.H., Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1227–1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 15.Murata H., Ihara Y., Nakamura H., Yodoi J., Sumikawa K., Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y., Shi W., Cui N., Wu Z., Jiang C. Oxidative stress inhibits vascular K(ATP) channels by S-glutathionylation. J. Biol. Chem. 2010;285:38641–38648. doi: 10.1074/jbc.M110.162578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang H., Viola H.M., Filipovska A., Hool L.C. Ca(v)1.2 calcium channel is glutathionylated during oxidative stress in guinea pig and ischemic human heart. Free Radical Biol. Med. 2011;51:1501–1511. doi: 10.1016/j.freeradbiomed.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Gallogly M.M., Shelton M.D., Qanungo S., Pai H.V., Starke D.W., Hoppel C.L., Lesnefsky E.J., Mieyal J.J. Glutaredoxin regulates apoptosis in cardiomyocytes via NFkappaB targets Bcl-2 and Bcl-xL: implications for cardiac aging. Antioxid Redox Signal. 2010;12:1339–1353. doi: 10.1089/ars.2009.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cross J.V., Templeton D.J. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen E.M., Mieyal J.J. Protein-thiol oxidation and cell death: regulatory role of glutaredoxins. Antioxid Redox Signal. 2012;17:1748–1763. doi: 10.1089/ars.2012.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diotte N.M., Xiong Y., Gao J., Chua B.H., Ho Y.S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Biophys. Acta. 2009;1793:427–438. doi: 10.1016/j.bbamcr.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 22.Hurd T.R., Requejo R., Filipovska A., Brown S., Prime T.A., Robinson A.J., Fearnley I.M., Murphy M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008;283:24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gravina S. Case Western Reserve University; Cleveland: 1993. Characterization and Kinetic Mechanism of Thiol-transferase. in Pharmacology. [Google Scholar]
- 24.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 25.Gravina S.A., Mieyal J.J. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32:3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 26.Starke D.W., Chen Y., Bapna C.P., Lesnefsky E.J., Mieyal J.J. Sensitivity of protein sulfhydryl repair enzymes to oxidative stress. Free Radical Biol. Med. 1997;23:373–384. doi: 10.1016/s0891-5849(97)00009-9. [DOI] [PubMed] [Google Scholar]
- 27.Hoppel C., DiMarco J.P., Tandler B. Riboflavin and rat hepatic cell structure and function. Mitochondrial oxidative metabolism in deficiency states. J. Biol. Chem. 1979;254:4164–4170. [PubMed] [Google Scholar]
- 28.Hoppel C.L., Kerr D.S., Dahms B., Roessmann U. Deficiency of the reduced nicotinamide adenine dinucleotide dehydrogenase component of complex I of mitochondrial electron transport. Fatal infantile lactic acidosis and hypermetabolism with skeletal–cardiac myopathy and encephalopathy. J. Clin. Invest. 1987;80:71–77. doi: 10.1172/JCI113066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puchowicz M.A., Varnes M.E., Cohen B.H., Friedman N.R., Kerr D.S., Hoppel C.L. Oxidative phosphorylation analysis: assessing the integrated functional activity of human skeletal muscle mitochondria—case studies. Mitochondrion. 2004;4:377–385. doi: 10.1016/j.mito.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Chance B., Williams G.R. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J. Biol. Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- 31.Gallogly M.M., Starke D.W., Leonberg A.K., Ospina S.M., Mieyal J.J. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry. 2008;47:11144–11157. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jao S.C., English Ospina S.M., Berdis A.J., Starke D.W., Post C.B., Mieyal J.J. Computational and mutational analysis of human glutaredoxin (thioltransferase): probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis. Biochemistry. 2006;45:4785–4796. doi: 10.1021/bi0516327. [DOI] [PubMed] [Google Scholar]
- 33.Judge S., Jang Y.M., Smith A., Hagen T., Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 34.Moghaddas S., Hoppel C.L., Lesnefsky E.J. Aging defect at the QO site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch. Biochem. Biophys. 2003;414:59–66. doi: 10.1016/s0003-9861(03)00166-8. [DOI] [PubMed] [Google Scholar]
- 35.Chen Q., Vazquez E.J., Moghaddas S., Hoppel C.L., Lesnefsky E.J. Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 36.Palmer J.W., Tandler B., Hoppel C.L. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- 37.Fannin S.W., Lesnefsky E.J., Slabe T.J., Hassan M.O., Hoppel C.L. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch. Biochem. Biophys. 1999;372:399–407. doi: 10.1006/abbi.1999.1508. [DOI] [PubMed] [Google Scholar]
- 38.Lesnefsky E.J., Gudz T.I., Moghaddas S., Migita C.T., Ikeda-Saito M., Turkaly P.J., Hoppel C.L. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J. Mol. Cell. Cardiol. 2001;33:37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- 39.Rouhier N., Unno H., Bandyopadhyay S., Masip L., Kim S.K., Hirasawa M., Gualberto J.M., Lattard V., Kusunoki M., Knaff D.B., Georgiou G., Hase T., Johnson M.K., Jacquot J.P. Functional, structural, and spectroscopic characterization of a glutathione-ligated [2Fe-2S] cluster in poplar glutaredoxin C1. Proc. Nat. Acad. Sci. U.S.A. 2007;104:7379–7384. doi: 10.1073/pnas.0702268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johansson C., Kavanagh K.L., Gileadi O., Oppermann U. Reversible sequestration of active site cysteines in a 2Fe–2S-bridged dimer provides a mechanism for glutaredoxin 2 regulation in human mitochondria. J. Biol. Chem. 2007;282:3077–3082. doi: 10.1074/jbc.M608179200. [DOI] [PubMed] [Google Scholar]
- 41.Berndt C., Hudemann C., Hanschmann E.M., Axelsson R., Holmgren A., Lillig C.H. How does iron–sulfur cluster coordination regulate the activity of human glutaredoxin 2? Antioxid Redox Signal. 2007;9:151–157. doi: 10.1089/ars.2007.9.151. [DOI] [PubMed] [Google Scholar]
- 42.Hoff K.G., Culler S.J., Nguyen P.Q., McGuire R.M, Silberg J.J., Smolke C.D. In vivo fluorescent detection of Fe–S clusters coordinated by human GRX2. Chem. Biol. 2009;16:1299–1308. doi: 10.1016/j.chembiol.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 43.Winterbourn C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 44.McCord J.M., Fridovich I. The reduction of cytochrome c by milk xanthine oxidase. J. Biol. Chem. 1968;243:5753–5760. [PubMed] [Google Scholar]
- 45.Kussmaul L., Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Nat. Acad. Sci. U.S.A. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raha S., Robinson B.H. Mitochondria, oxygen free radicals, and apoptosis. Am. J. Med. Genet. 2001;106:62–70. doi: 10.1002/ajmg.1398. [DOI] [PubMed] [Google Scholar]
- 47.Muller F.L., Liu Y., Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 48.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Handy D.E., Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–1367. doi: 10.1089/ars.2011.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suh J.H., Heath S.H., Hagen T.M. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radical Biol. Med. 2003;35:1064–1072. doi: 10.1016/s0891-5849(03)00468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ho Y.S., Xiong Y., Ho D.S., Gao J., Chua B.H., Pai H., Mieyal J.J. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radical Biol. Med. 2007;43:1299–1312. doi: 10.1016/j.freeradbiomed.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chrestensen C.A., Starke D.W., Mieyal J.J. Acute cadmium exposure inactivates thioltransferase (glutaredoxin), inhibits intracellular reduction of protein–glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 2000;275:26556–26565. doi: 10.1074/jbc.M004097200. [DOI] [PubMed] [Google Scholar]
- 53.Ferri K.F., Kroemer G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 2001;3:E255–263. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- 54.Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 55.Sabens Liedhegner E.A., Gao X.H., Mieyal J.J. Mechanisms of altered redox regulation in neurodegenerative diseases—focus on S-glutathionylation. Antioxid Redox Signal. 2012;16:543–566. doi: 10.1089/ars.2011.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herrmann J.M., Riemer J. The intermembrane space of mitochondria. Antioxid Redox Signal. 2010;13:1341–1358. doi: 10.1089/ars.2009.3063. [DOI] [PubMed] [Google Scholar]
- 57.Mitra S., Elliott S.J. Oxidative disassembly of the [2Fe–2S] cluster of human Grx2 and redox regulation in the mitochondria. Biochemistry. 2009;48:3813–3815. doi: 10.1021/bi900112m. [DOI] [PubMed] [Google Scholar]
- 58.Chen Q., Moghaddas S., Hoppel C.L., Lesnefsky E.J. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell Physiol. 2008;294:C460–466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 59.Finkel T. Radical medicine: treating ageing to cure disease. Nat. Rev. Mol. Cell Biol. 2005;6:971–976. doi: 10.1038/nrm1763. [DOI] [PubMed] [Google Scholar]
- 60.Al-Mehdi A.B., Pastukh V.M., Swiger B.M., Reed D.J., Patel M.R., Bardwell G.C., Pastukh V.V., Alexeyev M.F., Gillespie M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012:5. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kil I.S., Lee S.K., Ryu K.W., Woo H.A., Hu M.C., Bae S.H., Rhee S.G. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell. 2012;46:584–594. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 62.Rhee S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 63.Kuo C.F., Mashino T., Fridovich I. alpha, beta-Dihydroxyisovalerate dehydratase. A superoxide-sensitive enzyme. J. Biol. Chem. 1987;262:4724–4727. [PubMed] [Google Scholar]
- 64.Flint D.H., Tuminello J.F., Emptage M.H. The inactivation of Fe–S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 1993;268:22369–22376. [PubMed] [Google Scholar]
- 65.Hodgson W.G., Neaves A., Parker C.A. Detection of free radicals in sodium dithionite by paramagnetic resonance. Nature. 1956;178:489. (-489) [Google Scholar]
- 66.Archer S.L., Hampl V., Nelson D.P., Sidney E., Peterson D.A., Weir E.K. Dithionite increases radical formation and decreases vasoconstriction in the lung. Evidence that dithionite does not mimic alveolar hypoxia. Circ. Res. 1995;77:174–181. doi: 10.1161/01.res.77.1.174. [DOI] [PubMed] [Google Scholar]
- 67.Aviram I., Sharabani M. Kinetic studies of the reduction of neutrophil cytochrome b-558 by dithionite. Biochem. J. 1986;237:567–572. doi: 10.1042/bj2370567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hanson G.T., Aggeler R., Oglesbee D., Cannon M., Capaldi R.A., Tsien R.Y., Remington S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 69.Hwang C., Sinskey A.J., Lodish H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 70.Wu H., Lin L., Giblin F., Ho Y.S., Lou M.F. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radical Biol. Med. 2011;51:2108–2117. doi: 10.1016/j.freeradbiomed.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lillig C.H., Lonn M.E., Enoksson M., Fernandes A.P., Holmgren A. Short interfering RNA-mediated silencing of glutaredoxin 2 increases the sensitivity of HeLa cells toward doxorubicin and phenylarsine oxide. Proc. Nat. Acad. Sci. U.S.A. 2004;101:13227–13232. doi: 10.1073/pnas.0401896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferri A., Fiorenzo P., Nencini M., Cozzolino M., Pesaresi M.G., Valle C., Sepe S., Moreno S., Carri M.T. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 2010;19:4529–4542. doi: 10.1093/hmg/ddq383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Enoksson M., Fernandes A.P., Prast S., Lillig C.H., Holmgren A., Orrenius S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem. Biophys. Res. Commun. 2005;327:774–779. doi: 10.1016/j.bbrc.2004.12.067. [DOI] [PubMed] [Google Scholar]
- 74.Lee D.W., Kaur D., Chinta S.J., Rajagopalan S., Andersen J.K. A disruption in iron–sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: implications for Parkinson's disease. Antioxid Redox Signal. 2009;11:2083–2094. doi: 10.1089/ars.2009.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arvapalli R.K., Paturi S., Laurino J.P., Katta A., Kakarla S.K., Gadde M.K., Wu M., Rice K.M., Walker E.M., Wehner P., Blough E.R. Deferasirox decreases age-associated iron accumulation in the aging F344XBN rat heart and liver. Cardiovasc. Toxicol. 2010;10:108–116. doi: 10.1007/s12012-010-9068-9. [DOI] [PubMed] [Google Scholar]
- 76.Xu J., Jia Z., Knutson M.D., Leeuwenburgh C. Impaired iron status in aging research. Int. J. Mol. Sci. 2012;13:2368–2386. doi: 10.3390/ijms13022368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Finkel T., Holbrook N.J. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 78.Labunskyy V.M., Gladyshev V.N. Antioxid Redox Signal; 2012. Role of Reactive Oxygen Species-Mediated Signaling in Aging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Halliwell B. John Wiley & Sons, Ltd; 2001. Free Radicals and Other Reactive Species in Disease. in eLS. [Google Scholar]